
Inhibition of Autophagy Enhances the Antitumor Effect of Thioridazine in Acute Lymphoblastic Leukemia Cells

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Cell Viability Assays
2.4. Annexin V-FITC/PI Staining
2.5. Mitochondrial Transmembrane Potential (ΔΨ)
2.6. Lysosomal Staining
2.7. Western Blot Analysis
2.8. Statistical Analyses
3. Results
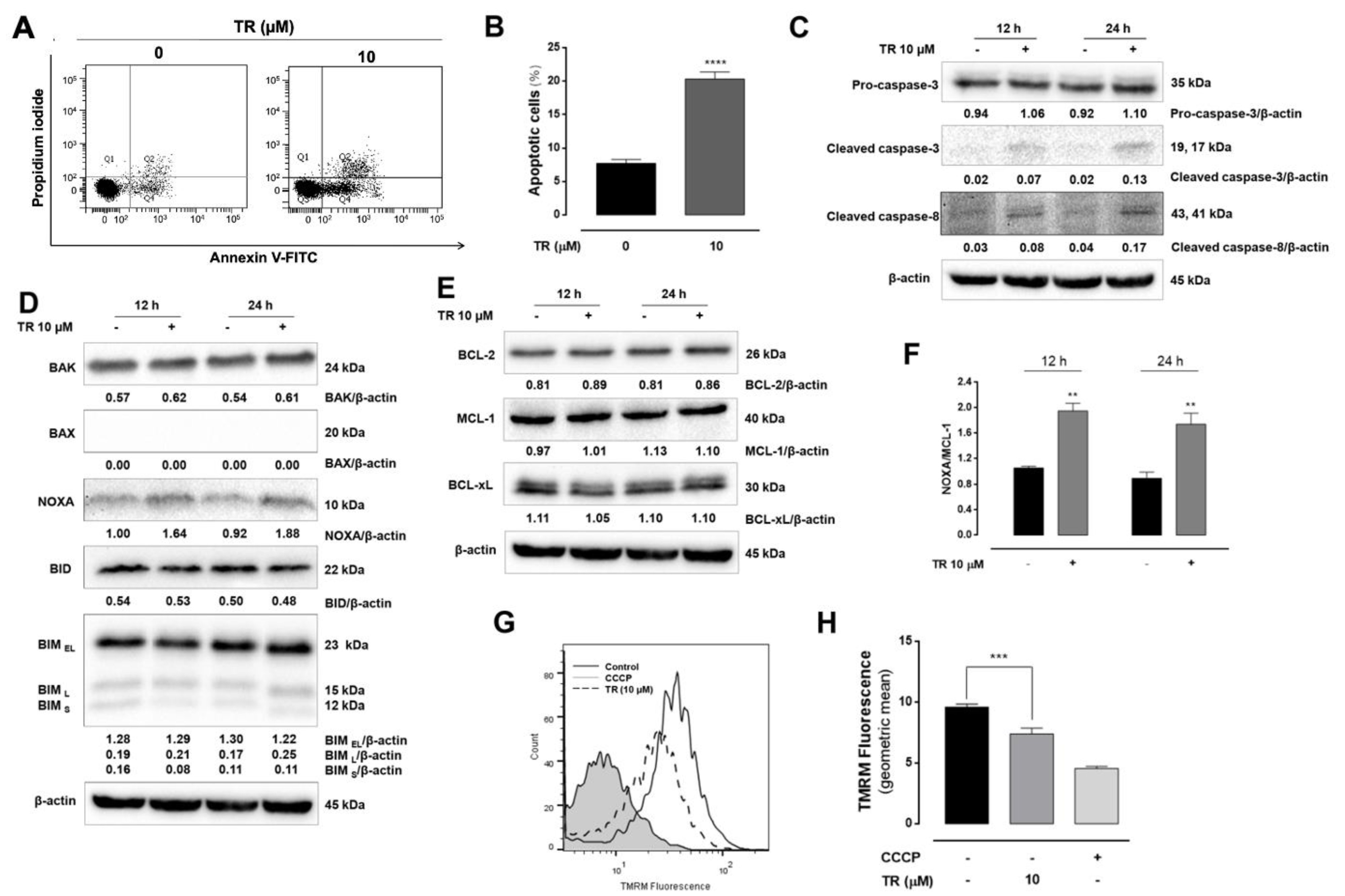
3.1. Thioridazine (TR) Exhibited Potent and Selective Cytotoxicity Against Human T-ALL Jurkat Cells
3.2. TR Induced Caspase-Dependent Apoptosis in Jurkat Cells
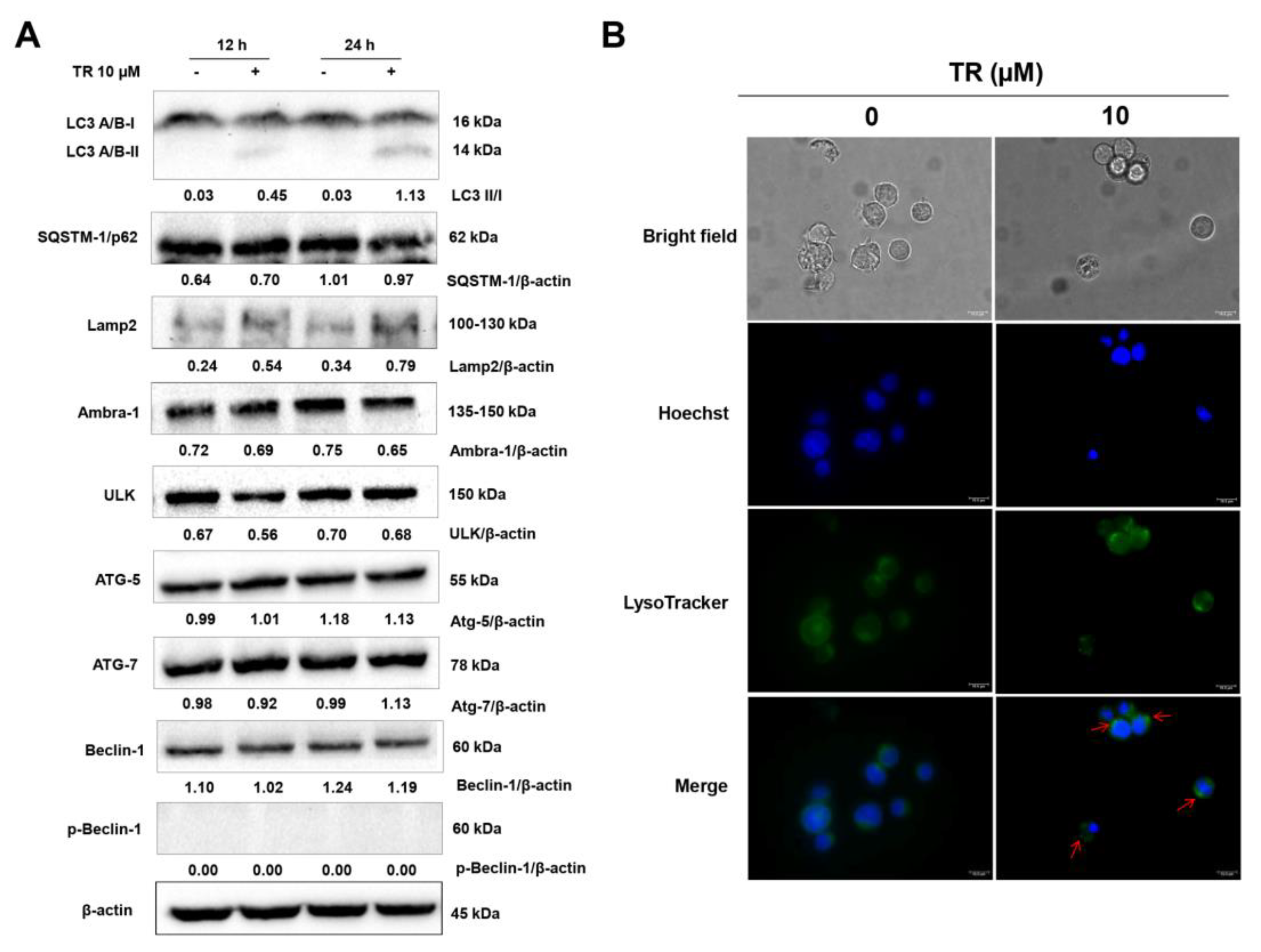
3.3. TR Stimulates Autophagy in Jurkat Cells
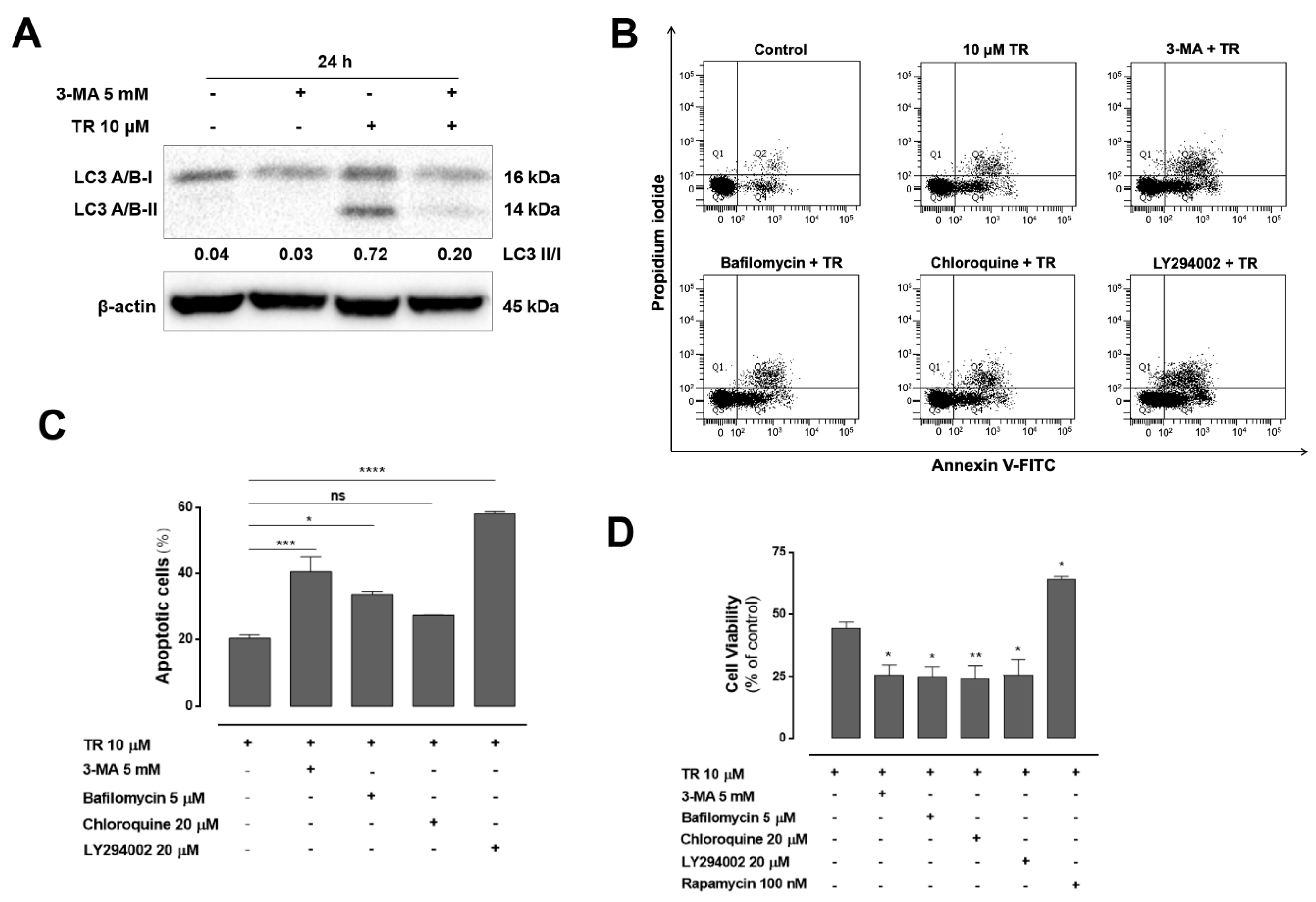
3.4. Inhibition of Autophagy Enhanced TR-Induced Apoptosis in Human Leukemia Jurkat Cells
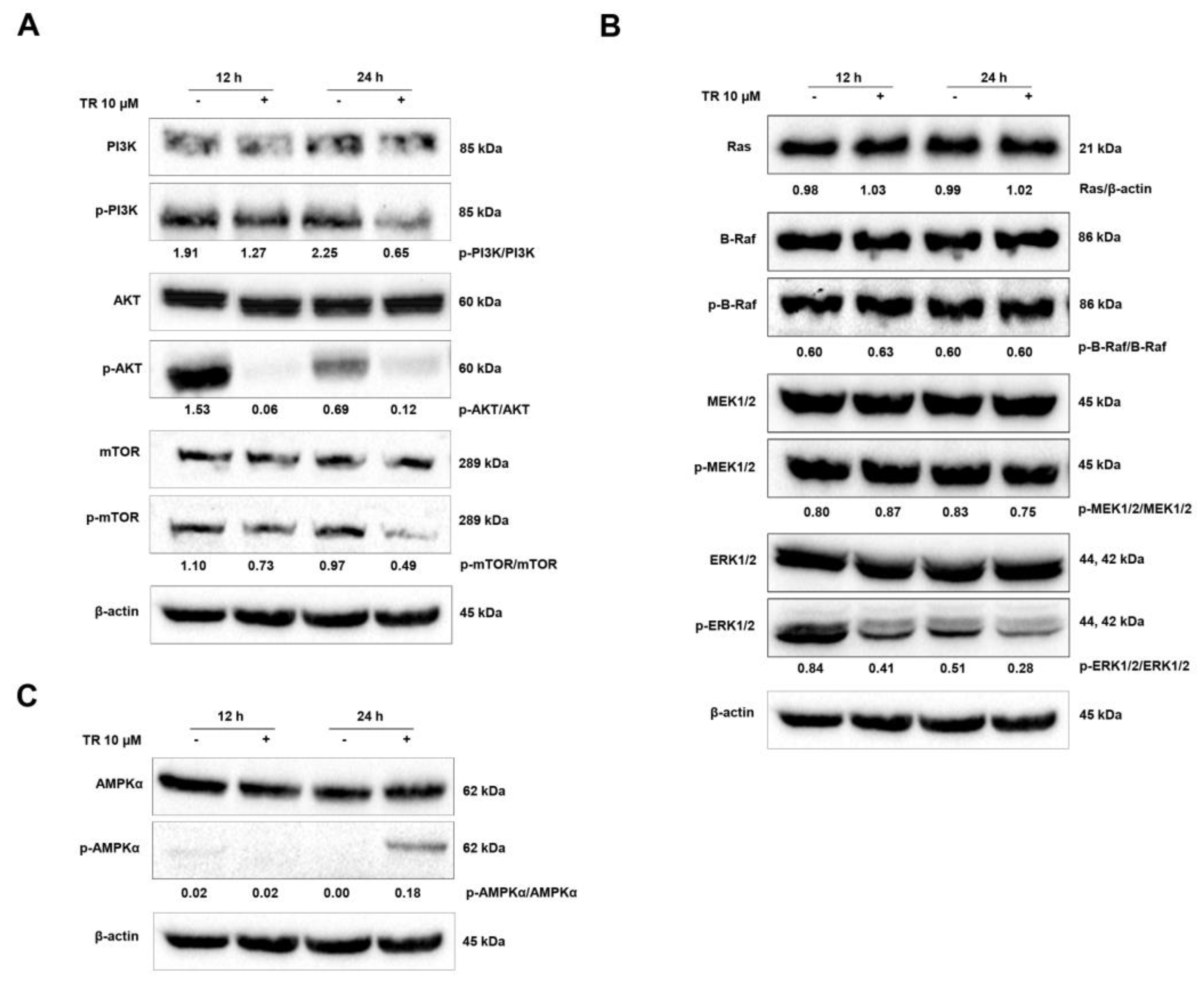
3.5. Modulation of Signaling Pathways Related to the Balance Between Proliferation and Death in Jurkat Cells by TR: PI3K/AKT/mTOR and Ras/Raf/MEK/ERK Cascades
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pui, C.H.; Robison, L.L.; Look, A.T. Acute lymphoblastic leukaemia. Lancet 2008, 371, 1030–1043. [Google Scholar] [CrossRef]
- Ferrando, A.A.; Neuberg, D.S.; Staunton, J.; Loh, M.L.; Huard, C.; Raimondi, S.C.; Behm, F.G.; Pui, C.H.; Downing, J.R.; Gilliland, D.G.; et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002, 1, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, H.E.; Ban, T.A. The history of the psychopharmacology of schizophrenia. Can. J. Psychiatry 1997, 42, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Min, K.J.; Seo, B.R.; Bae, Y.C.; Yoo, Y.H.; Kwon, T.K. Antipsychotic agent thioridazine sensitizes renal carcinoma Caki cells to TRAIL-induced apoptosis through reactive oxygen species-mediated inhibition of akt signaling and downregulation of Mcl-1 and c-FLIP(L). Cell Death Dis. 2014, 5, e1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga, B.; Csonka, A.; Csonka, A.; Molnar, J.; Amaral, L.; Spengler, G. Possible biological and clinical applications of phenothiazines. Anticancer Res. 2017, 37, 5983–5993. [Google Scholar] [PubMed] [Green Version]
- de Faria, P.A.; Bettanin, F.; Cunha, R.L.; Paredes-Gamero, E.J.; Homem-de-Mello, P.; Nantes, I.L.; Rodrigues, T. Cytotoxicity of phenothiazine derivatives associated with mitochondrial dysfunction: A structure-activity investigation. Toxicology 2015, 330, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Michał Otręba, M.; Kośmider, L. In vitro anticancer activity of fluphenazine, perphenazine and prochlorperazine. A review. J. Appl. Toxicol. 2021, 41, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Rho, S.B.; Kim, B.R.; Kang, S. A gene signature-based approach identifies thioridazine as an inhibitor of phosphatidylinositol-3’-kinase (PI3K)/AKT pathway in ovarian cancer cells. Gynecol. Oncol. 2011, 120, 121–127. [Google Scholar] [CrossRef]
- Kang, S.; Dong, S.M.; Kim, B.R.; Park, M.S.; Trink, B.; Byun, H.J.; Rho, S.B. Thioridazine induces apoptosis by targeting the PI3K/Akt/mTOR pathway in cervical and endometrial cancer cells. Apoptosis 2012, 17, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Zhelev, Z.; Ohba, H.; Bakalova, R.; Hadjimitova, V.; Ishikawa, M.; Shinohara, Y.; Baba, Y. Phenothiazines suppress proliferation and induce apoptosis in cultured leukemic cells without any influence on the viability of normal lymphocytes. Phenothiazines and leukemia. Cancer Chemother. Pharm. 2004, 53, 267–275. [Google Scholar] [CrossRef]
- Rai, S.; Tanaka, H.; Suzuki, M.; Espinoza, J.L.; Kumode, T.; Tanimura, A.; Yokota, T.; Oritani, K.; Watanabe, T.; Kanakura, Y.; et al. Chlorpromazine eliminates acute myeloid leukemia cells by perturbing subcellular localization of FLT3-ITD and KIT-D816V. Nat. Commun. 2020, 11, 4147. [Google Scholar] [CrossRef]
- Claudia Tregnago, C.; Da Ros, A.; Porcù, E.; Benetton, M.; Simonato, M.; Simula, L.; Borella, G.; Polato, K.; Minuzzo, S.; Borile, S.; et al. Thioridazine requires calcium influx to induce MLL-AF6-rearranged AML cell death. Blood Adv. 2020, 4, 4417–4429. [Google Scholar] [CrossRef]
- Buckley, N.A.; Whyte, I.M.; Dawson, A.H. Cardiotoxicity more common in thioridazine overdose than with other neuroleptics. J. Toxicol. Clin. Toxicol. 1995, 33, 199–204. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, J.; Fan, X.; Hu, S.; Zhou, F.; Dong, J.; Zhang, S.; Shang, Y.; Jiang, X.; Guo, H.; et al. Chaperone-mediated autophagy regulates proliferation by targeting RND3 in gastric cancer. Autophagy 2016, 12, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Xia, T.; Wang, J.; Wang, Y.; Wang, Y.; Cai, J.; Wang, M.; Chen, Q.; Song, J.; Yu, Z.; Huang, W.; et al. Inhibition of autophagy potentiates anticancer property of 20(S)-ginsenoside Rh2 by promoting mitochondria-dependent apoptosis in human acute lymphoblastic leukaemia cells. Oncotarget 2016, 7, 27336–27349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Brimmell, M.; Mendiola, R.; Mangion, J.; Packham, G. BAX frameshift mutations in cell lines derived from human haemopoietic malignancies are associated with resistance to apoptosis and microsatellite instability. Oncogene 1998, 16, 1803–1812. [Google Scholar] [CrossRef] [Green Version]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaravadi, R.K.; Thompson, C.B. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin. Cancer Res. 2007, 13, 7271–7279. [Google Scholar] [CrossRef] [Green Version]
- Franke, T.F.; Hornik, C.P.; Segev, L.; Shostak, G.A.; Sugimoto, C. PI3K/Akt and apoptosis: Size matters. Oncogene 2003, 22, 8983–8998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meloche, S.; Pouyssegur, J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 2007, 26, 3227–3239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Spengler, G.; Csonka, A.; Molnar, J.; Amaral, L. The anticancer activity of the old neuroleptic phenothiazine-type drug thioridazine. Anticancer Res. 2016, 36, 5701–5706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslostovar, L.; Boyd, A.L.; Almakadi, M.; Collins, T.J.; Leong, D.P.; Tirona, R.G.; Kim, R.B.; Julian, J.A.; Xenocostas, A.; Leber, B.; et al. A phase 1 trial evaluating thioridazine in combination with cytarabine in patients with acute myeloid leukemia. Blood Adv. 2018, 2, 1935–1945. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Ploner, C.; Kofler, R.; Villunger, A. Noxa: At the tip of the balance between life and death. Oncogene 2008, 27 (Suppl. 1), S84–S92. [Google Scholar] [CrossRef]
- Horing, E.; Montraveta, A.; Heine, S.; Kleih, M.; Schaaf, L.; Vohringer, M.C.; Esteve-Arenys, A.; Roue, G.; Colomer, D.; Campo, E.; et al. Dual targeting of MCL1 and NOXA as effective strategy for treatment of mantle cell lymphoma. Br. J. Haematol. 2017, 177, 557–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, N.L.; Derks, I.A.; Berk, E.; Spijker, R.; van Lier, R.A.; Eldering, E. The Noxa/Mcl-1 axis regulates susceptibility to apoptosis under glucose limitation in dividing t cells. Immunity 2006, 24, 703–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naumann, K.; Schmich, K.; Jaeger, C.; Kratz, F.; Merfort, I. Noxa/Mcl-1 balance influences the effect of the proteasome inhibitor MG-132 in combination with anticancer agents in pancreatic cancer cell lines. Anticancer Drugs 2012, 23, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Perlman, H.; Zhang, X.; Chen, M.W.; Walsh, K.; Buttyan, R. An elevated bax/bcl-2 ratio corresponds with the onset of prostate epithelial cell apoptosis. Cell Death Differ. 1999, 6, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Radogna, F.; Dicato, M.; Diederich, M. Cancer-type-specific crosstalk between autophagy, necroptosis and apoptosis as a pharmacological target. Biochem. Pharm. 2015, 94, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cheong, H.; Lu, C.; Lindsten, T.; Thompson, C.B. Therapeutic targets in cancer cell metabolism and autophagy. Nat. Biotechnol. 2012, 30, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528–539. [Google Scholar] [CrossRef]
- Zhu, S.; Cao, L.; Yu, Y.; Yang, L.; Yang, M.; Liu, K.; Huang, J.; Kang, R.; Livesey, K.M.; Tang, D. Inhibiting autophagy potentiates the anticancer activity of ifn1@/ifnalpha in chronic myeloid leukemia cells. Autophagy 2013, 9, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Lu, W.; Li, S.; Li, S.; Liu, J.; Xing, Y.; Zhang, S.; Zhou, J.Z.; Xing, H.; Xu, Y.; et al. Identification of JL1037 as a novel, specific, reversible lysine-specific demethylase 1 inhibitor that induce apoptosis and autophagy of AML cells. Oncotarget 2017, 8, 31901–31914. [Google Scholar] [CrossRef] [Green Version]
- Li, M.L.; Xu, Y.Z.; Lu, W.J.; Li, Y.H.; Tan, S.S.; Lin, H.J.; Wu, T.M.; Li, Y.; Wang, S.Y.; Zhao, Y.L. Chloroquine potentiates the anticancer effect of sunitinib on renal cell carcinoma by inhibiting autophagy and inducing apoptosis. Oncol. Lett. 2018, 15, 2839–2846. [Google Scholar] [CrossRef]
- Ozpolat, B.; Benbrook, D.M. Targeting autophagy in cancer management-strategies and developments. Cancer Manag. Res. 2015, 7, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Han, W.; Sun, J.; Feng, L.; Wang, K.; Li, D.; Pan, Q.; Chen, Y.; Jin, W.; Wang, X.; Pan, H.; et al. Autophagy inhibition enhances daunorubicin-induced apoptosis in K562 cells. PLoS ONE 2011, 6, e28491. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Zhong, L.; Liu, L.; Fang, Y.; Qi, X.; Cao, J.; Yang, B.; He, Q.; Ying, M. Autophagy contributes to dasatinib-induced myeloid differentiation of human acute myeloid leukemia cells. Biochem. Pharm. 2014, 89, 74–85. [Google Scholar] [CrossRef]
- Takahashi, H.; Inoue, J.; Sakaguchi, K.; Takagi, M.; Mizutani, S.; Inazawa, J. Autophagy is required for cell survival under L-asparaginase-induced metabolic stress in acute lymphoblastic leukemia cells. Oncogene 2017, 36, 4267–4276. [Google Scholar] [CrossRef] [Green Version]
- Tamburini, J.; Elie, C.; Bardet, V.; Chapuis, N.; Park, S.; Broet, P.; Cornillet-Lefebvre, P.; Lioure, B.; Ugo, V.; Blanchet, O.; et al. Constitutive phosphoinositide 3-kinase/Akt activation represents a favorable prognostic factor in de novo acute myelogenous leukemia patients. Blood 2007, 110, 1025–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Pi, C.; Wang, G. Inhibition of PI3K/Akt/mTOR pathway by apigenin induces apoptosis and autophagy in hepatocellular carcinoma cells. Biomed. Pharm. 2018, 103, 699–707. [Google Scholar] [CrossRef]
- Silva, A.; Yunes, J.A.; Cardoso, B.A.; Martins, L.R.; Jotta, P.Y.; Abecasis, M.; Nowill, A.E.; Leslie, N.R.; Cardoso, A.A.; Barata, J.T. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J. Clin. Investig. 2008, 118, 3762–3774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.Y.; Lee, K.S.; Choi, Y.K.; Lim, H.J.; Lee, H.G.; Lim, Y.; Lee, Y.H. The antipsychotic agent chlorpromazine induces autophagic cell death by inhibiting the Akt/mTOR pathway in human U-87MG glioma cells. Carcinogenesis 2013, 34, 2080–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.H.; Bai, L.Y.; Tsai, M.H.; Chu, P.C.; Chiu, C.F.; Chen, M.Y.; Chiu, S.J.; Chiang, J.H.; Weng, J.R. Pharmacological exploitation of the phenothiazine antipsychotics to develop novel antitumor agents-A drug repurposing strategy. Sci. Rep. 2016, 6, 27540. [Google Scholar] [CrossRef]
- Jhou, A.J.; Chang, H.C.; Hung, C.C.; Lin, H.C.; Lee, Y.C.; Liu, W.T.; Han, K.F.; Lai, Y.W.; Lin, M.Y.; Lee, C.H. Chlorpromazine, an antipsychotic agent, induces G2/M phase arrest and apoptosis via regulation of the PI3K/AKT/mTOR-mediated autophagy pathways in human oral cancer. Biochem. Pharm. 2021, 184, 114403. [Google Scholar] [CrossRef]
- Alameen, A.A.; Simioni, C.; Martelli, A.M.; Zauli, G.; Ultimo, S.; McCubrey, J.A.; Gonelli, A.; Marisi, G.; Ulivi, P.; Capitani, S.; et al. Healthy CD4+ T lymphocytes are not affected by targeted therapies against the PI3K/Akt/mTOR pathway in T-cell acute lymphoblastic leukemia. Oncotarget 2016, 7, 55690–55703. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, H.C.D.; Colturato-Kido, C.; Ferraz, L.S.; Costa, C.A.; Moraes, V.W.R.; Paredes-Gamero, E.J.; Tersariol, I.L.S.; Rodrigues, T. AMPK activation induced by promethazine increases NOXA expression and Beclin-1 phosphorylation and drives autophagy-associated apoptosis in chronic myeloid leukemia. Chem. Biol. Interact. 2020, 315, 108888. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Der, C.J. Targeting the RAF-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, A.F.; Braun, B.S.; Shannon, K.M. Targeting oncogenic Ras signaling in hematologic malignancies. Blood 2012, 120, 3397–3406. [Google Scholar] [CrossRef] [Green Version]
- Chung, E.; Kondo, M. Role of Ras/Raf/MEK/ERK signaling in physiological hematopoiesis and leukemia development. Immunol. Res. 2011, 49, 248–268. [Google Scholar] [CrossRef]
- Kohno, M.; Pouyssegur, J. Targeting the ERK signaling pathway in cancer therapy. Ann. Med. 2006, 38, 200–211. [Google Scholar] [CrossRef]
- Feifei, L.; Xiaotong, Y.; Meiyu, G.; Min, H. Targeting ERK, an Achilles’ Heel of the MAPK pathway, in cancer therapy. Acta Pharm. Sin. B 2018, 8, 552–562. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colturato-Kido, C.; Lopes, R.M.; Medeiros, H.C.D.; Costa, C.A.; Prado-Souza, L.F.L.; Ferraz, L.S.; Rodrigues, T. Inhibition of Autophagy Enhances the Antitumor Effect of Thioridazine in Acute Lymphoblastic Leukemia Cells. Life 2021, 11, 365. https://doi.org/10.3390/life11040365
Colturato-Kido C, Lopes RM, Medeiros HCD, Costa CA, Prado-Souza LFL, Ferraz LS, Rodrigues T. Inhibition of Autophagy Enhances the Antitumor Effect of Thioridazine in Acute Lymphoblastic Leukemia Cells. Life. 2021; 11(4):365. https://doi.org/10.3390/life11040365
Chicago/Turabian StyleColturato-Kido, Carina, Rayssa M. Lopes, Hyllana C. D. Medeiros, Claudia A. Costa, Laura F. L. Prado-Souza, Letícia S. Ferraz, and Tiago Rodrigues. 2021. "Inhibition of Autophagy Enhances the Antitumor Effect of Thioridazine in Acute Lymphoblastic Leukemia Cells" Life 11, no. 4: 365. https://doi.org/10.3390/life11040365