
Chronic Kidney Disease as a Systemic Inflammatory Syndrome: Update on Mechanisms Involved and Potential Treatment

 , , , , ,
, , , , , {kind=link}
{kind=link}
Abstract
:1. Introduction
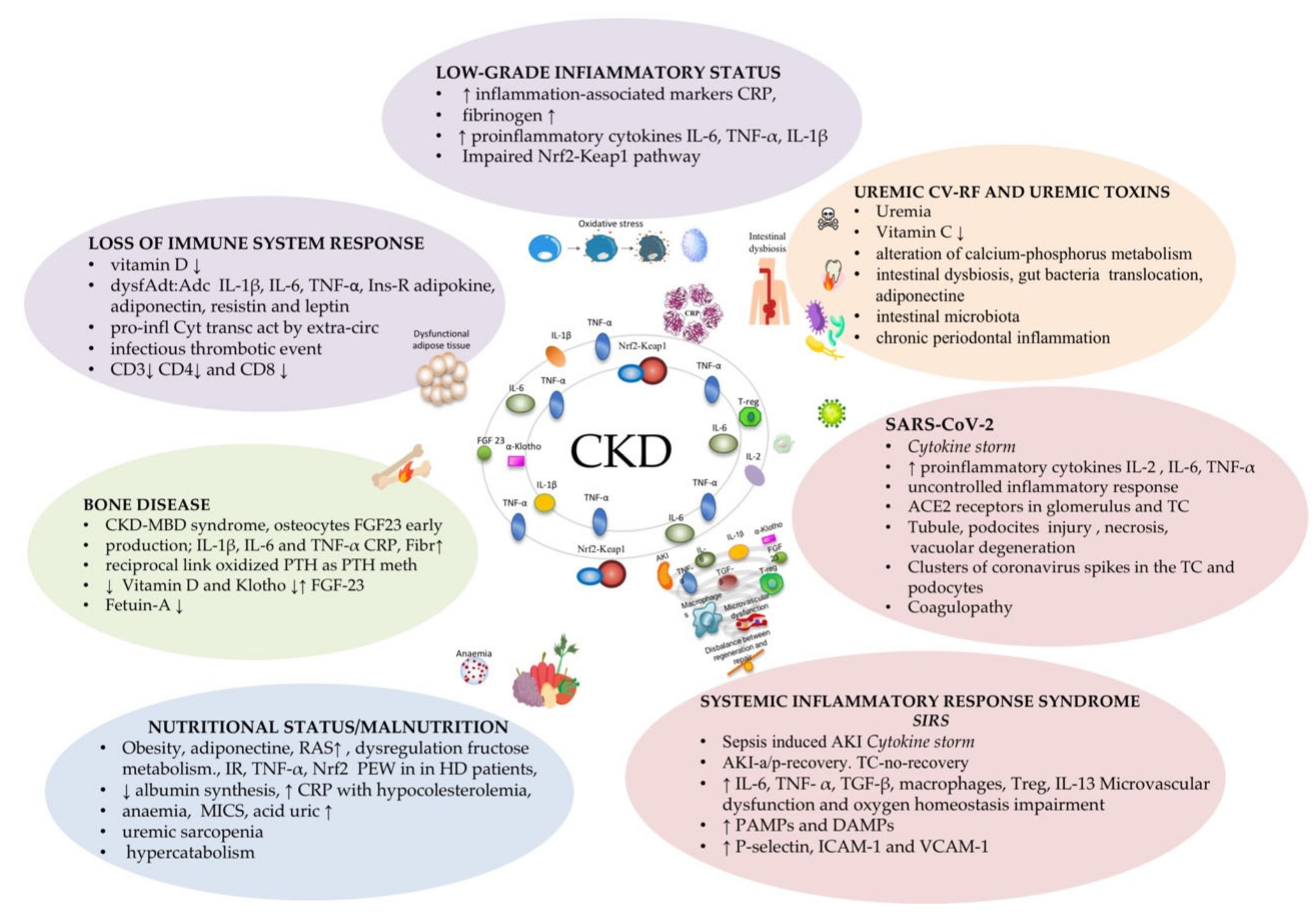
1.1. CKD and Systemic Inflammation
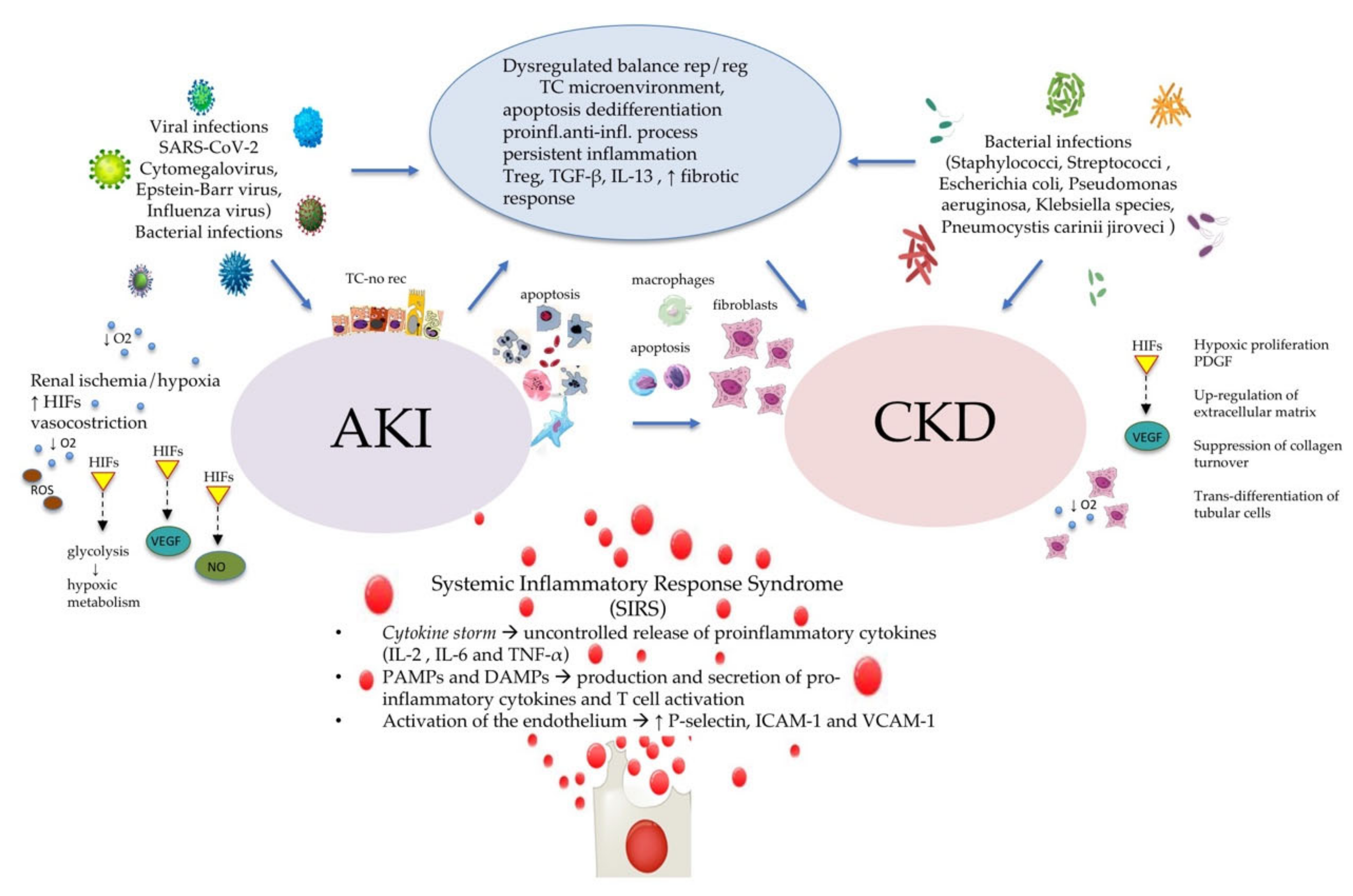
1.2. CKD, Inflammation, and AKI
1.3. CKD, Inflammation, and SARS-CoV-2
1.4. CKD Inflammation, and Bone Disease
1.5. CKD, Inflammation, and Nutritional Status/Malnutrition
1.6. CKD Inflammatory Status, and Treatment
1.7. Personal Experience
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lopes, J.A.; Raimundo, M. Metabolic syndrome, chronic kidney disease, and cardiovascular disease: A dynamic and life-threatening triad. Cardiol. Res. Pract. 2011, 1. [Google Scholar] [CrossRef] [Green Version]
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [Green Version]
- Liyanage, T.; Ninomiya, T.; Jha, V.; Neal, B.; Patrice, H.M.; Okpechi, I.; Zhao, M.H.; Lv, J.; Garg, A.X.; Knight, J.; et al. Worldwide access to treatment for end-stage kidney disease: A systematic review. Lancet 2015, 385, 1975–1982. [Google Scholar] [CrossRef]
- Kazancioǧlu, R. Risk factors for chronic kidney disease: An update. Kidney Int. Suppl. 2013, 3, 368–371. [Google Scholar] [CrossRef] [Green Version]
- Hsu, R.K.; Hsu, C.Y. The Role of Acute Kidney Injury in Chronic Kidney Disease. Semin. Nephrol. 2016, 36, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Liakopoulos, V.; Roumeliotis, S.; Gorny, X.; Dounousi, E.; Mertens, P.R. Oxidative Stress in Hemodialysis Patients: A Review of the Literature. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef]
- Levey, A.S.; Coresh, J. Chronic kidney disease. Lancet 2012, 379, 165–180. [Google Scholar] [CrossRef]
- Podkowińska, A.; Formanowicz, D. Chronic Kidney Disease as Oxidative Stress- and Inflammatory-Mediated Cardiovascular Disease. Antioxidants 2020, 9, 752. [Google Scholar] [CrossRef]
- Rossaint, J.; Oehmichen, J.; Van Aken, H.; Reuter, S.; Pavenstädt, H.J.; Meersch, M.; Unruh, M.; Zarbock, A. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J. Clin. Invest. 2016, 126, 962–974. [Google Scholar] [CrossRef] [Green Version]
- Rosengren, B.I.; Sagstad, S.J.; Karlsen, T.V.; Wiig, H. Isolation of interstitial fluid and demonstration of local proinflammatory cytokine production and increased absorptive gradient in chronic peritoneal dialysis. Am. J. Physiol. - Ren. Physiol. 2013, 304, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.M.; Albulescu, L.; Necula, L.G.; Mambet, C.; Anton, G.; Tanase, C. Inflammation-related mechanisms in chronic kidney disease prediction, progression, and outcome. J. Immunol. Res. 2018, 2018. [Google Scholar] [CrossRef]
- Noce, A.; Albanese, M.; Marrone, G.; Di Lauro, M.; Pietroboni Zaitseva, A.; Palazzetti, D.; Guerriero, C.; Paolino, A.; Pizzenti, G.; Di Daniele, F.; et al. Ultramicronized Palmitoylethanolamide (um-PEA): A New Possible Adjuvant Treatment in COVID-19 patients. Pharmaceuticals 2021, 14, 336. [Google Scholar] [CrossRef]
- Gupta, J.; Mitra, N.; Kanetsky, P.A.; Devaney, J.; Wing, M.R.; Reilly, M.; Shah, V.O.; Balakrishnan, V.S.; Guzman, N.J.; Girndt, M.; et al. Association between albuminuria, kidney function, and inflammatory biomarker profile in CKD in CRIC. Clin. J. Am. Soc. Nephrol. 2012, 7, 1938–1946. [Google Scholar] [CrossRef] [Green Version]
- Munoz Mendoza, J.; Isakova, T.; Cai, X.; Bayes, L.Y.; Faul, C.; Scialla, J.J.; Lash, J.P.; Chen, J.; He, J.; Navaneethan, S.; et al. Inflammation and elevated levels of fibroblast growth factor 23 are independent risk factors for death in chronic kidney disease. Kidney Int. 2017, 91, 711–719. [Google Scholar] [CrossRef] [Green Version]
- Honda, H.; Qureshi, A.R.; Heimbürger, O.; Barany, P.; Wang, K.; Pecoits-Filho, R.; Stenvinkel, P.; Lindholm, B. Serum albumin, C-reactive protein, interleukin 6, and fetuin a as predictors of malnutrition, cardiovascular disease, and mortality in patients with ESRD. Am. J. Kidney Dis. 2006, 47, 139–148. [Google Scholar] [CrossRef]
- Kim, H.J.; Vaziri, N.D. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am. J. Physiol. - Ren. Physiol. 2010, 298, 662–671. [Google Scholar] [CrossRef] [Green Version]
- Ori, Y.; Bergman, M.; Bessler, H.; Zingerman, B.; Levy-Drummer, R.S.; Gafter, U.; Salman, H. Cytokine secretion and markers of inflammation in relation to acidosis among chronic hemodialysis patients. Blood Purif. 2013, 35, 181–186. [Google Scholar] [CrossRef]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [Green Version]
- Dessì, M.; Noce, A.; Agnoli, A.; De Angelis, S.; Fuiano, L.; Tozzo, C.; Taccone-Gallucci, M.; Fuiano, G.; Federici, G. The usefulness of the prognostic inflammatory and nutritional index (PINI) in a haemodialysis population. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 811–815. [Google Scholar] [CrossRef]
- Noce, A.; Canale, M.P.; Capria, A.; Rovella, V.; Tesauro, M.; Splendiani, G.; Annicchiarico-Petruzzelli, M.; Manzuoli, M.; Simonetti, G.; Di Daniele, N. Coronary artery calcifications predict long term cardiovascular events in non diabetic Caucasian hemodialysis patients. Aging 2015, 7, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Morrone, L.F.; Bolasco, P.; Camerini, C.; Cianciolo, G.; Cupisti, A.; Galassi, A.; Mazzaferro, S.; Russo, D.; Russo, L.; Cozzolino, M. Vitamin D in patients with chronic kidney disease: A position statement of the Working Group “Trace Elements and Mineral Metabolism” of the Italian Society of Nephrology. J. Nephrol. 2016, 29, 305–328. [Google Scholar] [CrossRef]
- Pastore, A.; Noce, A.; Di Giovamberardino, G.; De Stefano, A.; Callà, C.; Zenobi, R.; Dessì, M.; Di Daniele, N. Homocysteine, cysteine, folate and vitamin B12 status in type 2 diabetic patients with chronic kidney disease. J. Nephrol. 2015, 28, 571–576. [Google Scholar] [CrossRef] [Green Version]
- Noce, A.; Marrone, G.; Rovella, V.; Cusumano, A.; Di Daniele, N.; Casasco, M. Beneficial effects of physical activity on uremic sarcopenia. Med. dello Sport 2018, 71, 370–392. [Google Scholar] [CrossRef]
- Taccone-Gallucci, M.; Noce, A.; Bertucci, P.; Fabbri, C.; Manca-di-Villahermosa, S.; Della-Rovere, F.R.; De Francesco, M.; Lonzi, M.; Federici, G.; Scaccia, F.; et al. Chronic treatment with statins increases the availability of selenium in the antioxidant defence systems of hemodialysis patients. J. Trace Elem. Med. Biol. 2010, 24, 27–30. [Google Scholar] [CrossRef] [Green Version]
- Ravarotto, V.; Simioni, F.; Pagnin, E.; Davis, P.A.; Calò, L.A. Oxidative stress – chronic kidney disease – cardiovascular disease: A vicious circle. Life Sci. 2018, 210, 125–131. [Google Scholar] [CrossRef]
- Anders, H.J.; Andersen, K.; Stecher, B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013, 83, 1010–1016. [Google Scholar] [CrossRef] [Green Version]
- Kshirsagar, A.V.; Craig, R.G.; Moss, K.L.; Beck, J.D.; Offenbacher, S.; Kotanko, P.; Klemmer, P.J.; Yoshino, M.; Levin, N.W.; Yip, J.K.; et al. Periodontal disease adversely affects the survival of patients with end-stage renal disease. Kidney Int. 2009, 75, 746–751. [Google Scholar] [CrossRef] [Green Version]
- Icardi, A.; Paoletti, E.; De Nicola, L.; Mazzaferro, S.; Russo, R.; Cozzolino, M. Renal anaemia and EPO hyporesponsiveness associated with vitamin D deficiency: The potential role of inflammation. Nephrol. Dial. Transplant. 2013, 28, 1672–1679. [Google Scholar] [CrossRef] [Green Version]
- Autier, P.; Boniol, M.; Pizot, C.; Mullie, P. Vitamin D status and ill health: A systematic review. Lancet Diabetes Endocrinol. 2014, 2, 76–89. [Google Scholar] [CrossRef]
- Murai, I.H.; Fernandes, A.L.; Sales, L.P.; Pinto, A.J.; Goessler, K.F.; Duran, C.S.C.; Silva, C.B.R.; Franco, A.S.; Macedo, M.B.; Dalmolin, H.H.H.; et al. Effect of a Single High Dose of Vitamin D3 on Hospital Length of Stay in Patients With Moderate to Severe COVID-19: A Randomized Clinical Trial. Jama 2021, 903, 1–8. [Google Scholar] [CrossRef]
- Chintam, K.; Chang, A.R. Strategies to Treat Obesity in Patients With CKD. Am. J. Kidney Dis. 2021, 77, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, P.; Díez, J.J. Adipose tissue in renal disease: Clinical significance and prognostic implications. Nephrol. Dial. Transplant. 2010, 25, 2066–2077. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Chonchol, M.B.; Byrne, C.D. CKD and nonalcoholic fatty liver disease. Am. J. kidney Dis. 2014, 64, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Marcuccilli, M.; Chonchol, M. NAFLD and chronic kidney disease. Int. J. Mol. Sci. 2016, 17, 562. [Google Scholar] [CrossRef] [Green Version]
- Satapathy, S.K.; Garg, S.; Chauhan, R.; Sakhuja, P.; Malhotra, V.; Sharma, B.C.; Sarin, S.K. Beneficial effects of tumor necrosis factor-α inhibition by pentoxifylline on clinical, biochemical, and metabolic parameters of patients with nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2004, 99, 1946–1952. [Google Scholar] [CrossRef]
- Laclair, R.; O’Neal, K.; Ofner, S.; Sosa, M.J.; Labarrere, C.A.; Moe, S.M. Precision of biomarkers to define chronic inflammation in CKD. Am. J. Nephrol. 2008, 28, 808–812. [Google Scholar] [CrossRef] [Green Version]
- Yeun, J.Y.; Levine, R.A.; Mantadilok, V.; Kaysen, G.A. C-reactive protein predicts all-cause and cardiovascular mortality in hemodialysis patients. Am. J. Kidney Dis. 2020, 35, 469–476. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Barany, P.; Heimbürger, O.; Pecoits-Filho, R.; Lindholm, B. Mortality, malnutrition, and atherosclerosis in ESRD: What is the role of interleukin-6? Kidney Int. Suppl. 2002, 61. [Google Scholar] [CrossRef] [Green Version]
- Ketteler, M.; Bongartz, P.; Westenfeld, R.; Wildberger, J.E.; Mahnken, A.H.; Böhm, R.; Metzger, T.; Wanner, C.; Jahnen-Dechent, W.; Floege, J. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: A cross-sectional study. Lancet 2003, 361, 827–833. [Google Scholar] [CrossRef]
- Belayev, L.Y.; Palevsky, P.M. The link between acute kidney injury and chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2014, 23, 149–154. [Google Scholar] [CrossRef]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute Kidney Injury and Chronic Kidney Disease as Interconnected Syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Wald, R.; Quinn, R.R.; Luo, J.; Li, P.; Scales, D.C.; Mamdani, M.M.; Ray, J.G. Chronic dialysis and death among survivors of acute kidney injury requiring dialysis. JAMA 2009, 302, 1179–1185. [Google Scholar] [CrossRef] [Green Version]
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int. Suppl. 2012, 2, 1–138.
- Chawla, L.S.; Amdur, R.L.; Amodeo, S.; Kimmel, P.L.; Palant, C.E. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int. 2011, 79, 1361–1369. [Google Scholar] [CrossRef] [Green Version]
- McMahon, G.M. Biomarkers in Nephrology. Am. J. kidney Dis. 2013, 62, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Umbro, I.; Gentile, G.; Tinti, F.; Muiesan, P.; Mitterhofer, A.P. Recent advances in pathophysiology and biomarkers of sepsis-induced acute kidney injury. J. Infect. 2016, 72, 131–142. [Google Scholar] [CrossRef]
- Quinto, B.M.R.; Iizuka, I.J.; Monte, J.C.M.; Santos, B.F.; Pereira, V.; Durão, M.S.; Dalboni, M.A.; Cendoroglo, M.; Santos, O.F.P.; Batista, M.C. TNF-α depuration is a predictor of mortality in critically ill patients under continuous veno-venous hemodiafiltration treatment. Cytokine 2015, 71, 255–260. [Google Scholar] [CrossRef] [Green Version]
- Malard, B.; Lambert, C.; Kellum, J.A. “In vitro comparison of the adsorption of inflammatory mediators by blood purification devices”: A misleading article for clinical practice? Intensive Care Med. Exp. 2019, 7. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1, O2, and the 3 PHDs: How animal cells signal hypoxia to the nucleus. Cell 2001, 107, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Gunaratnam, L.; Bonventre, J.V. HIF in kidney disease and development. J. Am. Soc. Nephrol. 2009, 20, 1877–1887. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, P. HIF-1: An Oxygen Response System with Special Relevance to the Kidney. J. Am. Soc. Nephrol. 2003, 14, 2712–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haase, V.H. The VHL/HIF oxygen-sensing pathway and its relevance to kidney disease. Kidney Int. 2006, 69, 1302–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norman, J.T.; Clark, I.M.; Garcia, P.L. Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int. 2000, 58, 2351–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leaf, D.E.; Christov, M. Dysregulated Mineral Metabolism in AKI. Semin. Nephrol. 2019, 39, 41–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Zhong, X.; Jin, J.; Li, J.; Meng, X.M. Potential targeted therapy and diagnosis based on novel insight into growth factors, receptors, and downstream effectors in acute kidney injury and acute kidney injury-chronic kidney disease progression. Signal Transduct. Target. Ther. 2020, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bozzette, S.A.; Sattler, F.R.; Chiu, J.; Wu, A.W.; Gluckstein, D.; Kemper, C.; Bartok, A.; Niosi, J.; Abramson, I.; Coffman, J.; et al. A controlled trial of early adjunctive treatment with corticosteroids for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome. California Collaborative Treatment Group. New English J. Med. 1990, 323, 1120–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, F.Q.; Assreuy, J.; Moss, D.W.; Rees, D.; Leal, L.M.; Moncada, S.; Carrier, M.; O’Donnell, C.A.; Liew, F.Y. Differential induction of nitric oxide synthase in various organs of the mouse during endotoxaemia: Role of TNF-alpha and IL-1-beta. N. Engl. J. Med. 1994, 81, 211–215. [Google Scholar]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the Eye of the Cytokine Storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [Green Version]
- Terzi, I.; Papaioannou, V.; Papanas, N.; Dragoumanis, C.; Petala, A.; Theodorou, V.; Gioka, T.; Vargemezis, V.; Maltezos, E.; Pneumatikos, I. Alpha1-microglobulin as an early biomarker of sepsis-associated acute kidney injury: A prospective cohort study. Hippokratia 2014, 18, 262–268. [Google Scholar]
- Cho, S.Y.; Choi, J.H. Biomarkers of Sepsis. Infect. Chemother. 2014, 46, 1–12. [Google Scholar] [CrossRef]
- Haase, M.; Bellomo, R.; Devarajan, P.; Schlattmann, P.; Haase-Fielitz, A.; Bagshaw, S.M.; Bogle, R.; Changchun, C.; Constantin, J.M.; Cruz, D.; et al. Accuracy of Neutrophil Gelatinase-Associated Lipocalin (NGAL) in Diagnosis and Prognosis in Acute Kidney Injury: A Systematic Review and Meta-analysis. Am. J. Kidney Dis. 2009, 54, 1012–1024. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhao, S.; Teng, T.; Abdalla, A.E.; Zhu, W.; Xie, L.; Wang, Y.; Guo, X. Systematic Comparison of Two Animal-to-Human Transmitted Human Coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses 2020, 12, 244. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Luo, R.; Wang, K.; Zhang, M.; Wang, Z.; Dong, L.; Li, J.; Yao, Y.; Ge, S.; Xu, G. Kidney impairment is associated with in-hospital death of COVID-19 patients. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.; Hui, D.; Wu, A.; Chan, P.; Cameron, P.; Joynt, G.; Ahuja, A.T.; van der Gast, C. A Major Outbreak of Severe Acute Respiratory Syndrome in Hong Kong. N. Engl. J. Med. 2003, 348, 1986–1994. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hua, S.; Ming, Y.; Cheng, W.; Li-Xia, Y.; Fang, T.; Zhu, H.-Y. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020, 98, 219–227. [Google Scholar]
- Yan, Z.; Meng, X.; Shulan, Z.; Peng, X.; Wei, C.; Wei, J.; Huan, C.; Xin, D.; Hua, Z.; Hongmin, Z. Coagulopathy and Antiphospholipid Antibodies in Patients with Covid-19. N. Engl. J. Med. 2020, 38, 1–3. [Google Scholar]
- Akalin, E.; Azzi, Y.; Bartash, R.; Seethamraju, H.; Parides, M.; Hemmige, V.; Ross, M. Covid-19 and Kidney Transplantation. N. Engl. J. Med. 2020, 328, 2475–2477. [Google Scholar] [CrossRef]
- Naicker, S.; Yang, C.W.; Hwang, S.J.; Liu, B.C.; Chen, J.H.; Jha, V. The Novel Coronavirus 2019 epidemic and kidneys. Kidney Int. 2020, 97, 824–828. [Google Scholar] [CrossRef]
- Cozzolino, M.; Ureña-Torres, P.; Vervloet, M.G.; Brandenburg, V.; Bover, J.; Goldsmith, D.; Larsson, T.E.; Massy, Z.A.; Mazzaferro, S. Is chronic kidney disease-mineral bone disorder (CKD-MBD) really a syndrome? Nephrol. Dial. Transplant. 2014, 29, 1815–1820. [Google Scholar] [CrossRef] [Green Version]
- Mazzaferro, S.; Bagordo, D.; De Martini, N.; Pasquali, M.; Rotondi, S.; Tartaglione, L.; Stenvinkel, P. Inflammation, Oxidative Stress, and Bone in Chronic Kidney Disease in the Osteoimmunology Era. Calcif. Tissue Int. 2021, 108, 452–460. [Google Scholar] [CrossRef]
- Moe, S.; Drüeke, T.; Cunningham, J.; Goodman, W.; Martin, K.; Olgaard, K.; Ott, S.; Sprague, S.; Lameire, N.; Eknoyan, G. Definition, evaluation, and classification of renal osteodystrophy: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006, 69, 1945–1953. [Google Scholar] [CrossRef] [Green Version]
- Cozzolino, M.; Mazzaferro, S. The fibroblast growth factor 23: A new player in the field of cardiovascular, bone and renal disease. Curr. Vasc. Pharmacol. 2010, 8, 404–411. [Google Scholar] [CrossRef]
- Grabner, A.; Mazzaferro, S.; Cianciolo, G.; Krick, S.; Capelli, I.; Rotondi, S.; Ronco, C.; La Manna, G.; Faul, C. Fibroblast Growth Factor 23: Mineral Metabolism and beyond. Contrib. Nephrol. 2017, 190, 83–95. [Google Scholar] [CrossRef]
- Isakova, T.; Xie, H.; Yang, W.; Xie, D.; Anderson, A.H.; Scialla, J.; Wahl, P.; Gutiérrez, O.M.; Steigerwalt, S.; He, J.; et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA J. Am. Med. Assoc. 2011, 305, 2432–2439. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.; Shah, A.; Gutierrez, O.; Ankers, E.; Monroy, M.; Tamez, H.; Steele, D.; Chang, Y.; Camargo, C.A.; Tonelli, M.; et al. Vitamin D levels and early mortality among incident hemodialysis patients. Kidney Int. 2007, 72, 1004–1013. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Grabner, A.; Yanucil, C.; Schramm, K.; Czaya, B.; Krick, S.; Czaja, M.J.; Bartz, R.; Abraham, R.; Di Marco, G.S.; et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 2016, 90, 985–996. [Google Scholar] [CrossRef] [Green Version]
- Scialla, J.J.; Xie, H.; Rahman, M.; Anderson, A.H.; Isakova, T.; Ojo, A.; Zhang, X.; Nessel, L.; Hamano, T.; Grunwald, J.E.; et al. Fibroblast growth factor-23 and cardiovascular events in CKD. J. Am. Soc. Nephrol. 2014, 25, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Mazzaferro, S.; Tartaglione, L.; Rotondi, S.; Bover, J.; Goldsmith, D.; Pasquali, M. News on Biomarkers in CKD-MBD. Semin. Nephrol. 2014, 34, 598–611. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Kopple, J.D. Relative contributions of nutrition and inflammation to clinical outcome in dialysis patients. Am. J. Kidney Dis. 2001, 38, 1343–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enia, G.; Sicuso, C.; Alati, G.; Zoccali, C.; Pustorino, D.; Biondo, A. Subjective global assessment of nutrition in dialysis patients. Nephrol. Dial. Transplant. 1993, 8, 1094–1098. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.; Muscaritoli, M.; Andreozzi, P.; Sgreccia, A.; De Leo, S.; Mazzaferro, S.; Mitterhofer, A.P.; Pasquali, M.; Protopapa, P.; Spagnoli, A.; et al. Sarcopenia and cardiovascular risk indices in patients with chronic kidney disease on conservative and replacement therapy. Nutrition 2019, 62, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Moorthi, R.N.; Avin, K.G. Clinical relevance of sarcopenia in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2017, 26, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Bauersachs, J.; Jaisser, F.; Toto, R. Mineralocorticoid receptor activation and mineralocorticoid receptor antagonist treatment in cardiac and renal diseases. Hypertension 2015, 65, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Rocha, R.; Stier, C.T.; Kifor, I.; Ochoa-Maya, M.R.; Rennke, H.G.; Williams, G.H.; Adler, G.K. Aldosterone: A mediator of myocardial necrosis and renal arteriopathy. Endocrinology 2000, 141, 3871–3878. [Google Scholar] [CrossRef]
- Quinkler, M.; Zehnder, D.; Eardley, K.S.; Lepenies, J.; Howie, A.J.; Hughes, S.V.; Cockwell, P.; Hewison, M.; Stewart, P.M. Increased expression of mineralocorticoid effector mechanisms in kidney biopsies of patients with heavy proteinuria. Circulation 2005, 112, 1435–1443. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.; Joharapurkar, A.; Jain, M. Role of mineralocorticoid receptor antagonists in kidney diseases. Drug Dev. Res. 2020, 1–23. [Google Scholar] [CrossRef]
- Orena, S.; Maurer, T.S.; She, L.; Eudy, R.; Bernardo, V.; Dash, D.; Loria, P.; Banker, M.E.; Tugnait, M.; Okerberg, C.V.; et al. PF-03882845, a non-steroidal mineralocorticoid receptor antagonist, prevents renal injury with reduced risk of hyperkalemia in an animal model of nephropathy. Front. Pharmacol. 2013, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zitt, E.; Eller, K.; Huber, J.M.; Kirsch, A.H.; Tagwerker, A.; Mayer, G.; Rosenkranz, A.R. The selective mineralocorticoid receptor antagonist eplerenone is protective in mild anti-GBM glomerulonephritis. Int. J. Clin. Exp. Pathol. 2011, 4, 606–615. [Google Scholar]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Perkovic, V.; de Zeeuw, D.; Mahaffey, K.W.; Fulcher, G.; Erondu, N.; Shaw, W.; Barrett, T.D.; Weidner-Wells, M.; Deng, H.; Matthews, D.R.; et al. Canagliflozin and renal outcomes in type 2 diabetes: Results from the CANVAS Program randomised clinical trials. Lancet Diabetes Endocrinol. 2018, 6, 691–704. [Google Scholar] [CrossRef]
- Garvey, W.T.; Van Gaal, L.; Leiter, L.A.; Vijapurkar, U.; List, J.; Cuddihy, R.; Ren, J.; Davies, M.J. Effects of canagliflozin versus glimepiride on adipokines and inflammatory biomarkers in type 2 diabetes. Metabolism. 2018, 85, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.A.; Tan, L. Empagliflozin and Canagliflozin Attenuate Inflammatory Cytokines Interferon-Λ, Tumor Necrosis Factor-Α, Interleukin-6: Possible Mechanism of Decreasing Cardiovascular Risk in Diabetes Mellitus. J. Am. Coll. Cardiol. 2018, 71, A1830. [Google Scholar] [CrossRef]
- Kawanami, D.; Matoba, K.; Takeda, Y.; Nagai, Y.; Akamine, T.; Yokota, T.; Sango, K.; Utsunomiya, K. SGLT2 inhibitors as a therapeutic option for diabetic nephropathy. Int. J. Mol. Sci. 2017, 18, 83. [Google Scholar] [CrossRef]
- Terami, N.; Ogawa, D.; Tachibana, H.; Hatanaka, T.; Wada, J.; Nakatsuka, A.; Eguchi, J.; Sato Horiguchi, C.; Nishii, N.; Yamada, H.; et al. Long-term treatment with the sodium glucose cotransporter 2 inhibitor, dapagliflozin, ameliorates glucose homeostasis and diabetic nephropathy in db/db mice. PLoS One 2014, 9, 1–13. [Google Scholar] [CrossRef]
- Dekkers, C.C.J.; Petrykiv, S.; Laverman, G.D.; Cherney, D.Z.; Gansevoort, R.T.; Heerspink, H.J.L. Effects of the SGLT-2 inhibitor dapagliflozin on glomerular and tubular injury markers. Diabetes Obes. Metab. 2018, 20, 1988–1993. [Google Scholar] [CrossRef] [Green Version]
- Noce, A.; Marrone, G.; Di Lauro, M.; Urciuoli, S.; Pietroboni Zaitseva, A.; Wilson Jones, G.; Di Daniele, N.; Romani, A. Cardiovascular Protection of Nephropathic Male Patients by Oral Food Supplements. Cardiovasc. Ther. 2020, 2020. [Google Scholar] [CrossRef]
- Raimann, J.G.; Levin, N.W.; Craig, R.G.; Sirover, W.; Kotanko, P.; Handelman, G. Is Vitamin C Intake too Low in Dialysis Patients? Semin. Dial. 2013, 26, 1–5. [Google Scholar] [CrossRef]
- Gillis, K.; Stevens, K.K.; Bell, E.; Patel, R.K.; Jardine, A.G.; Morris, S.T.W.; Schneider, M.P.; Delles, C.; Mark, P.B. Ascorbic acid lowers central blood pressure and asymmetric dimethylarginine in chronic kidney disease. Clin. Kidney J. 2018, 11, 532–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noce, A.; Bocedi, A.; Campo, M.; Marrone, G.; Di Lauro, M.; Cattani, G.; Di Daniele, N.; Romani, A. A pilot study of a natural food supplement as new possible therapeutic approach in chronic kidney disease patients. Pharmaceuticals 2020, 13, 148. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.; Petramala, L.; Muscaritoli, M.; Cianci, R.; Mazzaferro, S.; Mitterhofer, A.P.; Pasquali, M.; D’Ambrosio, V.; Carta, M.; Ansuini, M.; et al. Α-Lipoic Acid in Patients With Autosomal Dominant Polycystic Kidney Disease. Nutrition 2020, 71. [Google Scholar] [CrossRef] [PubMed]
- Kojima, I.; Tanaka, T.; Inagi, R.; Kato, H.; Yamashita, T.; Sakiyama, A.; Ohneda, O.; Takeda, N.; Sata, M.; Miyata, T.; et al. Protective role of hypoxia-inducible factor-2α against ischemic damage and oxidative stress in the kidney. J. Am. Soc. Nephrol. 2007, 18, 1218–1226. [Google Scholar] [CrossRef]
- Kalisvaart, M.; Schlegel, A.; Umbro, I.; De Haan, J.E.; Scalera, I.; Polak, W.G.; Ijzermans, J.N.M.; Mirza, D.F.; Perera, M.T.P.R.; Isaac, J.I.; et al. The Impact of Combined Warm Ischemia Time on Development of Acute Kidney Injury in Donation after Circulatory Death Liver Transplantation: Stay Within the Golden Hour. Transplantation 2018, 102, 783–793. [Google Scholar] [CrossRef]
- Umbro, I.; Tinti, F.; Scalera, I.; Evison, F.; Gunson, B.; Sharif, A.; Ferguson, J.; Muiesan, P.; Mitterhofer, A.P. Acute kidney injury and post-reperfusion syndrome in liver transplantation. World J. Gastroenterol. 2016, 22, 9314–9323. [Google Scholar] [CrossRef]
- Lai, S.; Mazzaferro, S.; Mitterhofer, A.P.; Bonci, E.; Marotta, P.G.; Pelligra, F.; Murciano, M.; Celani, C.; Troiani, P.; Cimino, G.; et al. Renal involvement and metabolic alterations in adults patients affected by cystic fibrosis. J. Transl. Med. 2019, 17, 1–7. [Google Scholar] [CrossRef]
- Romani, A.; Bernini, R.; Noce, A.; Urciuoli, S.; Lauro, M.D.; Zaitseva, A.P.; Marrone, G.; Daniele, N. Di Potential beneficial effects of extra virgin olive oils characterized by high content in minor polar compounds in nephropathic patients: A pilot study. Molecules 2020, 25, 757. [Google Scholar] [CrossRef]
- Noce, A.; Marrone, G.; Urciuoli, S.; Di Daniele, F.; Di Lauro, M.; Zaitseva, A.P.; Di Daniele, N.; Romani, A. Usefulness of extra virgin olive oil minor polar compounds in the management of chronic kidney disease patients. Nutrients 2021, 13, 581. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2013, 3, 1–150. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tinti, F.; Lai, S.; Noce, A.; Rotondi, S.; Marrone, G.; Mazzaferro, S.; Di Daniele, N.; Mitterhofer, A.P. Chronic Kidney Disease as a Systemic Inflammatory Syndrome: Update on Mechanisms Involved and Potential Treatment. Life 2021, 11, 419. https://doi.org/10.3390/life11050419
Tinti F, Lai S, Noce A, Rotondi S, Marrone G, Mazzaferro S, Di Daniele N, Mitterhofer AP. Chronic Kidney Disease as a Systemic Inflammatory Syndrome: Update on Mechanisms Involved and Potential Treatment. Life. 2021; 11(5):419. https://doi.org/10.3390/life11050419
Chicago/Turabian StyleTinti, Francesca, Silvia Lai, Annalisa Noce, Silverio Rotondi, Giulia Marrone, Sandro Mazzaferro, Nicola Di Daniele, and Anna Paola Mitterhofer. 2021. "Chronic Kidney Disease as a Systemic Inflammatory Syndrome: Update on Mechanisms Involved and Potential Treatment" Life 11, no. 5: 419. https://doi.org/10.3390/life11050419