
Advances in Cereal Crop Genomics for Resilience under Climate Change


Abstract
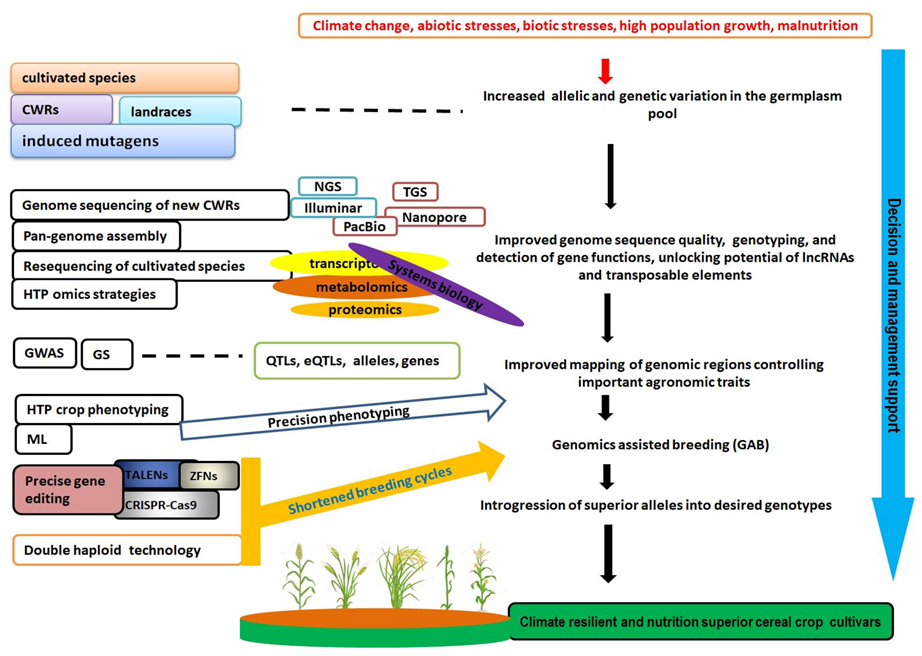
:1. Introduction
2. De Novo Domestication of Crop Wild Relatives and Better Exploitation of Orphan Crop Species
3. Advances in DNA Sequencing Technologies Accelerating Traits Discovery and Decoding Crop Species’ Whole Genomes

{kind=link}
| Species Name | Ploid Level | Genome Size | Assembled Genome (%) | 1 Repeat Elements (%) | GC % | Genes | 2 Sequencing Strategy | 3 Public Year | References |
|---|---|---|---|---|---|---|---|---|---|
| Triticum aestivum | 2n = 6x = 42 (AABBDD) allopolyploid | ~17 Gb | 14.5 Gb (85.29) | 85.00 | 48.25 | 107,891 | De novo WGS + BAC assemblies | 2018 | [12,86,92] |
| 4 Triticum urartu | 2n = 6x = 42 (AABBDD) | 4.94 Gb | 3.92 Gb (79.35) | 66.88 | 46.00 | 34,879 | WGS + Illumina | 2013 | [12,93] |
| Oryza sativa | 2n = 2x = 24 | 389 Mb | 370 Mb (95.12) | ~51.00 | ~43.58–43.73 | 35,679 | BAC PMs + Sanger seq. | 2005 | [12,35,82] |
| Zea mays | 2n = 2x = 20 | 2.3 Gb | 2.048 Gb (89.04) | 85.00 | 46.91 | 47,800 | BAC PMs + BAC seq. | 2009 | [35,83,86] |
| Secale cereale | 2n = 2x = 14, RR | 7.86 Gb | 7.74 Gb (98.47) | 90.31 | 45.89 | 45,596 | PacBio + short read Illumina + Hi-C + Bio-Nano | 2021 | [94] |
| Pearl millet | 2n = 2x = 14 | ~1.79 Gb | 1.76 Gb (98.32) | 68.16 | 47.90 | 38,579 | WGS + BAC | 2017 | [73] |
| Sorghum bicolor (v1) | 2n = 2x = 20 | ~730 Mb | 625.6 Mb (85.70) | ~63.00 | 44.50 | ~27,640 | WGS + BACs + Sanger | 2009 | [35,84] |
| Sorghum bicolor (v3) | 2n = 2x = 20 | ~700 Mb | 655.2 Mb (93.60) | 62.70 | 44.50 | 34,211 | Deep WG short read seq. + Sanger + BAC PMs | 2018 | [90] |
| Eleusine coracana | 2n = 4x = 36 (AABB) | 1.45 Gb | 1.19 Gb (82.31) | 49.92 | 44.80 | 85,243 | WGS + Illumina paired-end | 2017 | [12,95] |
| Hordeum vulgare | 2n = 2x = 14 | 5.1 Gb | 4.56 Gb (89.41) | ~84.00 | 44.40 | 26,159 | WGS | 2012 | [35,96] |
| Setaria italica | 2n = 2x = 18 | ~490 Mb | ~423 Mb (86.33) | ~46.30 | 46.17 | 38,801 | WGS + NGS | 2012 | [12,75] |
| Setaria italica | 2n = 2x = 18 | ~510 Mb | ~400 Mb (78.43) | ~40.00 | 46.17 | 24,000– 29,000 | WGS + Sanger + Illumina + BAC end seq. | 2012 | [12,35,74] |
| Eragrotis tef | 2C = 2n = 4x = 40 | 772 Mb | 672 Mb (87.05) | 22.40 | 45.50 | 28,113 | Illumina HighSeq 2000 single and paired-end | 2014 | [12,32] |
| Digitaria exilis | 2n = 4x = 36 | 701.66 Mb | 655.72 Mb (91.5) | 49.00 | - | 57,021 | Deep seq. of short reads + Illumina paired-end + Hi-C + Bionano optical map | 2020 | [32,97] |
4. Approaches in Mapping of Genomic Regions Controlling Variation of Quantitatively Inherited Traits
5. Broadening Crop Genetic Diversity through Mutagenesis
6. Use of Sequence Specific Nucleases for Precise Gene Editing for Crop Improvements
7. Double Haploid Technique as a Tool for Accelerated Crop Breeding for Climate Resilience
8. The Integral Role of Crop Phenotyping in Complementing Crop Genotyping
9. Unlocking the Roles of Plant Long Non-Coding RNAs (lncRNAs) in Regulating Plant Stress Responses and Adaptation
10. Pan-Genomics, Transposable Elements, and Machine Learning Hold Promise for Crop Improvement Getting into the Future
10.1. Pan-Genomics Facilitating Better Understanding and Utilization of Broader Crop Genetic Diversity for Accelerated Crop Improvement
10.2. Transposable Elements as Research Target for Decoding Crop Genomes and Understanding Crop Responses to Biotic and Abiotic Stresses
10.3. Machine Learning as a Powerful Tool for Gene Function Prediction and High-Throughput Field and Stress Phenotyping
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bevan, M.; Waugh, R. Applying plant genomics to crop improvement. Genome Biol. 2007, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Scheben, A.; Yuan, Y.; Edwards, D. Advances in genomics for adapting crops to climate change. Curr. Plant Biol. 2016, 6, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Hendre, P.S.; Muthemba, S.; Kariba, R.; Muchugi, A.; Fu, Y.; Chang, Y.; Song, B.; Liu, H.; Liu, M.; Liao, X.; et al. African Orphan Crops Consortium (AOCC): Status of developing genomic resources for African orphan crops. Planta 2019, 250, 989–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, D.; Borelli, T.; Beltrame, D.M.; Oliveira, C.N.; Coradin, L.; Wasike, V.W.; Wasilwa, L.; Mwai, J.; Manjella, A.; Samarasinghe, G.W.; et al. The potential of neglected and underutilized species for improving diets and nutrition. Planta 2019, 250, 709–729. [Google Scholar] [CrossRef]
- Sarwar, M.H.; Sarwar, M.F.; Sarwar, M.; Qadri, N.A.; Moghal, S. The importance of cereals (Poacea: Gramineae) nutrition in human health: A review. J. Cereals Oilseeds 2013, 4, 32–35. [Google Scholar] [CrossRef]
- Fatima, Z.; Ahmed, M.; Hussain, M.; Abbas, G.; Ul-Allah, S.; Ahmad, S.; Ahmed, N.; Ali, M.A.; Sarwar, G.; ul Haque, E.; et al. The fingerprints of climate warming on cereal crops phenology and adaptation options. Sci. Rep. 2020, 10, 1–21. [Google Scholar] [CrossRef]
- Macauley, H.; Ramadjita, T. Cereal Crops: Rice, Maize, Millet, Sorghum, Wheat: Background Paper, Feeding Africa, 21–23 October 2015, Dakar, Senegal; The African Development Bank Group and the African Union: Abidjan, Ivory Coast, 2015; pp. 1–31. [Google Scholar]
- Shiferaw, B.; Prasanna, B.M.; Hellin, J.; Bänziger, M. Crops that feed the world 6. Past successes and future challenges to the role played by maize in global food security. Food Secur. 2011, 3, 307–327. [Google Scholar] [CrossRef] [Green Version]
- Godfray, H.C.J.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food security: The challenge of feeding 9 billion people. Science 2010, 327, 812–818. [Google Scholar] [CrossRef] [Green Version]
- Gärtner, P. Cereal Crops Fighting the Climate Chaos. (20 January 2021). Available online: https://phys.org/news/2021-01-cereal-crops-climate-chaos.html (accessed on 15 March 2021).
- Goron, T.L.; Raizada, M.N. Genetic diversity and genomic resources available for the small millet crops to accelerate a New Green Revolution. Front. Plant Sci. 2015, 6, 157. [Google Scholar] [CrossRef] [Green Version]
- Mohanta, T.K.; Bashir, T.; Hashem, A.; Abd_Allah, E.F. Systems biology approach in plant abiotic stresses. Plant Physiol. Biochem. 2017, 121, 58–73. [Google Scholar] [CrossRef]
- Ahmed, K.F.; Wang, G.; Yu, M.; Koo, J.; You, L. Potential impact of climate change on cereal crop yield in West Africa. Clim. Chang. 2015, 133, 321–334. [Google Scholar] [CrossRef]
- Wang, J.; Vanga, S.K.; Saxena, R.; Orsat, V.; Raghavan, V. Effect of climate change on the yield of cereal crops: A review. Climate 2018, 6, 41. [Google Scholar] [CrossRef] [Green Version]
- Gustin, G. Climate Change Could Lead to Major Crop Failures in World’s Biggest Corn Regions: Politics and Policy. Inside Climate News. (11 June 2018). Available online: https://insideclimatenews.org/news/11062018/climate-change-research-food-security-agriculture-impacts-corn-vegetables-crop-prices/ (accessed on 26 February 2021).
- Reynolds, M.P.; Quilligan, E.; Aggarwal, P.K.; Bansal, K.C.; Cavalieri, A.J.; Chapman, S.C.; Chapotin, S.M.; Datta, S.K.; Duveiller, E.; Gill, K.S.; et al. An integrated approach to maintaining cereal productivity under climate change. Glob. Food Secur. 2016, 8, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Ahsan, F.; Chandio, A.A.; Fang, W. Climate change impacts on cereal crops production in Pakistan. Int. J. Clim. Chang. Strateg. Manag. 2020, 12, 257–269. [Google Scholar] [CrossRef]
- Li, M. Climate Change to Adversely Impact Grain Production in China by 2030; IFPRI: Washington, DC, USA, 2018. [Google Scholar]
- Eigenbrode, S.D.; Binns, W.P.; Huggins, D.R. Confronting climate change challenges to dryland cereal production: A call for collaborative, transdisciplinary research, and producer engagement. Front. Ecol. Evol. 2018, 5, 164. [Google Scholar] [CrossRef] [Green Version]
- Pourkheirandish, M.; Golicz, A.A.; Bhalla, P.L.; Singh, M.B. Global role of crop genomics in the face of climate change. Front. Plant Sci. 2020, 11, 922. [Google Scholar] [CrossRef]
- Qaim, M. Globalisation of Agrifood Systems and Sustainable Nutrition. Proc. Nutr. Soc. 2017, 76, 12–21. [Google Scholar] [CrossRef]
- Qaim, M. Role of new plant breeding technologies for food security and sustainable agricultural development. Appl. Econ. Perspect. Policy 2020, 42, 129–150. [Google Scholar] [CrossRef]
- Kole, C.; Muthamilarasan, M.; Henry, R.; Edwards, D.; Sharma, R.; Abberton, M.; Batley, J.; Bentley, A.; Blakeney, M.; Bryant, J.; et al. Application of genomics-assisted breeding for generation of climate resilient crops: Progress and prospects. Front. Plant Sci. 2015, 6, 563. [Google Scholar] [CrossRef] [Green Version]
- Kahane, R.; Hodgkin, T.; Jaenicke, H.; Hoogendoorn, C.; Hermann, M.; Hughes, J.D.A.; Padulosi, S.; Looney, N. Agrobiodiversity for food security, health and income. Agron. Sustain. Dev. 2013, 33, 671–693. [Google Scholar] [CrossRef] [Green Version]
- Dawson, I.K.; Powell, W.; Hendre, P.; Bančič, J.; Hickey, J.M.; Kindt, R.; Hoad, S.; Hale, I.; Jamnadass, R. The role of genetics in mainstreaming the production of new and orphan crops to diversify food systems and support human nutrition. New Phytol. 2019, 224, 37–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilian, B.; Dempewolf, H.; Guarino, L.; Werner, P.; Coyne, C.; Warburton, M.L. Crop Science special issue: Adapting agriculture to climate change: A walk on the wild side. Crop Sci. 2020, 61, 32–36. [Google Scholar] [CrossRef]
- Ghatak, A.; Chaturvedi, P.; Bachmann, G.; Valledor, L.; Ramšak, Ž.; Bazargani, M.M.; Bajaj, P.; Jegadeesan, S.; Li, W.; Sun, X.; et al. Physiological and Proteomic Signatures Reveal Mechanisms of Superior Drought Resilience in Pearl Millet Compared to Wheat. Front. Plant Sci. 2021, 11, 600278. [Google Scholar] [CrossRef]
- Ray, D.K.; Mueller, N.D.; West, P.C.; Foley, J.A. Yield trends are insufficient to double global crop production by 2050. PLoS ONE 2013, 8, e66428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biello, D. Cereal killer: Climate Change Stunts Growth of Global Crop Yields. Sci. Am. Retrieved Jan. 2011, 4, 2012. [Google Scholar]
- Gao, C. Genome engineering for crop improvement and future agriculture. Cell 2021, 184, 1621–1635. [Google Scholar] [CrossRef]
- Mabhaudhi, T.; Chimonyo, V.G.P.; Hlahla, S.; Massawe, F.; Mayes, S.; Nhamo, L.; Modi, A.T. Prospects of orphan crops in climate change. Planta 2019, 250, 695–708. [Google Scholar] [CrossRef] [Green Version]
- Cannarozzi, G.; Plaza-Wüthrich, S.; Esfeld, K.; Larti, S.; Wilson, Y.S.; Girma, D.; de Castro, E.; Chanyalew, S.; Blösch, R.; Farinelli, L.; et al. Genome and transcriptome sequencing identifies breeding targets in the orphan crop tef (Eragrostis tef). BMC Genom. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Liu, H.; Liu, M.; Liao, X.; Sahu, S.K.; Fu, Y.; Song, B.; Cheng, S.; Kariba, R.; Muthemba, S.; et al. The draft genomes of five agriculturally important African orphan crops. GigaScience 2019, 8, giy152. [Google Scholar] [CrossRef]
- Rodríguez, J.P.; Rahman, H.; Thushar, S.; Singh, R.K. Healthy and resilient cereals and pseudo-cereals for marginal agriculture: Molecular advances for improving nutrient bioavailability. Front Genet. 2020, 11, 49. [Google Scholar] [CrossRef] [Green Version]
- Bevan, M.W.; Uauy, C. Genomics reveals new landscapes for crop improvement. Genome Biol. 2013, 14, 206. Available online: http://genomebiology.com/2013/14/6/206 (accessed on 16 March 2021). [CrossRef] [Green Version]
- Campos-de Quiroz, H. Plant genomics: An overview. Biol. Res. 2002, 35, 385–399. [Google Scholar] [CrossRef]
- Terryn, N.; Rouzé, P.; Van Montagu, M. Plant genomics. FEBS Lett. 1999, 452, 3–6. [Google Scholar] [CrossRef]
- Akpınar, B.A.; Lucas, S.J.; Budak, H. Genomics approaches for crop improvement against abiotic stress. Sci. World J. 2013, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Prasad, A.; Muthamilarasan, M.; Parida, S.K.; Prasad, M. Breeding and biotechnological interventions for trait improvement: Status and prospects. Planta 2020, 252, 1–18. [Google Scholar] [CrossRef]
- Abdeeva, I.; Abdeev, R.; Bruskin, S.; Piruzian, E. Transgenic plants as a tool for plant functional genomics. In Transgenic Plants-Advances and Limitations; IntechOpen: London, UK, 2012; pp. 259–284. [Google Scholar]
- Singh, B.; Salaria, N.; Thakur, K.; Kukreja, S.; Gautam, S.; Goutam, U. Functional Genomic Approaches to Improve Crop Plant Heat Stress Tolerance [version 1; peer review: 2 approved, 1 approved with reservations]. F1000Research 2019, 8, 1721. [Google Scholar] [CrossRef]
- Bohra, A.; Chand Jha, U.; Godwin, I.D.; Kumar Varshney, R. Genomic interventions for sustainable agriculture. Plant Biotechnol. J. 2020, 18, 2388–2405. [Google Scholar] [CrossRef]
- Bansal, K.C.; Lenka, S.K.; Mondal, T.K. Genomic resources for breeding crops with enhanced abiotic stress tolerance. Plant Breed. 2014, 133, 1–11. [Google Scholar] [CrossRef]
- Kamenya, S.N.; Mikwa, E.O.; Song, B.; Odeny, D.A. Genetics and breeding for climate change in Orphan crops. Theor. Appl. Genet. 2021, 1–29. [Google Scholar] [CrossRef]
- Gupta, A.; Rico-Medina, A.; Caño-Delgado, A.I. The physiology of plant responses to drought. Science 2020, 368, 266–269. [Google Scholar] [CrossRef]
- Kumar, A.; Tomer, V.; Kaur, A.; Kumar, V.; Gupta, K. Millets: A solution to agrarian and nutritional challenges. Agric. Food Secur. 2018, 7, 1–15. [Google Scholar] [CrossRef]
- Ananda, G.K.S.; Myrans, H.; Norton, S.L.; Gleadow, R.; Furtado, A.; Henry, R.J. Wild Sorghum as a Promising Resource for Crop Improvement. Front. Plant Sci. 2020, 11, 1108. [Google Scholar] [CrossRef]
- Choudhary, M.; Singh, V.; Muthusamy, V.; Wani, S.H. Harnessing crop wild relatives for crop improvement. LS-An Int. J. Life Sci. 2017, 6, 73–85. [Google Scholar] [CrossRef]
- Tuberosa, R.; Salvi, S. Genomics-based approaches to improve drought tolerance of crops. Trends Plant Sci. 2006, 11, 405–412. [Google Scholar] [CrossRef]
- Brozynska, M.; Furtado, A.; Henry, R.J. Genomics of crop wild relatives: Expanding the gene pool for crop improvement. Plant Biotechnol. J. 2016, 14, 1070–1085. [Google Scholar] [CrossRef]
- Kofsky, J.; Zhang, H.; Song, B.-H. The Untapped Genetic Reservoir: The Past, Current, and Future Applications of the Wild Soybean (Glycine soja). Front. Plant Sci. 2018, 9, 949. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.W.; Garg, V.; Roorkiwal, M.; Golicz, A.A.; Edwards, D.; Varshney, R.K. Super-pangenome by integrating the wild side of a species for accelerated crop improvement. Trends Plant Sci. 2020, 25, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.C.; de Wet, M.J.; Harlan, J.R. Morphology of Saccharum- Sorghum hybrid derivatives. Am. J. Bot. 1978, 65, 936–942. [Google Scholar] [CrossRef]
- Jannoo, N.; Grivet, L.; Chantret, N.; Garsmeur, O.; Glaszmann, J.-C.; Arruda, P.; D’Hont, A. Orthologous comparison in a gene-rich region among grasses reveals stability in the sugarcane polyploid genome. Plant J. 2007, 50, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S.L.; Shapter, F.M.; Henry, R.J.; Cordeiro, G.; Izquierdo, L.; Lee, L.S. Domestication to crop improvement: Genetic resources for Sorghum and Saccharum (Andropogoneae). Ann. Bot. 2007, 100, 975–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammadov, J.; Buyyarapu, R.; Guttikonda, S.K.; Parliament, K.; Abdurakhmonov, I.Y.; Kumpatla, S.P. Wild Relatives of Maize, Rice, Cotton, and Soybean: Treasure Troves for Tolerance to Biotic and Abiotic Stresses. Front. Plant Sci. 2018, 9, 886. [Google Scholar] [CrossRef]
- Warburton, M.L.; Rauf, S.; Marek, L.; Hussain, M.; Ogunola, O.; de Jesus Sanchez Gonzalez, J. The use of crop wild relatives in maize and sunflower breeding. Crop Sci. 2017, 57, 1227–1240. [Google Scholar] [CrossRef]
- Yumurtaci, A. Utilization of wild relatives of wheat, barley, maize and oat in developing abiotic and biotic stress tolerant new varieties. Emir. J. Food Agric. 2015, 27, 1–23. [Google Scholar] [CrossRef]
- Sharma, D.; Khulbe, R.K.; Pal, R.S.; Bettanaika, J.; Kant, L. Wild Progenitor and Landraces Led Genetic Gain in the Modern-Day Maize (Zea mays L.). In Landraces-Traditional Variety and Natural Breed; IntechOpen: London, UK, 2021; pp. 1–16. [Google Scholar] [CrossRef]
- Abrouk, M.; Ahmed, H.I.; Cubry, P.; Šimoníková, D.; Cauet, S.; Pailles, Y.; Bettgenhaeuser, J.; Gapa, L.; Scarcelli, N.; Couderc, M.; et al. Fonio millet Genome Unlocks African Orphan Crop Diversity for Agriculture in a Changing Climate. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- International Plant Genetic Resources Institute (IPGRI). Neglected and Underutilized Plant Species: Strategic Action Plan of the International Plant Genetic Resources Institute; IPGRI: Rome, Italy, 2002; pp. 1–28. [Google Scholar]
- Padulosi, S.; Hoeschle-Zeledon, I. Underutilized plant species: What are they? LEISA-LEUSDEN- 2004, 20, 5–6. [Google Scholar]
- Chivenge, P.; Mabhaudhi, T.; Modi, A.T.; Mafongoya, P. The potential role of neglected and underutilised crop species as future crops under water scarce conditions in Sub-Saharan Africa. Int. J. Environ. Res. Public Health 2015, 12, 5685–5711. [Google Scholar] [CrossRef] [Green Version]
- Varshney, R.K.; Ribaut, J.M.; Buckler, E.S.; Tuberosa, R.; Rafalski, J.A.; Langridge, P. Can genomics boost productivity of orphan crops? Nat. Biotechnol. 2012, 30, 1172–1176. [Google Scholar] [CrossRef]
- Gregory, P.J.; Mayes, S.; Hui, C.H.; Jahanshiri, E.; Julkifle, A.; Kuppusamy, G.; Kuan, H.W.; Lin, T.X.; Massawe, F.; Suhairi, T.A.; et al. Crops For the Future (CFF): An overview of research efforts in the adoption of underutilised species. Planta 2019, 250, 979–988. [Google Scholar] [CrossRef] [Green Version]
- Dansi, A.; Vodouhè, R.; Azokpota, P.; Yedomonhan, H.; Assogba, P.; Adjatin, A.; Loko, Y.L.; Dossou-Aminon, I.; Akpagana, K. Diversity of the neglected and underutilized crop species of importance in Benin. Sci. World J. 2012, 19, 932947. [Google Scholar] [CrossRef] [Green Version]
- Mayes, S.; Ho, W.K.; Chai, H.H.; Gao, X.; Kundy, A.C.; Mateva, K.I.; Zahrulakmal, M.; Hahiree, M.K.I.M.; Kendabie, P.; Licea, L.C.; et al. Bambara groundnut: An exemplar underutilised legume for resilience under climate change. Planta 2019, 250, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Voytas, D.F.; Gao, C. Precision genome engineering and agriculture: Opportunities and regulatory challenges. PLoS Biol. 2014, 12, e1001877. [Google Scholar] [CrossRef]
- Lata, C.; Gupta, S.; Prasad, M. Foxtail millet: A model crop for genetic and genomic studies in bioenergy grasses. Crit. Rev. Biotechnol. 2013, 33, 328–343. [Google Scholar] [CrossRef]
- Rao, P.P.; Birthal, P.S.; Reddy, B.V.; Rai, K.N.; Ramesh, S. Diagnostics of sorghum and pearl millet grains-based nutrition in India. Int. Sorghum Millets News Lett. 2006, 247, 93–96. [Google Scholar]
- Vadez, V.; Hash, T.; Bidinger, F.R.; Kholova, J. Phenotyping pearl millet for adaptation to drought. Front. Physiol. 2012, 3, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.K.; Singh, R.B.; Pujarula, V.L.; Bollam, S.; Pusuluri, M.; Chellapilla, T.S.; Yadav, R.S.; Gupta, R. Genome-Wide Association Studies and Genomic Selection in Pearl Millet: Advances and Prospects. Front. Genet. 2020, 10, 1389. [Google Scholar] [CrossRef]
- Varshney, R.K.; Shi, C.; Thudi, M.; Mariac, C.; Wallace, J.; Qi, P.; Zhang, H.; Zhao, Y.; Wang, X.; Rathore, A.; et al. Pearl millet genome sequence provides a resource to improve agronomic traits in arid environments. Nat. Biotech. 2017, 35, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Bennetzen, J.L.; Schmutz, J.; Wang, H.; Percifield, R.; Hawkins, J.; Pontaroli, A.C.; Estep, M.; Feng, L.; Vaughn, J.N.; Grimwood, J.; et al. Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 2012, 30, 555–561. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Liu, X.; Quan, Z.; Cheng, S.; Xu, X.; Pan, S.; Xie, M.; Zeng, P.; Yue, Z.; Wang, W.; et al. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotechnol. 2012, 30, 549–554. [Google Scholar] [CrossRef] [Green Version]
- Batley, J.; Edwards, D. The application of genomics and bioinformatics to accelerate crop improvement in a changing climate. Curr. Opin. Plant Biol. 2016, 30, 78–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, R.K.; Hoisington, D.A.; Tyagi, A.K. Advances in cereal genomics and applications in crop breeding. Trends Biotechnol. 2006, 24, 490–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Lin, F.; An, D.; Wang, W.; Huang, R. Genome Sequencing and Assembly by Long Reads in Plants. Genes 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanger, F.; Coulson, A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 1975, 94, 441–448. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flagel, L.E.; Blackman, B.K. The First Ten Years of Plant Genome Sequencing and Prospects for the Next Decade. In Plant Genome Diversity; Wendel, J., Greilhuber, J., Dolezel, J., Leitch, I., Eds.; Springer: Vienna, Austria, 2012; Volume 1, pp. 1–15. [Google Scholar] [CrossRef]
- Project, International Rice Genome Sequencing. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [Green Version]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The sorghum bicolor genome and the diversifi cation of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef] [Green Version]
- Metzker, M.L. Emerging technologies in DNA sequencing. Genome Res. 2005, 15, 1767–1776. [Google Scholar] [CrossRef] [Green Version]
- Michael, T.P.; Jackson, S. The first 50 plant genomes. Plant Genome 2013, 6, 547–562. [Google Scholar] [CrossRef] [Green Version]
- Michael, T.P.; VanBuren, R. Progress, challenges and the future of crop genomes. Curr. Opin. Plant Biol. 2015, 24, 71–81. [Google Scholar] [CrossRef]
- McNally, K.L.; Mauleon, R.P.; Chebotarov, D.; Klassen, S.P.; Kohli, A.; Ye, G.; Leung, H.; Hamilton, R.S.; Wing, R.A. Mass genome sequencing of crops and wild relatives to accelerate crop breeding: The digital rice genebank. In IOP Conference Series, Proceedings of the Earth and Environmental Science, 1 March 2020, Bogor, Indonesia; IOP Publishing: Bristol, UK, 2020; Volume 482, p. 012005. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- McCormick, R.F.; Truong, S.K.; Sreedasyam, A.; Jenkins, J.; Shu, S.; Sims, D.; Kennedy, M.; Amirebrahimi, M.; Weers, B.D.; McKinley, B.; et al. The Sorghum bicolor reference genome: Improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant J. 2018, 93, 338–354. [Google Scholar] [CrossRef] [Green Version]
- Cooper, E.A.; Brenton, Z.W.; Flinn, B.S.; Jenkins, J.; Shu, S.; Flowers, D.; Luo, F.; Wang, Y.; Xia, P.; Barry, K.; et al. A new reference genome for Sorghum bicolor reveals high levels of sequence similarity between sweet and grain genotypes: Implications for the genetics of sugar metabolism. BMC Genom. 2019, 20, 1–13. [Google Scholar] [CrossRef] [Green Version]
- International Wheat Genome Sequencing Consortium (IWGSC). Wheat Genome: Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361. [Google Scholar] [CrossRef] [Green Version]
- Ling, H.Q.; Zhao, S.; Liu, D.; Wang, J.; Sun, H.; Zhang, C.; Fan, H.; Li, D.; Dong, L.; Tao, Y.; et al. Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 2013, 496, 87–90. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Wang, L.; Yang, J.; He, H.; Jin, H.; Li, X.; Ren, T.; Ren, Z.; Li, F.; Han, X.; et al. A high-quality genome assembly highlights rye genomic characteristics and agronomically important genes. Nat. Genet. 2021, 1–11. [Google Scholar] [CrossRef]
- Hittalmani, S.; Mahesh, H.B.; Shirke, M.D.; Biradar, H.; Uday, G.; Aruna, Y.R.; Lohithaswa, H.C.; Mohanrao, A. Genome and transcriptome sequence of finger millet (Eleusine coracana (L.) Gaertn.) provides insights into drought tolerance and nutraceutical properties. BMC Genom. 2017, 18, 1–16. [Google Scholar] [CrossRef]
- International Barley Genome Sequencing Consortium. A physical, genetic and functional sequence assembly of the barley genome. Nature 2012, 491, 711. [Google Scholar] [CrossRef]
- Wang, X.; Chen, S.; Ma, X.; Yssel, A.E.; Chaluvadi, S.R.; Johnson, M.S.; Gangashetty, P.; Hamidou, F.; Sanogo, M.D.; Zwaenepoel, A.; et al. Genome sequence and genetic diversity analysis of an under-domesticated orphan crop, white fonio (Digitaria exilis). GigaScience 2021, 10, giab013. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genome Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Berlin, K.; Koren, S.; Chin, C.S. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol. 2015, 33, 623–630. [Google Scholar] [CrossRef]
- MPerez-de-Castro, A.; Vilanova, S.; Cañizares, J.; Pascual, L.; MBlanca, J.; JDiez, M.; Prohens, J.; Picó, B. Application of genomic tools in plant breeding. Curr. Genom. 2012, 13, 179–195. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Lu, Z.; Xu, G.; Wang, Y.; Jin, B. Analysis and comprehensive comparison of PacBio and nanopore-based RNA sequencing of the Arabidopsis transcriptome. Plant Methods 2020, 16, 1–3. [Google Scholar] [CrossRef]
- Shi, J.; Ma, X.; Zhang, J.; Zhou, Y.; Liu, M.; Huang, L.; Sun, S.; Zhang, X.; Gao, X.; Zhan, W.; et al. Chromosome confirmation capture resolved near complete genome assembly of broomcorn millet. Nat. Commun. 2019, 10, 464. [Google Scholar] [CrossRef] [Green Version]
- Benevenuto, J.; Ferrão, L.F.V.; Amadeu, R.R.; Munoz, P. How can a high-quality genome assembly help plant breeders? Gigascience 2019, 8, giz068. [Google Scholar] [CrossRef]
- Alqudah, A.M.; Sallam, A.; Baenziger, P.S.; Börner, A. GWAS: Fast-forwarding gene identification and characterization in temperate Cereals: Lessons from Barley—A review. J. Adv. Res. 2020, 22, 119–135. [Google Scholar] [CrossRef]
- Beyene, Y.; Semagn, K.; Crossa, J.; Mugo, S.; Atlin, G.N.; Tarekegne, A.; Meisel, B.; Sehabiague, P.; Vivek, B.S.; Oikeh, S.; et al. Improving maize grain yield under drought stress and non-stress environments in sub-Saharan Africa using marker-assisted recurrent selection. Crop Sci. 2016, 56, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, P.F.; Badu-Apraku, B.; Gracen, V.E.; Danquah, E.Y.; Garcia-Oliveira, A.L.; Asante, M.D.; Afriyie-Debrah, C.; Gedil, M. Identification of quantitative trait loci for grain yield and other traits in tropical maize under high and low soil-nitrogen environments. Crop Sci. 2018, 58, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Cattivelli, L.; Rizza, F.; Badeck, F.W.; Mazzucotelli, E.; Mastrangelo, A.M.; Francia, E.; Marè, C.; Tondelli, A.; Stanca, A.M. Drought tolerance improvement in crop plants: An integrated view from breeding to genomics. Field Crops Res. 2008, 105, 1–14. [Google Scholar] [CrossRef]
- Mousavi-Derazmahalleh, M.; Bayer, P.E.; Hane, J.K.; Valliyodan, B.; Nguyen, H.T.; Nelson, M.N.; Erskine, W.; Varshney, R.K.; Papa, R.; Edwards, D. Adapting legume crops to climate change using genomic approaches. Plant Cell Environ. 2019, 42, 6–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, M.; Wani, S.H.; Kumar, P.; Bagaria, P.K.; Rakshit, S.; Roorkiwal, M.; Varshney, R.K. QTLian breeding for climate resilience in cereals: Progress and prospects. Funct. Integr. Genom. 2019, 19, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, H.M.; Azeem, F.; Tahir, N.; Iqbal, M.S. QTL mapping for crop improvement against abiotic stresses in cereals. J. Anim. Plant Sci. 2018, 28, 1558–1573. [Google Scholar]
- Mir, R.R.; Zaman-Allah, M.; Sreenivasulu, N.; Trethowan, R.; Varshney, R.K. Integrated genomics, physiology and breeding approaches for improving drought tolerance in crops. Theor. Appl. Genet. 2012, 125, 625–645. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Qin, F. Genetic dissection of maize drought tolerance for trait improvement. Mol. Breed. 2021, 41, 1–3. [Google Scholar] [CrossRef]
- Gupta, S.M.; Arora, S.; Mirza, N.; Pande, A.; Lata, C.; Puranik, S.; Kumar, J.; Kumar, A. Finger Millet: A “Certain” Crop for an “Uncertain” Future and a Solution to Food Insecurity and Hidden Hunger under Stressful Environments. Front. Plant Sci. 2017, 8, 643. [Google Scholar] [CrossRef] [Green Version]
- Nepolean, T.; Kaul, J.; Mukri, G.; Mittal, S. Genomics-Enabled Next-Generation Breeding Approaches for Developing System-Specific Drought Tolerant Hybrids in Maize. Front. Plant Sci. 2018, 9, 361. [Google Scholar] [CrossRef]
- Maazou, A.R.S.; Tu, J.L.; Qiu, J.; Liu, Z.Z. Breeding for drought tolerance in maize (Zea mays L.). Am. J. Plant Sci. 2016, 7, 1858–1870. [Google Scholar] [CrossRef] [Green Version]
- Pang, Y.; Liu, C.; Wang, D.; Amand, P.S.; Bernardo, A.; Li, W.; He, F.; Li, L.; Wang, L.; Yuan, X.; et al. High-Resolution Genome-Wide Association Study Identifies Genomic Regions and Candidate Genes for Important Agronomic Traits in Wheat. Mol. Plant 2020, 13, 1311–1327. [Google Scholar] [CrossRef]
- Shamshad, M.; Sharma, A. The usage of genomic selection strategy in plant breeding. Next Gener. Plant Breed. 2018, 26, 93. [Google Scholar]
- Huang, X.; Han, B. Natural variations and genome-wide association studies in crop plants. Annu. Rev. Plant Biol. 2014, 65, 531–551. [Google Scholar] [CrossRef]
- Rafalski, J.A. Association genetics in crop improvement. Curr. Opin. Plant Biol. 2010, 13, 174–180. [Google Scholar] [CrossRef]
- Jain, M.; Moharana, K.C.; Shankar, R.; Kumari, R.; Garg, R. Genome wide discovery of DNA polymorphisms in rice cultivars with contrasting drought and salinity stress response and their functional relevance. Plant Biotechnol. J. 2014, 12, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in ricelandraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, Y.; Wei, X.; Li, C.; Wang, A.; Zhao, Q.; Li, W.; Guo, Y.; Deng, L.; Zhu, C.; et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat. Genet. 2012, 44, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Pham, A.T.; Maurer, A.; Pillen, K.; Brien, C.; Dowling, K.; Berger, B.; Eglinton, J.K.; March, T.J. Genome-wide association of barley plant growth under drought stress using a nested association mapping population. BMC Plant Biol. 2019, 19, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Huang, X.; Zhi, H.; Zhao, Y.; Zhao, Q.; Li, W.; Chai, Y.; Yang, L.; Liu, K.; Lu, H.; et al. A haplotype map of genomic variations and genome-wide association studies of agronomic traits in foxtail millet (Setaria italica). Nat. Genet. 2013, 45, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.P.; Ramu, P.; Deshpande, S.P.; Hash, C.T.; Shah, T.; Upadhyaya, H.D.; Riera- Lizarazu, O.; Brown, P.J.; Acharya, C.B.; Mitchell, S.E.; et al. Population genomic and genome-wide association studies of agro climatic traits in sorghum. Proc. Natl. Acad. Sci. 2013, 110, 453–458. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, H.; Hu, X.; Li, K.; Liu, Z.; Wu, Y.; Huang, C. Improving Genomic Selection with Quantitative Trait Loci and Nonadditive Effects Revealed by Empirical Evidence in Maize. Front. Plant Sci. 2019, 10, 1129. [Google Scholar] [CrossRef] [PubMed]
- Voss-Fels, K.P.; Cooper, M.; Hayes, B.J. Accelerating crop genetic gains with genomic selection. Theor. Appl. Genet. 2019, 132, 669–686. [Google Scholar] [CrossRef]
- Spindel, J.; Begum, H.; Akdemir, D.; Virk, P.; Collard, B.; Redoña, E.; Atlin, G.; Jannink, J.-L.; McCouch, S.R. Genomic selection and association mapping in rice (Oryza sativa): Effect of trait genetic architecture, training population composition, marker number and statistical model on accuracy of rice genomic selection in elite, tropical rice breeding lines. PLoS Genet. 2015, 11, e1004982. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Liu, X.; Fu, J.; Wang, H.; Wang, J.; Huang, C.; Prasanna, B.M.; Olsen, M.S.; Wang, G.; Zhang, A. Enhancing genetic gain through genomic selection: From livestock to plants. Plant Commun. 2020, 1, 100005. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Y.; Hu, Z.; Xu, C. Genomic selection methods for crop improvement: Current status and prospects. Crop J. 2018, 6, 330–340. [Google Scholar] [CrossRef]
- Tong, H.; Nikoloski, Z. Machine learning approaches for crop improvement: Leveraging phenotypic and genotypic big data. J. Plant Physiol. 2021, 153354. [Google Scholar] [CrossRef]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Heffner, E.L.; Sorrells, M.E.; Jannink, J.L. Genomic selection for crop improvement. Crop Sci. 2009, 49, 1–12. [Google Scholar] [CrossRef]
- Sikora, P.; Chawade, A.; Larsson, M.; Olsson, J.; Olsson, O. Mutagenesis as a tool in plant genetics, functional genomics, and breeding. Int. J. Plant Genom. 2011, 2011, 314829. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Amatriaín, M.; Cuesta-Marcos, A.; Hayes, P.M.; Muehlbauer, G.J. Barley genetic variation: Implications for crop improvement. Brief. Funct. Genom. 2014, 13, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Jankowicz-Cieslak, J.; Mba, C.; Till, B.J. Mutagenesis for crop breeding and functional genomics. In Biotechnologies for Plant Mutation Breeding; Springer: Cham, Switzerland, 2017; pp. 3–18. [Google Scholar]
- Li, J.; Yang, J.; Li, Y.; Ma, L. Current strategies and advances in wheat biology. Crop J. 2020, 8, 879–891. [Google Scholar] [CrossRef]
- Singh, R.; Tiwari, R.; Sharma, D.; Tiwari, V.; Sharma, I. Mutagenesis for wheat improvement in the genomics era. J. Wheat Res. (JWR) 2014, 6, 120–125. [Google Scholar]
- Kharkwal, M.C.; Shu, Q.Y. The role of induced mutations in world food security. Induced plant mutations in the genomics era. Food Agric. Organ. United Nations Rome 2009, 2009, 33–38. [Google Scholar]
- Ahmar, S.; Gill, R.A.; Jung, K.-H.; Faheem, A.; Qasim, M.U.; Mubeen, M.; Zhou, W. Conventional and Molecular Techniques from Simple Breeding to Speed Breeding in Crop Plants: Recent Advances and Future Outlook. Int. J. Mol. Sci. 2020, 21, 2590. [Google Scholar] [CrossRef] [Green Version]
- Pathirana, R. Plant mutation breeding in agriculture. Plant Sci. Rev. 2011, 6, 107–126. [Google Scholar] [CrossRef]
- Joint FAO/International Atomic Energy Agency (IAEA) Programme of Nuclear Techniques in Agriculture. Mutant Variety Database (MVD). 2016. Available online: https://www.mvd.iaea.org/ (accessed on 29 March 2021).
- Yamaguchi, I.; Otobe, C.K.; Yanagisawa, T. Breeding of 2 waxy wheat [Triticum aestivum] cultivars, Akebono-mochi and Ibuki-mochi, and their main features. Bull. Natl. Inst. Crop Sci. 2003, 3, 21–33. [Google Scholar]
- Wanga, M.A.; Kumar, A.A.; Kangueehi, G.N.; Shimelis, H.; Horn, L.N.; Sarsu, F.; Andowa, J.F. Breeding sorghum using induced mutations: Future prospect for Namibia. Am. J. Plant Sci. 2018, 9, 2696. [Google Scholar] [CrossRef] [Green Version]
- Kurowska, M.; Daszkowska-Golec, A.; Gruszka, D.; Marzec, M.; Szurman, M.; Szarejko, I.; Maluszynski, M. TILLING-a shortcut in functional genomics. J. Appl. Genet. 2011, 52, 371–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruzangohar, M.; Kalashyan, E.; Kalambettu, P.; Ens, J.; Wiebe, K.; Pozniak, C.J.; Tricker, P.J.; Baumann, U. Novel Informatic Tools to Support Functional Annotation of the durum wheat genome. Front. Plant Sci. 2019, 10, 1244. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hao, L.; Parry, M.A.; Phillips, A.L.; Hu, Y.G. Progress in TILLING as a tool for functional genomics and improvement of crops. J. Integr. Plant Biol. 2014, 56, 425–443. [Google Scholar] [CrossRef] [Green Version]
- McCallum, C.; Henikoff, S.; Colbert, T. Fred Hutchinson Cancer Research Center, Assignee. Reverse Genetic Strategy for Identifying Functional Mutations in Genes of Known Sequences. U.S. Patent Application US 10/240,456, 18 March 2004. [Google Scholar]
- Bettgenhaeuser, J.; Krattinger, S.G. Rapid gene cloning in cereals. Theor. Appl. Genet. 2019, 132, 699–711. [Google Scholar] [CrossRef] [Green Version]
- Saintenac, C.; Lee, W.S.; Cambon, F.; Rudd, J.J.; King, R.C.; Marande, W.; Powers, S.J.; Bergès, H.; Phillips, A.L.; Uauy, C.; et al. Wheat receptor-kinase-like protein Stb6 controls gene-for-gene resistance to fungal pathogen Zymoseptoria tritici. Nat. Genet. 2018, 50, 368–374. [Google Scholar] [CrossRef]
- Irshad, A.; Guo, H.; Zhang, S.; Liu, L. TILLING in cereal crops for allele expansion and mutation detection by using modern sequencing technologies. Agronomy 2020, 10, 405. [Google Scholar] [CrossRef] [Green Version]
- Ram, H.; Soni, P.; Salvi, P.; Gandass, N.; Sharma, A.; Kaur, A.; Sharma, T.R. Insertional mutagenesis approaches and their use in rice for functional genomics. Plants 2019, 8, 310. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Kim, C.K.; Kang, M.; Ji, S.U.; Yoon, U.H.; Kim, Y.H.; Lee, G.S. A Gene Functional Study of Rice Using Ac/Ds Insertional Mutant Population. Plant Breed. Biotech. 2018, 6, 313–320. [Google Scholar] [CrossRef]
- Springer, P.S. Gene traps: Tools for plant development and genomics. Plant Cell 2000, 12, 1007–1020. [Google Scholar] [CrossRef]
- Hiei, Y.; Ohta, S.; Komari, T.; Kumashiro, T. Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J. 1994, 6, 271–282. [Google Scholar] [CrossRef] [Green Version]
- Ratanasut, K.; Rod-In, W.; Sujipuli, K. In planta Agrobacterium-mediated transformation of rice. Rice Sci. 2017, 24, 181–186. [Google Scholar] [CrossRef]
- Cheng, X.; Sardana, R.K.; Altosaar, I. Rice Transformation by Agrobacterium Infection. In Recombinant Proteins from Plants. Methods in Biotechnology; Cunningham, C., Porter, A.J.R., Eds.; Humana Press: Totowa, NJ, USA, 1998; Volume 3, pp. 1–9. [Google Scholar]
- Wu, J.-L.; Wu, C.; Lei, C.; Baraoidan, M.; Bordeos, A.; Madamba, M.R.S.; Ramos-Pamplona, M.; Mauleon, R.; Portugal, A.; Ulat, V.J.; et al. Chemical- and irradiation-induced mutants of indica rice IR64 for forward and reverse genetics. Plant Mol. Biol. 2005, 59, 85–97. [Google Scholar] [CrossRef]
- Hwang, H.H.; Yu, M.; Lai, E.M. Agrobacterium-Mediated Plant Transformation: Biology and Applications. Arab. Book 2017, 15. [Google Scholar] [CrossRef] [Green Version]
- Voytas, D.F. Plant genome engineering with sequence-specific nucleases. Annu. Rev. Plant Biol. 2013, 64, 327–350. [Google Scholar] [CrossRef]
- Sun, Y.; Li, J.; Xia, L. Precise genome modification via sequence-specific nucleases-mediated gene targeting for crop improvement. Front. Genet. 2016, 7, 1928. [Google Scholar] [CrossRef] [Green Version]
- Weeks, D.P.; Spalding, M.H.; Yang, B. Use of designer nucleases for targeted gene and genome editing in plants. Plant Biotechnol. J. 2016, 14, 483–495. [Google Scholar] [CrossRef]
- Hilscher, J.; Bürstmayr, H.; Stoger, E. Targeted modification of plant genomes for precision crop breeding. Biotechnol. J. 2017, 12, 1–20. [Google Scholar] [CrossRef]
- Zhang, Y.; Massel, K.; Godwin, I.D.; Gao, C. Applications and potential of genome editing in crop improvement. Genome Biol. 2018, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ansari, W.A.; Chandanshive, S.U.; Bhatt, V.; Nadaf, A.B.; Vats, S.; Katara, J.L.; Sonah, H.; Deshmukh, R. Genome editing in cereals: Approaches, applications and challenges. Int. J. Mol. Sci. 2020, 21, 4040. [Google Scholar] [CrossRef] [PubMed]
- Jun, R.E.N.; Xixun, H.U.; Kejian, W.A.N.G.; Chun, W.A.N.G. Development and application of CRISPR/Cas system in rice. Rice Sci. 2019, 26, 69–76. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [Green Version]
- Miglani, G.S. Genome editing in crop improvement: Present scenario and future prospects. J. Crop Improv. 2017, 31, 453–559. [Google Scholar] [CrossRef]
- Mladenov, E.; Iliakis, G. Induction and repair of DNA double strand breaks: The increasing spectrum of non-homologous end joining pathways. Mutat. Res. 2011, 711, 61–72. [Google Scholar] [CrossRef]
- Bhutia, K.L.; Tyagi, W. Use of Sequence Specific Nucleases for Site Specific Modification of Plant Genome for Crop Improvement. Int. J. Agric. Sci. Res. (IJASR) 2017, 7, 491–502. [Google Scholar]
- Cristea, S.; Freyvert, Y.; Santiago, Y.; Holmes, M.C.; Urnov, F.D.; Gregory, P.D.; Cost, G.J. In vivo cleavage of transgene donors promotes nuclease-mediated targeted integration. Biotechnol. Bioeng. 2013, 110, 871–880. [Google Scholar] [CrossRef]
- Verma, P.; Tandon, R.; Yadav, G.; Gaur, V. Structural aspects of DNA repair and recombination in crop improvement. Front. Genet. 2020, 11, 574549. [Google Scholar] [CrossRef]
- Puchta, H.; Fauser, F. Synthetic nucleases for genome engineering in plants: Prospects for a bright future. Plant J. 2014, 78, 727–741. [Google Scholar] [CrossRef]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell. Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Ryan, R.P.; Vorhölter, F.J.; Potnis, N.; Jones, J.B.; Van Sluys, M.A.; Bogdanove, A.J.; Dow, J.M. Pathogenomics of Xanthomonas: Understanding bacterium-plant interactions. Nat. Rev. Microbiol. 2011, 9, 344–355. [Google Scholar] [CrossRef]
- Üstün, S.; Börnke, F. Interactions of Xanthomonas type-III effector proteins with the plant ubiquitin and ubiquitin-like pathways. Front. Plant Sci. 2014, 5, 736. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, N.; Mukhtar, Z. Genetic manipulations in crops: Challenges and opportunities. Genomics 2017, 109, 494–505. [Google Scholar] [CrossRef]
- Khan, Z.; Khan, S.H.; Mubarik, M.S.; Sadia, B.; Ahmad, A. Use of TALEs and TALEN technology for genetic improvement of plants. Plant Mol. Biol. Rep. 2017, 35, 1–19. [Google Scholar] [CrossRef]
- Ng, W.A.; Ma, A.; Chen, M.; Reed, B.H. A method for rapid selection of randomly induced mutations in a gene of interest using CRISPR/Cas9 mediated activation of gene expression. G3 Genes Genomes Genet. 2020, 10, 1893–1901. [Google Scholar] [CrossRef] [Green Version]
- Razzaq, A.; Saleem, F.; Kanwal, M.; Mustafa, G.; Yousaf, S.; Imran Arshad, H.M.; Hameed, M.K.; Khan, M.S.; Joyia, F.A. Modern trends in plant genome editing: An inclusive review of the CRISPR/Cas9 toolbox. Int. J. Mol. Sci. 2019, 20, 4045. [Google Scholar] [CrossRef] [Green Version]
- Raza, A.; Tabassum, J.; Kudapa, H.; Varshney, R.K. Can omics deliver temperature resilient ready-to-grow crops? Crit. Rev. Biotechnol. 2021, 1–24. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; Van Der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Song, G.; Jia, M.; Chen, K.; Kong, X.; Khattak, B.; Xie, C.; Li, A.; Mao, L. CRISPR/Cas9: A powerful tool for crop genome editing. Crop J. 2016, 4, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Valavanidis, A. Nobel Prize in Chemistry 2020. Discovery of CRISPR-Cas9 Genetic Scissors. A revolutionary genome editing technology that can cut any DNA molecule at a predetermined site (10 November 2020). Sci. Rev. 2020, 1–33. Available online: www.chem-tox-ecotox.org/ScientificReviews1 (accessed on 30 March 2021).
- Boglioli, E.; Richard, M. Rewriting the book of life: A new era in precision gene editing. Working Paper. Boston Consult. Group (BCG) 2015, 1–27. [Google Scholar]
- Nadakuduti, S.S.; Enciso-Rodríguez, F. Advances in Genome Editing With CRISPR Systems and Transformation Technologies for Plant DNA Manipulation. Front. Plant Sci. 2021, 11, 637159. [Google Scholar] [CrossRef]
- Mao, Y.; Zhang, H.; Xu, N.; Zhang, B.; Gou, F.; Zhu, J.K. Application of the CRISPR–Cas system for efficient genome engineering in plants. Mol. Plant. 2013, 6, 2008–2011. [Google Scholar] [CrossRef] [Green Version]
- Schaeffer, S.M.; Nakata, P.A. CRISPR/Cas9-mediated genome editing and gene replacement in plants: Transitioning from lab to field. Plant Sci. 2015, 240, 130–142. [Google Scholar] [CrossRef]
- Joung, J.; Konermann, S.; Gootenberg, J.S.; Abudayyeh, O.O.; Platt, R.J.; Brigham, M.D.; Sanjana, N.E.; Zhang, F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017, 12, 828–863. [Google Scholar] [CrossRef] [Green Version]
- Campenhout, C.V.; Cabochette, P.; Veillard, A.C.; Laczik, M.; Zelisko-Schmidt, A.; Sabatel, C.; Dhainaut, M.; Vanhollebeke, B.; Gueydan, C.; Kruys, V. Guidelines for optimized gene knockout using CRISPR/Cas9. BioTechniques 2019, 66, 95–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Yang, Y.; Qin, R.; Li, H.; Qiu, C.; Li, L. Rapid improvement of grain weight via highly efficient CRISPR/Cas9-mediated multiplex genome editing in rice. J. Genet. Genom. 2016, 43, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, D.; Alptekin, B.; Budak, H. CRISPR/Cas9 genome editing in wheat. Funct. Integr. Genom. 2017, 18, 31–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Gao, H.; Wang, H.; Lafitte, H.R.; Archibald, R.L.; Yang, M.; Hakimi, S.M.; Mo, H.; Habben, J.E. ARGOS8 variants generated by CRISPR-Cas9 improve maize grain yield under field drought stress conditions. Plant Biotechnol. J. 2017, 15, 207–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaul, T.; Sony, S.K.; Verma, R.; Motelb, K.F.A.; Prakash, A.T.; Eswaran, M.; Bharti, J.; Nehra, M.; Kaul, R. Revisiting CRISPR/Cas-mediated crop improvement: Special focus on nutrition. J. Biosci. 2020, 45, 1–37. [Google Scholar] [CrossRef]
- Wang, F.; Wang, C.; Liu, P.; Lei, C.; Hao, W.; Gao, Y.; Liu, Y.G.; Zhao, K. Enhanced rice blast resistance by CRISPR/Cas9-targeted mutagenesis of the ERF transcription factor gene OsERF922. PLoS ONE 2016, 11, e0154027. [Google Scholar] [CrossRef]
- Butt, H.; Eid, A.; Ali, Z.; Atia, M.A.; Mokhtar, M.M.; Hassan, N.; Lee, C.M.; Bao, G.; Mahfouz, M.M. Efficient CRISPR/Cas9-mediated genome editing using a chimeric single-guide RNA molecule. Front. Plant Sci. 2017, 8, 1441. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Jiao, G.; Liu, Z.; Zhang, X.; Li, J.; Guo, X.; Du, W.; Du, J.; Francis, F.; Zhao, Y.; et al. Generation of high-amylose rice through CRISPR/Cas9-mediated targeted mutagenesis of starch branching enzymes. Front. Plant Sci. 2017, 8, 298. [Google Scholar] [CrossRef]
- Li, M.; Li, X.; Zhou, Z.; Wu, P.; Fang, M.; Pan, X.; Lin, Q.; Luo, W.; Wu, G.; Li, H. Reassessment of the four yield-related genes Gn1a, DEP1, GS3, and IPA1 in rice using a CRISPR/Cas9 system. Front. Plant Sci. 2016, 7, 377. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Liu, Y.; Wang, F.; Li, T.; Chen, Z.; Kong, D.; Bi, J.; Zhang, F.; Luo, X.; Wang, J.; et al. Enhanced rice salinity tolerance via CRISPR/Cas9-targeted mutagenesis of the OsRR22 gene. Mol. Breed. 2019, 39, 47. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Zheng, Y.; Xiao, K.; Wei, Y.; Zhu, Y.; Cai, Q.; Chen, L.; Xie, H.; Zhang, J. OsPRX2 contributes to stomatal closure and improves potassium deficiency tolerance in rice. Biochem. Biophys. Res. Commun. 2018, 495, 461–467. [Google Scholar] [CrossRef]
- Liang, Z.; Chen, K.; Li, T.; Zhang, Y.; Wang, Y.; Zhao, Q.; Liu, J.; Zhang, H.; Liu, C.; Ran, Y.; et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nat. Commun. 2017, 8, 14261. [Google Scholar] [CrossRef]
- Zhang, Y.; Bai, Y.; Wu, G.; Zou, S.; Chen, Y.; Gao, C.; Tang, D. Simultaneous modification of three homoeologs of TaEDR1 by genome editing enhances powdery mildew resistance in wheat. Plant J. 2017, 91, 714. [Google Scholar] [CrossRef] [Green Version]
- Shan, Q.; Wang, Y.; Li, J.; Gao, C. Genome editing in rice and wheat using the CRISPR/Cas system. Nat. Protoc. 2014, 9, 2395. [Google Scholar] [CrossRef]
- Svitashev, S.; Young, J.K.; Schwartz, C.; Gao, H.; Falco, S.C.; Cigan, A.M. Targeted mutagenesis, precise gene editing, and site-specific gene insertion in maize using Cas9 and guide RNA. Plant Physiol. 2015, 169, 931–945. [Google Scholar] [CrossRef] [Green Version]
- Prado, J.R.; Segers, G.; Voelker, T.; Carson, D.; Dobert, R.; Phillips, J.; Cook, K.; Cornejo, C.; Monken, J.; Grapes, L.; et al. Genetically engineered crops: From idea to product. Annu. Rev. Plant Biol. 2014, 65, 769–790. [Google Scholar] [CrossRef] [Green Version]
- Edmeades, G.O. Progress in Achieving and Delivering Drought Tolerance in Maize—An Update; ISAA: Ithaca, NY, USA, 2013; pp. 1–39. [Google Scholar]
- Zenda, T.; Liu, S.; Duan, H. Adapting Cereal Grain Crops to Drought Stress: 2020 and Beyond. In Abiotic Stress in Plants; IntechOpen: London, UK, 2020; pp. 1–30. [Google Scholar] [CrossRef]
- Hickey, L.T.; Hafeez, A.N.; Robinson, H.; Jackson, S.A.; Leal-Bertioli, S.C.; Tester, M.; Gao, C.; Godwin, I.D.; Hayes, B.J.; Wulff, B.B. Breeding crops to feed 10 billion. Nat. Biotechnol. 2019, 37, 744–754. [Google Scholar] [CrossRef] [Green Version]
- Khan, T.N.; Meldrum, A.; Croser, J.S. Pea: Overview. Encyclopedia of Food Grains, 2nd ed.; Wrigley, C., Corke, H., Seetharaman, K., Faubion, J., Eds.; Academic Press: London, UK, 2016; Volume 1, pp. 324–333. [Google Scholar] [CrossRef]
- Yan, G.; Liu, H.; Wang, H.; Lu, Z.; Wang, Y.; Mullan, D.; Hamblin, J.; Liu, C. Accelerated generation of selfed pure line plants for gene identification and crop breeding. Front. Plant Sci. 2017, 8, 1786. [Google Scholar] [CrossRef] [Green Version]
- Rajcan, I.; Boersma, J.G.; Shaw, E.J. Plant Systems/Plant Genetic Techniques: Plant Breeder’s Toolbox. In Comprehensive Biotechnology, 2nd ed.; Moo-Young, M., Ed.; Elservier: Amsterdam, The Netherlands, 2011; Volume 4, pp. 133–147. [Google Scholar] [CrossRef]
- Royo, C.; Elias, E.M.; Manthey, F.A. Durum Wheat Breeding. In Cereals. Handbook of Plant Breeding; Carena, M., Ed.; Springer: New York, YK, USA, 2009; Volume 3, pp. 199–226. [Google Scholar] [CrossRef]
- Gupta, S.K. Brassicas. Breeding Oilseed Crops for Sustainable Production; Academic Press: Amsterdam, The Netherlands, 2016; pp. 33–53. [Google Scholar] [CrossRef]
- Yang, J.; Liu, Z.; Chen, Q.; Qu, Y.; Tang, J.; Lübberstedt, T.; Li, H. Mapping of QtL for Grain Yield components Based on a DH population in Maize. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Hussain, B.; Kha, M.; Ali, Q.; Shaukat, S. Double haploid production is the best method for genetic improvement and genetic studies of wheat. Int. J. Agro Vet. Med. Sci. 2012, 6, 216–228. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, S.L.; Britt, A.B.; Tripathi, L.; Sharma, S.; Upadhyaya, H.D.; Ortiz, R. Haploids: Constraints and opportunities in plant breeding. Biotechnol. Adv. 2015, 33, 812–829. [Google Scholar] [CrossRef]
- Li, H.; Singh, R.P.; Braun, H.; Pfeiffer, W.H.; Wang, J. Doubled haploids versus conventional breeding in CIMMYT wheat breeding programs. Crop Sci. 2013, 53, 74–83. [Google Scholar] [CrossRef]
- Asif, M. Progress and Opportunities of Doubled Haploid Production; Springer Briefs in Plant Sciences; Springer International Publishing: Cham, Switzerland, 2013; Volume 6, pp. 1–75. [Google Scholar] [CrossRef]
- Forster, B.P.; Thomas, W.T.B. Doubled haploids in genetics and plant breeding. Plant Breed Rev. 2005, 25, 57–88. [Google Scholar] [CrossRef]
- Weber, D.F. Today’s use of haploids in corn plant breeding. Adv. Agron. 2014, 123, 123–144. [Google Scholar] [CrossRef]
- Uliana Trentin, H.; Frei, U.K.; Lübberstedt, T. Breeding maize maternal haploid inducers. Plants 2020, 9, 614. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, B.M.; Cairns, J.E.; Zaidi, P.H.; Beyene, Y.; Makumbi, D.; Gowda, M.; Magorokosho, C.; Zaman-Allah, M.; Olsen, M.; Das, A.; et al. Beat the stress: Breeding for climate resilience in maize for the tropical rainfed environments. Theor. Appl. Genet. 2021, 1–24. [Google Scholar] [CrossRef]
- Setter, T.L. Analysis of constituents for phenotyping drought tolerance in crop improvement. Front. Physiol. 2012, 3, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanem, M.E.; Marrou, H.; Sinclair, T.R. Physiological phenotyping of plants for crop improvement. Trends Plant Sci. 2015, 20, 139–144. [Google Scholar] [CrossRef]
- Fiorani, F.; and Schurr, U. Future scenarios for plant phenotyping. Annu. Rev. Plant Biol. 2013, 64, 267–291. [Google Scholar] [CrossRef] [Green Version]
- Violle, C.; Navas, M.L.; Vile, D.; Kazakou, E.; Fortunel, C.; Hummel, I.; Garnier, E. Let the concept of trait be functional! Oikos 2007, 116, 882–892. [Google Scholar] [CrossRef]
- Yang, W.; Feng, H.; Zhang, X.; Zhang, J.; Doonan, J.H.; Batchelor, W.D.; Xiong, L.; Yan, J. Crop phenomics and high-throughput phenotyping: Past decades, current challenges, and future perspectives. Mol. Plant 2020, 13, 187–214. [Google Scholar] [CrossRef] [Green Version]
- Araus, J.L.; Cairns, J.E. Field high-throughput phenotyping: The new crop breeding frontier. Trends Plant Sci. 2014, 19, 52–61. [Google Scholar] [CrossRef]
- Großkinsky, D.K.; Svensgaard, J.; Christensen, S.; Roitsch, T. Plant phenomics and the need for physiological phenotyping across scales to narrow the genotype-to-phenotype knowledge gap. J. Exp. Bot. 2015, 66, 5429–5440. [Google Scholar] [CrossRef] [Green Version]
- Oladosu, Y.; Rafii, M.Y.; Samuel, C.; Fatai, A.; Magaji, U.; Kareem, I.; Kamarudin, Z.S.; Muhammad, I.; Kolapo, K. Drought resistance in rice from conventional to molecular breeding: A Review. Int. J. Mol. Sci. 2019, 20, 3519. [Google Scholar] [CrossRef] [Green Version]
- Calleja-Cabrera, J.; Boter, M.; Oñate-Sánchez, L.; Pernas, M. Root growth adaptation to climate change in crops. Front. Plant Sci. 2020, 11, 544. [Google Scholar] [CrossRef]
- Khadka, K.; Earl, H.J.; Raizada, M.N.; Navabi, A. A physio-morphological trait-based approach for breeding drought tolerant wheat. Front. Plant Sci. 2020, 11, 715. [Google Scholar] [CrossRef]
- Araus, J.L.; Sanchez, C.; Edmeades, G.O. Phenotyping maize for adaptation to drought. In Drought Phenotyping in Crops: From Theory to Practice CGIAR Generation Challenge Program; Monneveux, P., Ribaut, J.M., Eds.; CIMMYT: Texcoco, Mexico, 2011; pp. 263–283. [Google Scholar]
- Sinclair, T.R. Challenges in breeding for yield increase for drought. Trends Plant Sci. 2011, 16, 289–293. [Google Scholar] [CrossRef]
- Sinclair, T.R.; Purcell, L.C.; Sneller, C.H. Crop transformation and the challenge to increase yield potential. Trends Plant Sci. 2004, 9, 70–75. [Google Scholar] [CrossRef]
- Panguluri, S.K.; Kumar, A.A. Phenotyping for Plant Breeding; Springer: New York, NY, USA, 2016. [Google Scholar]
- Mir, R.R.; Reynolds, M.; Pinto, F.; Khan, M.A.; Bhat, M.A. High-throughput phenotyping for crop improvement in the genomics era. Plant Sci. 2019, 282, 60–72. [Google Scholar] [CrossRef]
- Hussain, S.; Mubeen, M.; Ahmad, A.; Akram, W.; Hammad, H.M.; Ali, M.; Masood, N.; Amin, A.; Farid, H.U.; Sultana, S.R.; et al. Using GIS tools to detect the land use/land cover changes during forty years in Lodhran district of Pakistan. Environ. Sci. Pollut. Res. 2019. [Google Scholar] [CrossRef]
- Fahlgren, N.; Gehan, M.A.; Baxter, I. Lights, camera, action: High-throughput plant phenotyping is ready for a close-up. Curr. Opin. Plant. Biol. 2015, 24, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Barker III, J.; Zhang, N.; Sharon, J.; Steeves, R.; Wang, X.; Wei, Y.; Poland, J. Development of a field-based high-throughput mobile phenotyping platform. Comput. Electron. Agric. 2016, 122, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhang, Q.; Huang, D. A Review of Imaging Techniques for Plant Phenotyping. Sensors 2014, 14, 20078–20111. [Google Scholar] [CrossRef]
- Badigannavar, A.; Teme, N.; de Oliveira, A.C.; Li, G.; Vaksmann, M.; Viana, V.E.; Ganapathi, T.R.; Sarsu, F. Physiological, genetic and molecular basis of drought resilience in sorghum [Sorghum bicolor (L.) Moench]. Ind. J. Plant Physiol. 2018, 23, 670–688. [Google Scholar] [CrossRef]
- Fischer, K.S.; Fukai, S.; Kumar, A.; Leung, H.; Jongdee, B. Phenotyping rice for adaptation to drought. In Drought Phenotyping in Crops: From Theory to Practice: CGIAR Generation Challenge Program; Monneveux, P., Ribaut, J.M., Eds.; CIMMYT: Texcoco, Mexico, 2011; pp. 215–243. [Google Scholar]
- Monneveux, P.; Jing, R.; Misra, S.C. Phenotyping wheat for adaptation to drought using physiological traits. Front. Physiol. 2012, 3, 429. [Google Scholar] [CrossRef] [Green Version]
- Passioura, J.B. Phenotyping for drought tolerance in grain crops: When is it useful to breeders? Funct. Plant Biol. 2012, 39, 851–859. [Google Scholar] [CrossRef]
- Wang, D.; Fahad, S.; Saud, S.; Kamran, M.; Khan, A.; Khan, M.N.; Hammad, H.M.; Nasim, W. Morphological acclimation to agronomic manipulation in leaf dispersion and orientation to promote “Ideotype” breeding: Evidence from 3D visual modeling of “super” rice (Oryza sativa L.). Plant Physiol. Biochem. 2019, 135, 499–510. [Google Scholar] [CrossRef]
- Mutka, A.M.; Bart, R.S. Image-based phenotyping of plant disease symptoms. Front. Plant Sci. 2015, 5, 734. [Google Scholar] [CrossRef] [Green Version]
- Tardieu, F.; Cabrera-Bosquet, L.; Pridmore, T.; Bennett, M. Plant phenomics, from sensors to knowledge. Curr. Biol. 2017, 27, R770–R783. [Google Scholar] [CrossRef]
- Singh, B.; Mishra, S.; Bohra, A.; Joshi, R.; Siddique, K.H. Crop phenomics for abiotic stress tolerance in crop plants. In Biochemical, Physiological and Molecular Avenues for Combating Abiotic Stress Tolerance in Plants; Wani, S.H., Ed.; Academic Press: Amsterdam, The Netherlands, 2018; pp. 277–296. [Google Scholar] [CrossRef]
- Liu, X.; Hao, L.; Li, D.; Zhu, L.; Hu, S. Long non-coding RNAs and their biological roles in plants. Genom. Proteom. Bioinf. 2015, 13, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huanca-Mamani, W.; Arias-Carrasco, R.; Cárdenas-Ninasivincha, S.; Rojas-Herrera, M.; Sepúlveda-Hermosilla, G.; Caris-Maldonado, J.C.; Bastías, E.; Maracaja-Coutinho, V. Long non-coding RNAs responsive to salt and boron stress in the hyper-arid Lluteno maize from Atacama Desert. Genes 2018, 9, 170. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Zhang, Y.; Chen, X.; Chen, Y. Plant noncoding RNAs: Hidden players in development and stress responses. Annu. Rev. Cell Dev. Biol. 2019, 35, 407–431. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Meng, X.; Dobrovolskaya, O.B.; Orlov, Y.L.; Chen, M. Non-coding RNAs and their roles in stress response in plants. Genom. Proteom. Bioinf. 2017, 15, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Dinger, M.E.; Pang, K.C.; Mercer, T.R.; Crowe, M.L.; Grimmond, S.M.; Mattick, J.S. NRED: A database of long noncoding RNA expression. Nucleic Acids Res. 2009, 37, D122–D126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, U.C.; Nayyar, H.; Jha, R.; Khurshid, M.; Zhou, M.; Mantri, N.; Siddique, K.H. Long non-coding RNAs: Emerging players regulating plant abiotic stress response and adaptation. BMC Plant Biol. 2020, 20, 1–20. [Google Scholar] [CrossRef]
- Megha, S.; Basu, U.; Rahman, M.H.; Kav, N.N. The role of long non-coding RNAs in abiotic stress tolerance in plants. In Elucidation of Abiotic Stress Signaling in Plants; Springer: New York, NY, USA, 2015; pp. 93–106. [Google Scholar] [CrossRef]
- Wierzbicki, A.T.; Haag, J.R.; Pikaard, C.S. Noncoding transcription by RNA polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell 2008, 135, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Eichten, S.R.; Shimizu, R.; Petsch, K.; Yeh, C.T.; Wu, W.; Chettoor, A.M.; Givan, S.A.; Cole, R.A.; Fowler, J.E.; et al. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014, 15, R40. [Google Scholar] [CrossRef] [Green Version]
- Di, C.; Yuan, J.; Wu, Y.; Li, J.; Lin, H.; Hu, L.; Zhang, T.; Qi, Y.; Gerstein, M.B.; Guo, Y.; et al. Characterization of stress-responsive lncRNAs in Arabidopsis Thaliana by Integrating Expression, Epigenetic and Structural Features. Plant J. 2014, 80, 848–861. [Google Scholar] [CrossRef]
- Zhang, M.; Zhao, H.; Xie, S.; Chen, J.; Xu, Y.; Wang, K.; Zhao, H.; Guan, H.; Hu, X.; Jiao, Y.; et al. Extensive, clustered parental imprinting of protein-coding and noncoding RNAs in developing maize endosperm. Proc. Natl. Acad. Sci. USA 2011, 108, 20042–20047. [Google Scholar] [CrossRef] [Green Version]
- Chekanova, J.A. Long non-coding RNAs and their functions in plants. Curr. Opin. Plant Biol. 2015, 27, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Böhmdorfer, G.; Wierzbicki, A.T. Control of chromatin structure by long noncoding RNA. Trends Cell Biol. 2015, 25, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Han, Z.; Guo, Q.; Liu, Y.; Zheng, Y.; Wu, F.; Jin, W. Identification of maize long non-coding RNAs responsive to drought stress. PLoS ONE. 2014, 9, e98958. [Google Scholar] [CrossRef] [Green Version]
- Amaral, P.P.; Dinger, M.E.; Mattick, J.S. Non-coding RNAs in homeostasis, disease and stress responses: An evolutionary perspective. Brief. Funct. Genom. 2013, 12, 254–278. [Google Scholar] [CrossRef] [Green Version]
- Li, J.R.; Liu, C.C.; Sun, C.H.; Chen, Y.T. Plant stress RNA-seq nexus: A stress-specific transcriptome database in plant cells. BMC Genom. 2018, 19, 966. [Google Scholar] [CrossRef] [Green Version]
- Pang, J.; Zhang, X.; Ma, X.; Zhao, J. Spatio-temporal transcriptional dynamics of maize long non-coding RNAs responsive to drought stress. Genes 2019, 10, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.; Xie, S.; Liu, Y.; Yi, F.; Yu, J. Genome-wide annotation of genes and noncoding RNAs of foxtail millet in response to simulated drought stress by deep sequencing. Plant Mol. Biol. 2013, 83, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Chung, P.J.; Jung, H.; Jeong, D.H.; Ha, S.H.; Choi, Y.D.; Kim, J.K. Transcriptome profiling of drought responsive noncoding RNAs and their target genes in rice. BMC Genom. 2016, 17, 563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, M.; Wang, Y.; Yao, Y.; Song, N.; Hu, Z.; Qin, D.; Xie, C.; Peng, H.; Ni, Z.; Sun, Q. Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing. BMC Plant Biol. 2011, 11, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Budak, H.; Kaya, S.B.; Cagirici, H.B. Long non-coding RNA in plants in the era of reference sequences. Front. Plant Sci. 2020, 11, 276. [Google Scholar] [CrossRef] [Green Version]
- Wani, S.H.; Kumar, V.; Khare, T.; Tripathi, P.; Shah, T.; Ramakrishna, C.; Aglawe, S.; Mangrauthia, S.K. miRNA applications for engineering abiotic stress tolerance in plants. Biologia 2020, 75, 1–19. [Google Scholar] [CrossRef]
- Tao, Y.; Zhao, X.; Mace, E.; Henry, R.; Jordan, D. Exploring and Exploiting Pan-genomics for Crop Improvement. Mol. Plant 2019, 12, 156–169. [Google Scholar] [CrossRef] [Green Version]
- Edwards, D.; Batley, J. Plant Genomics and Climate Change||The Impact of Genomics Technology on Adapting Plants to Climate Change. Plant Genom. Clim. Chang. 2016, 173–178. [Google Scholar] [CrossRef]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef]
- Sebat, J.; Lakshmi, B.; Troge, J.; Alexander, J.; Young, J.; Lundin, P.; Månér, S.; Massa, H.; Walker, M.; Chi, M.; et al. Large-scale copy number polymorphism in the human genome. Science 2004, 305, 525–528. [Google Scholar] [CrossRef] [Green Version]
- Pinkel, D.; Segraves, R.; Sudar, D.; Clark, S.; Poole, I.; Kowbel, D.; Collins, C.; Kuo, W.L.; Chen, C.; Zhai, Y.; et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 1998, 20, 207–211. [Google Scholar] [CrossRef]
- Saxena, R.K.; Edwards, D.; Varshney, R.K. Structural variations in plant genomes. Brief. Funct. Genom. 2014, 13, 296–307. [Google Scholar] [CrossRef] [Green Version]
- Danilevicz, M.F.; Fernandez, C.G.T.; Marsh, J.I.; Bayer, P.E.; Edwards, D. Plant pangenomics: Approaches, applications and advancements. Curr. Opin. Plant Biol. 2020, 54, 18–25. [Google Scholar] [CrossRef]
- Tranchant-Dubreuil, C.; Rouard, M.; Sabot, F. Plant pangenome: Impacts on phenotypes and evolution. Annu. Plant Rev. Online 2018, 453–478. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, C.N.; Foerster, J.M.; Johnson, J.M.; Sekhon, R.S.; Muttoni, G.; Vaillancourt, B.; Peñagaricano, F.; Lindquist, E.; Pedraza, M.A.; Barry, K.; et al. Insights into the maize pan-genome and pan-transcriptome. Plant Cell 2014, 26, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Schatz, M.C.; Maron, L.G.; Stein, J.C.; Wences, A.H.; Gurtowski, J.; Biggers, E.; Lee, H.; Kramer, M.; Antoniou, E.; Ghiban, E.; et al. Whole genome de novo assemblies of three divergent strains of rice, Oryza sativa, document novel gene space of aus and indica. Genome Biol. 2014, 15, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Feng, Q.; Lu, H.; Li, Y.; Wang, A.; Tian, Q.; Zhan, Q.; Lu, Y.; Zhang, L.; Huang, T.; et al. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 2018, 50, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef]
- Montenegro, J.D. The pangenome of hexaploid bread wheat. Plant J. 2017, 90, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Song, J.M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Golicz, A.A.; Batley, J.; Edwards, D. Towards plant pangenomics. Plant Biotechnol. J. 2016, 14, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Coletta, R.D.; Qiu, Y.; Ou, S.; Hufford, M.B.; Hirsch, C.N. How the pan-genome is changing crop genomics and improvement. Genome Biol. 2021, 22, 1–19. [Google Scholar] [CrossRef]
- Computational Pan-Genomics Consortium. Computational pan-genomics: Status, promises and challenges. Brief Bioinform. 2018, 19, 118–135. [Google Scholar] [CrossRef] [Green Version]
- Zuo, W.; Chao, Q.; Zhang, N.; Ye, J.; Tan, G.; Li, B.; Xing, Y.; Zhang, B.; Liu, H.; Fengler, K.A.; et al. A maize wall-associated kinase confers quantitative resistance to head smut. Nat. Genet. 2015, 47, 151–157. [Google Scholar] [CrossRef]
- Gamuyao, R.; Chin, J.H.; Pariasca-Tanaka, J.; Pesaresi, P.; Catausan, S.; Dalid, C.; Slamet-Loedin, I.; Tecson-Mendoza, E.M.; Wissuwa, M.; Heuer, S. The protein kinase Pstol1 from traditional rice confers tolerance of phosphorus deficiency. Nature 2012, 488, 535–539. [Google Scholar] [CrossRef]
- Makalowski, W.; Gotea, V.; Pande, A.; Makalowski, I. Transposable elements: Classification, identification, and their use as a tool for comparative genomics. In Evolutionary Genomics Methods in Molecular Biology; Anisimova, M., Ed.; Humana: New York, NY, USA, 2019; Volume 1910, pp. 177–207. [Google Scholar] [CrossRef] [Green Version]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2017, 8, 973–982. [Google Scholar] [CrossRef]
- Dubin, M.J.; Scheid, O.M.; Becker, C. Transposons: A blessing curse. Curr. Opin. Plant Biol. 2018, 42, 23–29. [Google Scholar] [CrossRef]
- Gaut, B.S.; d’Ennequin, M.L.T.; Peek, A.S.; Sawkins, M.C. Maize as a model for the evolution of plant nuclear genomes. Proc. Natl. Acad. Sci. USA 2000, 97, 7008–7015. [Google Scholar] [CrossRef] [Green Version]
- Elliott, T.A.; Gregory, T.R. What’s in a genome? The C-value enigma and the evolution of eukaryotic genome content. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140331. [Google Scholar] [CrossRef]
- Lönnig, W.E.; Saedler, H. Chromosome rearrangements and transposable elements. Annu. Rev. Genet. 2002, 36, 389–410. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yu, C.; Krishnaswamy, L.; Peterson, T. Transposable elements as catalysts for chromosome rearrangements. Methods Mol. Biol. 2011, 315–326. [Google Scholar] [CrossRef]
- Jiang, N.; Ferguson, A.A.; Slotkin, R.K.; Lisch, D. Pack-Mutator-like transposable elements (Pack-MULEs) induce directional modification of genes through biased insertion and DNA acquisition. Proc. Natl. Acad. Sci. USA 2011, 108, 1537–1542. [Google Scholar] [CrossRef] [Green Version]
- Fedoroff, N.V. Transposable elements, epigenetics, and genome evolution. Science 2012, 338, 758–767. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Ferguson, A.A.; Jiang, N. What makes up plant genomes: The vanishing line between transposable elements and genes. Biochim. Biophys. Acta 2016, 1859, 366–380. [Google Scholar] [CrossRef] [Green Version]
- Ariel, F.D.; Manavella, P.A. When junk DNA turns functional: Transposon-derived noncoding RNAs in plants. J. Exp. Bot. 2021. [Google Scholar] [CrossRef]
- Lisch, D. How important are transposons for plant evolution? Nat. Rev. Genet. 2013, 14, 49–61. [Google Scholar] [CrossRef]
- Anderson, S.N.; Stitzer, M.C.; Brohammer, A.B.; Zhou, P.; Noshay, J.M.; O’Connor, C.H.; Hirsch, C.D.; Ross-Ibarra, J.; Hirsch, C.N.; Springer, N.M. Transposable Elements Contribute to Dynamic Genome Content in Maize. Plant J. 2019, 100, 1052–1065. [Google Scholar] [CrossRef]
- Makarevitch, I.; Waters, A.J.; West, P.T.; Stitzer, M.; Hirsch, C.N.; Ross-Ibarra, J.; Springer, N.M. Transposable elements contribute to activation of maize genes in response to abiotic stress. PLoS Genet. 2015, 11, e1004915. [Google Scholar] [CrossRef] [Green Version]
- Yokosho, K.; Yamaji, N.; Fujii-Kashino, M.; Ma, J.F. Retrotransposon-mediated aluminum tolerance through enhanced expression of the citrate transporter OsFRDL4. Plant Physiol. 2016, 172, 2327–2336. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Cimen, E.; Singh, N.; Buckler, E. Deep learning for plant genomics and crop improvement. Curr. Opin. Plant Biol. 2020, 54, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Ganapathysubramanian, B.; Singh, A.K.; Sarkar, S. Machine learning for high-throughput stress phenotyping in plants. Trends Plant Sci. 2016, 21, 110–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Scheben, A.; Edwards, D. Advances in integrating genomics and bioinformatics in the plant breeding pipeline. Agriculture 2018, 8, 75. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, J. Supervised and unsupervised machine learning algorithms. Mach. Learn. Mastery 2016, 16. Available online: https://machinelearningmastery.com/supervised-and-unsupervised-machine-learning-algorithms/ (accessed on 20 April 2021).
- Mahood, E.H.; Kruse, L.H.; Moghe, G.D. Machine learning: A powerful tool for gene function prediction in plants. Appl. Plant Sci. 2020, 8, e11376. [Google Scholar] [CrossRef]
- Mejía-Guerra, M.K.; Buckler, E.S. A k-mer grammar analysis to uncover maize regulatory architecture. BMC Plant Biol. 2019, 19, 103. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yin, Z.; Manley, P.; Burken, J.; Shakoor, G.; Fahlgren, N.; Mockler, T. Early drought plant stress detection with bi-directional long-term memory networks. Photogramm. Eng. Remote. Sens. 2018, 84, 459–468. [Google Scholar] [CrossRef]
- Ghosal, S.; Blystone, D.; Singh, A.K.; Ganapathysubramanian, B.; Singh, A.; Sarkar, S. An explainable deep machine vision framework for plant stress phenotyping. Proc. Natl. Acad. Sci. USA 2018, 115, 4613–4618. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Zhang, H.; Lin, L.; Wang, H.; Gao, Y.; Zhao, L.; Chen, Y.P.P.; Chen, R.; Gu, L. A similarity searching system for biological phenotype images using deep convolutional encoder-decoder architecture. Curr. Bioinform. 2019, 14, 628–639. [Google Scholar] [CrossRef]
- Libbrecht, M.W.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Esposito, S.; Carputo, D.; Cardi, T.; Tripodi, P. Applications and trends of machine learning in genomics and phenomics for next-generation breeding. Plants 2020, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Zhang, H.H.; Wang, X. Machine learning for big data analytics in plants. Trends Plant Sci. 2014, 19, 798–808. [Google Scholar] [CrossRef]
- Xu, C.; Jackson, S.A. Machine learning and complex biological data. Genome Biol. 2019, 20, 76. [Google Scholar] [CrossRef]
- Kwon, M.S.; Lee, B.T.; Lee, S.Y.; Kim, H.U. Modeling regulatory networks using machine learning for systems metabolic engineering. Curr. Opin. Biotechnol. 2020, 65, 63–170. [Google Scholar] [CrossRef]
- Ni, Y.; Aghamirzaie, D.; Elmarakeby, H.; Collakova, E.; Li, S.; Grene, R.; Heath, L.S. A machine learning approach to predict gene regulatory networks in seed development in Arabidopsis. Front. Plant Sci. 2016, 7, 1936. [Google Scholar] [CrossRef] [Green Version]
- Korani, W.; Clevenger, J.P.; Chu, Y.; Ozias-Akins, P. Machine learning as an effective method for identifying true single nucleotide polymorphisms in polyploid plants. Plant Genome 2019, 12, 180023. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Bodner, G.; Rewald, B. Phenotyping: Using machine learning for improved pairwise genotype classification based on root traits. Front. Plant Sci. 2016, 7, 1864. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, M.G.; Manuel, V.; Diego, G.; Milton, V.; Henry, R.; Animesh, A. Machine learning for high-throughput field phenotyping and image processing provides insight into the association of above and below-ground traits in cassava (Manihot esculenta Crantz). Plant Methods 2020, 16, 1–19. [Google Scholar] [CrossRef]
- Long, P.; Zhang, L.; Huang, B.; Chen, Q.; Liu, H. Integrating genome sequence and structural data for statistical learning to predict transcription factor binding sites. Nucleic Acids Res. 2020, 48, 12604–12617. [Google Scholar] [CrossRef]
- Sun, L.; Liu, H.; Zhang, L.; Meng, J. lncRScan-SVM: A tool for predicting long non-coding RNAs using support vector machine. PLoS ONE 2015, 10, e0139654. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, J.; Wei, Z.; Hakonarson, H. DeepPolyA: A convolutional neural network approach for polyadenylation site prediction. IEEE Access 2018, 6, 24340–24349. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 2017, 12, 2478. [Google Scholar] [CrossRef]
- Li, Y.; Shi, W.; Wasserman, W.W. Genome-wide prediction of cis-regulatory regions using supervised deep learning methods. BMC Bioinform. 2018, 19, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crane-Droesch, A. Machine learning methods for crop yield prediction and climate change impact assessment in agriculture. Environ. Res. Lett. 2018, 13, 114003. [Google Scholar] [CrossRef] [Green Version]

| Species Name | Mutant Name | Parent | Mutant Development Type (and Mutation Induction Type Used) | 1 Trait Category | 2 Description of Specific Traits Improved | 3 Reg. Year | Country | References |
|---|---|---|---|---|---|---|---|---|
| Oryza sativa L. | Sinar 1 | Sintanur | Gamma irradiation | Y, QNR | Higher yield and higher aromatic value than parent | 2020 | Indonesia | [142] |
| Oryza sativa L. | Sinar 2 | Sintanur | Gamma irradiation | Y, BST, QNR | High yield, higher aromatic value, and higher disease resistance to BLB diseases | 2020 | Indonesia | [142] |
| Oryza sativa L. | Zhefu 802 | Simei No. 2 | Gamma irradiation | BST, Y, A, QNR | Higher rice blast resistance, higher yield, early maturity, good grain quality | 1990 | China | [139,141,142] |
| Triticum aestivum L. | Akebono-mochi | Kanto No. 107 | Hybridization with mutant obtained by EMS chemical treatment | QNR | Amylose free, lower pasting temperature, higher peak viscosity, and higher breakdown than for non-waxy wheat | 2000 | Japan | [142,143] |
| Triticum aestivum L. | Binagom-1 | L-880 (NIAB, Pakhistan) | Direct use of an induced mutant | AST, Y | Has higher salinity tolerance, higher yield | 2016 | Bangladesh | [142] |
| Triticum aestivum L. | Darkhan-172 | Darkhan-95 | Chemical mutageneis using sodium azide | Y, A | Higher yield, early maturity | 2018 | Mongolia | [142] |
| Hordeum vulgare L. | Centenario | Buenavista | Gamma irradiation (333 Gy) | A | Altered maturity, seed production traits | 2006 | Peru | [142] |
| Hordeum vulgare L. | Cruiser | Valticky, Diamant | Hybridization with mutant variety Diamant obtained by irradiation of seeds with X-rays (100 Gy) | A | Improved growth habit (erectoid type) | 2001 | Germany | [142] |
| Hordeum vulgare L. | Phenix | Kharkivskiy 99 (mutant) | Hybridization with mutant Kharkivskiy 99 | AST | Improved drought tolerance | 2000 | Ukraine | [142] |
| Zea mays L. | Kneja 627 | PCM4658 | Hybridization with mutant (from the cross PCM4658 × Mo17) | Y, A | Improved grain (seed) yield, late maturity | 2009 | Bulgaria | [142] |
| Zea mays L. | P26 | F1 P1 3747 SC M3 | Treatment with fast neutrons (7.5 Gy) | A | Agronomic and botanic traits (combining ability) | 2001 | Hungary | [142] |
| Zea mays L. | Longfuyu 3 | Fu2691 × 8008 | Direct use of an induced mutant | BST | Improved resistance to bacterial diseases | 2007 | China | [142] |
| Setaria sp. | Jingu 21 | Jinfen 52 | Gamma irradiation (350 Gy) | Y, QNR | Improved grain (seed) yield, improved culinary quality | 2000 | China | [142] |
| Panicum miliaceum L. | Cheget | Mutant parents not specified. | Hybridization with two chemo mutants | AST, BST | Improved drought tolerance, improved smut resistance | 1993 | Russia | [142] |
| Sorghum bicolor L. | Fambe | CSM388 | Direct use of an induced mutant, gamma irradiation (300 Gy) | AST, Y | Resistance to lodging, high grain yield (increased number of grains per panicle) | 1998 | Mali | [142] |
| Sorghum bicolor L. | PAHAT | - | Direct use of an induced mutant, gamma irradiation | Y, A, QNR | High yielding, semi dwarfness, early maturity, grain quality (protein, tannin, starch) | 2013 | Indonesia | [142,144] |
| Sorghum bicolor L. | Samurai 1 | Zh-30 | Direct use of an induced mutant, gamma irradiation (0.3 Other) | Y, A, BST, QNR, AST | High yield, improved food processing quality, improved biomass, lodging resistance, resistance to midrib rot disease, large seed size | 2014 | Indonesia | [142] |
| Crop Species | Delivery Mode 1 | Target Gene/s | DNA Repair Type 2 | sgRNA Promoter | Cas9 Promoter 3 | Vector Used | Trait Targeted for Improvement | References |
|---|---|---|---|---|---|---|---|---|
| Oryza sativa L. | EP | ERF922 | NHEJ | OsU6a | Ubi | C-ERF922 | Enhanced rice blast resistance | [196] |
| AMT | ALS | HR | OsU3 | Ubi | pCXUN-Cas9-gRNA1-gRNA2-armed donor vector | Improved herbicides resistance | [197] | |
| EP | SBEIIb | NHEJ | OsU3 | Ubi | pCXUN-Cas9 | High amylose content | [198] | |
| EP, AMT | Gn1a, GS3, DEP1 | NHEJ | OsU6a | Ubi | pYLCRISPR/Cas9(I) | Improved grain number, larger grain size, and dense erect panicles | [199] | |
| AMT | RR22 | NHEJ | OsU6a | Ubi | pYLCRSPR/Cas9 Pubi-H | Enhanced salt tolerance | [200] | |
| AMT | PRX2 | NHEJ | OsPRX2 | - | pCAMBIA1301 | Improved potassium deficiency tolerance | [201] | |
| Triticum aestivum L. | BMT | GW2 | NHEJ | TaU6 | Ubi | pET28a-Cas9-His | Increased grain weight and protein content | [202] |
| BMT | EDR1 | NHEJ | TaU6 | Ubi | pJIT163-Ubi-Cas9 | Increased powdery mildew resistance | [203] | |
| AMT | MLO | NHEJ | TaU6 | Ubi | pUC-T vector (CWBIO) | Increased mildew resistance | [204] | |
| AMT | DREB2 and ERF3 | NHEJ | TaU6 | - | pJIT163-2NLSCas9 | Improved drought resistance | [193] | |
| Zea mays L. | AMT | ARGOS8 | HR | ZmU6 | Ubi | sgRNA-Cas9 | Improved grain yield under drought stress tolerance | [194] |
| AT | ALS2 | HR | ZmU1 | Ubi | UBI-Cas9 T-DNA vector | Improved resistance to herbicides | [205] | |
| BMT | LIG1, M26, 45, ALS1 | HR | ZmU6 | Ubi | Cas9 DNA vector | Enhanced herbicide resistance and male sterility | [205] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zenda, T.; Liu, S.; Dong, A.; Duan, H. Advances in Cereal Crop Genomics for Resilience under Climate Change. Life 2021, 11, 502. https://doi.org/10.3390/life11060502
Zenda T, Liu S, Dong A, Duan H. Advances in Cereal Crop Genomics for Resilience under Climate Change. Life. 2021; 11(6):502. https://doi.org/10.3390/life11060502
Chicago/Turabian StyleZenda, Tinashe, Songtao Liu, Anyi Dong, and Huijun Duan. 2021. "Advances in Cereal Crop Genomics for Resilience under Climate Change" Life 11, no. 6: 502. https://doi.org/10.3390/life11060502