
Evaluation of Conserved RNA Secondary Structures within and between Geographic Lineages of Zika Virus


Abstract
:1. Introduction
2. Materials and Methods
2.1. Genomic Data Source
2.2. Genomic Alignments According to the Geographical Origin
2.3. Phylogenomics Analysis
2.4. Prediction of Conserved Secondary Structures
3. Results
3.1. Phylogenomics Analysis
3.2. Prediction of Conserved Secondary Structures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

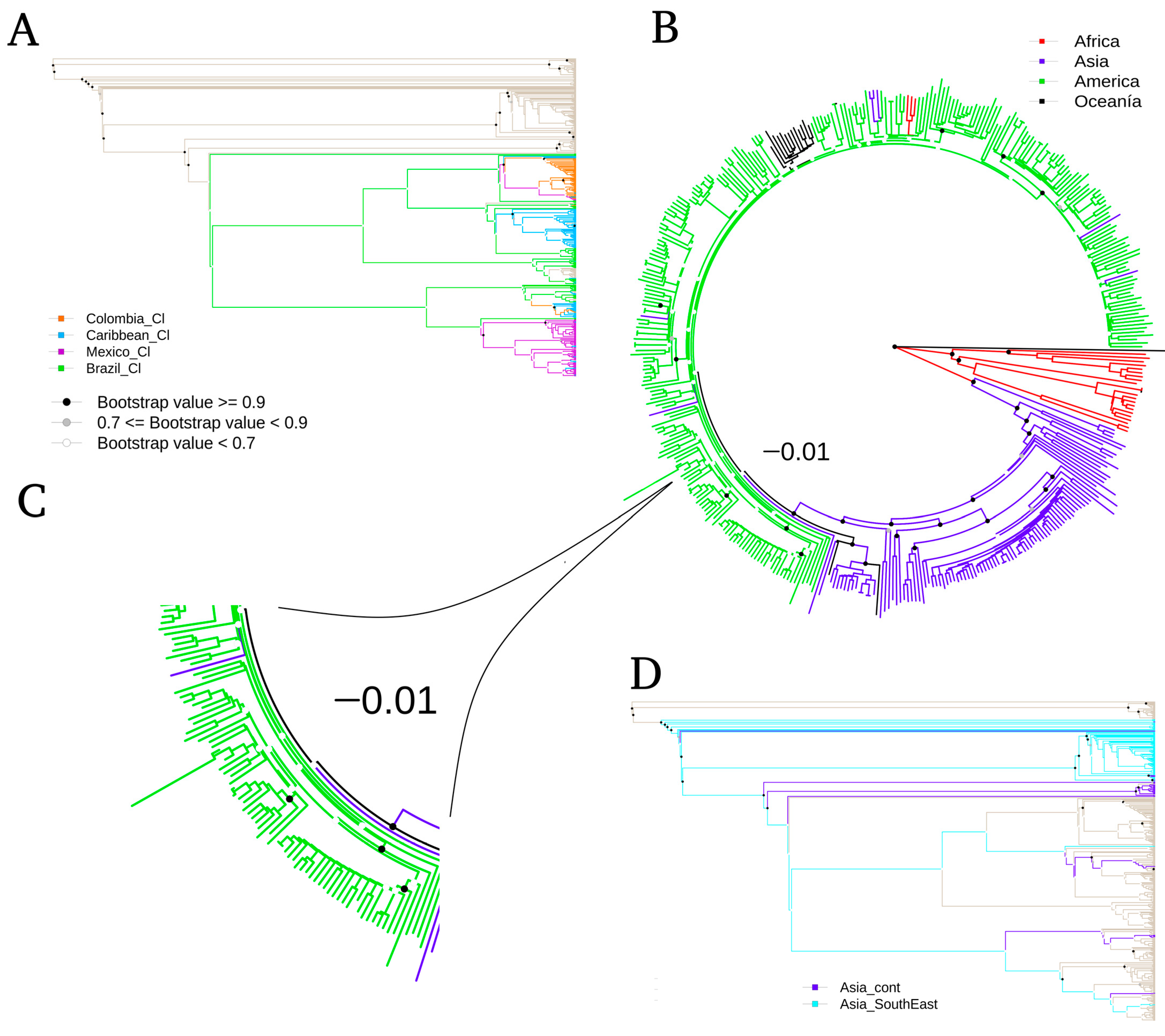{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
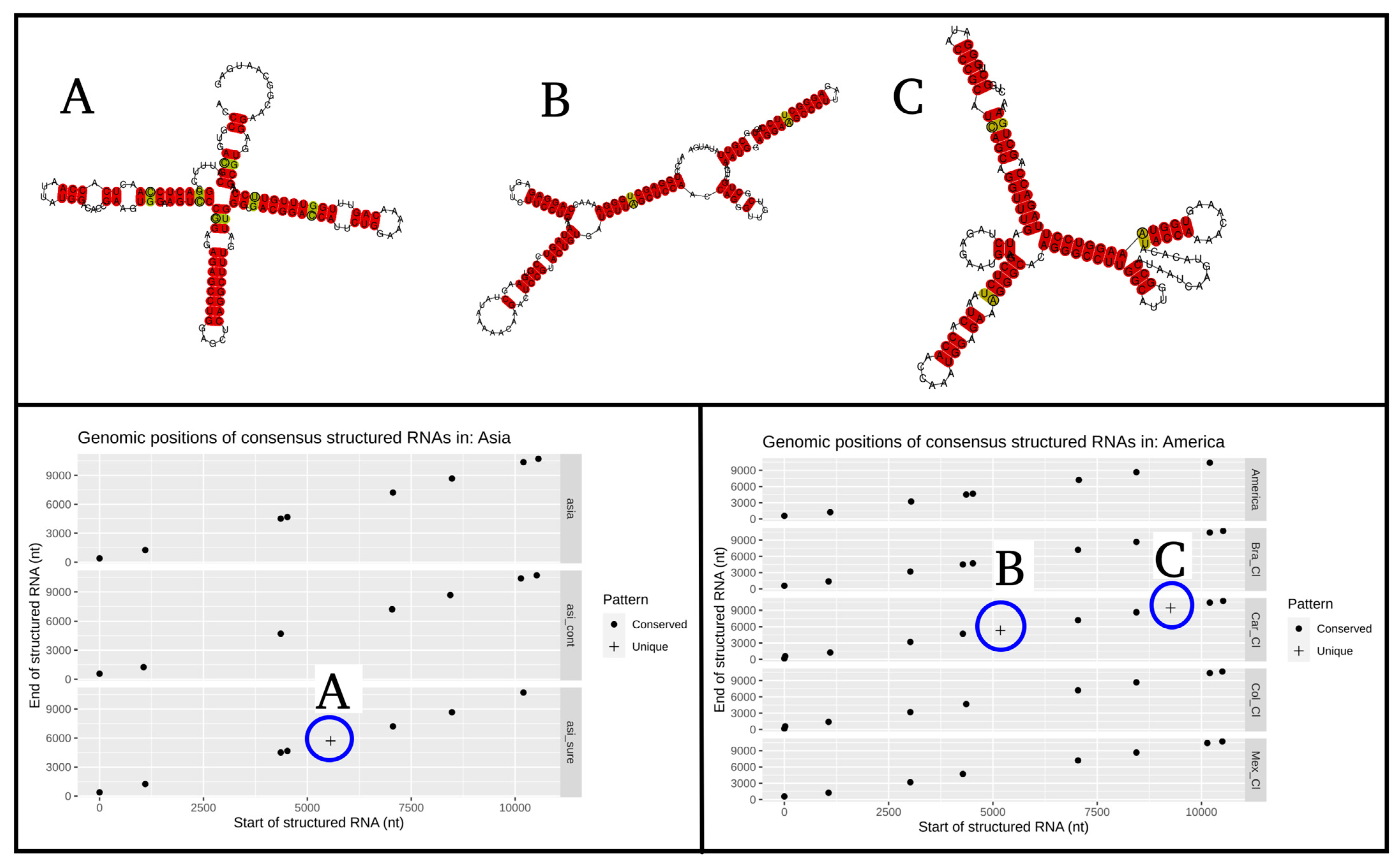{kind=link}
| Region | Countries (n seqs) |
|---|---|
| Africa | Uganda (10), Cape verde (3), Cetral African Republic (3), Guinea (1), Nigeria (1), Senegal (6) |
| Asia_Cont | China (20), Japan (4), South Korea (1), Taiwan (2), India (1) |
| Asia_Southeast | Cambodia (2), Indonesia (1), Malaysia (3), Philippines (1), Singapore (57), Thailand (14) |
| Cl_Brazil | Brazil (54), Argentina (1), Ecuador (3) |
| Cl_Caribbean | Cuba (2), Dominican Republic (12), USA (34), French Guiana (2), Canada (2), Haiti (11), Guadeloupe (7), Puerto Rico (16), Suriname (3), Martinique (1) |
| Cl_Colombia | Colombia (40), Panama (10), Peru (2) |
| Cl_Mexico | Mexico (35), Honduras (14), Nicaragua (16), Guatemala (1) |
| Oceania | Australia (1), French Polynesia (13) |
References
- Wang, L.; Valderramos, S.; Wu, A.; Ouyang, S.; Li, C.; Brasil, P.; Bonaldo, M.; Coates, T.; Nielsen-Saines, K.; Jiang, T.; et al. From Mosquitos to Humans: Genetic Evolution of Zika Virus. Cell Host Microbe 2016, 19, 561–565. [Google Scholar] [CrossRef] [Green Version]
- Oehler, E.; Watrin, L.; Larre, P.; Leparc-Goffart, I.; Lastère, S.; Valour, F.; Baudouin, L.; Mallet, H.; Musso, D.; Ghawche, F. Zika virus infection complicated by Guillain-Barré syndrome—Case report, French Polynesia, December 2013. Eurosurveillance 2014, 19, 20720. [Google Scholar] [CrossRef] [Green Version]
- Fauci, A.; Morens, D. Zika Virus in the Americas—Yet Another Arbovirus Threat. N. Engl. J. Med. 2016, 374, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Ventura, C.; Maia, M.; Bravo-Filho, V.; Góis, A.; Belfort, R. Zika virus in Brazil and macular atrophy in a child with mi-crocephaly. Lancet 2016, 387, 228. [Google Scholar] [CrossRef] [Green Version]
- Metsky, H.; Matranga, C.; Wohl, S.; Schaffner, S.; Freije, C.; Winnicki, S.; West, K.; Qu, J.; Baniecki, M.; Gladden-Young, A.; et al. Zika virus evolution and spread in the Americas. Nature 2017, 546, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, S.; Costa, F.; Garcia-Blanco, M.; Ko, A.; Ribeiro, G.; Saade, G.; Shi, P.; Vasilakis, N. Zika virus: History, emergence, biology, and prospects for control. Antivir. Res. 2016, 130, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Gubler, D. Zika Virus. Clin. Microbiol. Rev. 2016, 29, 487–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, L.; Jamieson, D.; Powers, A.; Honein, M. Zika Virus. N. Engl. J. Med. 2016, 374, 1552–1563. [Google Scholar] [CrossRef]
- Kuno, G.; Chang, G. Full-length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Arch. Virol. 2007, 152, 687–696. [Google Scholar] [CrossRef]
- Rodenhuis-Zybert, I.; Wilschut, J.; Smit, J. Dengue virus life cycle: Viral and host factors modulating infectivity. Cell. Mol. Life Sci. 2010, 67, 2773–2786. [Google Scholar] [CrossRef] [PubMed]
- Romero-López, C.; Berzal-Herranz, A. Unmasking the information encoded as structural motifs of viral RNA genomes: A potential antiviral target. Rev. Med. Virol. 2013, 23, 340–354. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Sanlés, A.; Ríos-Marco, P.; Romero-López, C.; Berzal-Herranz, A. Functional Information Stored in the Conserved Structural RNA Domains of Flavivirus Genomes. Front. Microbiol. 2017, 8, 546. [Google Scholar] [CrossRef]
- Manokaran, G.; Finol, E.; Wang, C.; Gunaratne, J.; Bahl, J.; Ong, E.; Tan, H.; Sessions, O.; Ward, A.; Gubler, D.; et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 2015, 350, 217–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, B.; Laurence, H.; Massey, A.; Costantino, D.; Xie, X.; Yang, Y.; Shi, P.; Nix, J.; Beckham, J.; Kieft, J. Zika virus produces noncoding RNAs using a multi-pseudoknot structure that confounds a cellular exonuclease. Science 2016, 354, 1148–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villordo, S.; Gamarnik, A. Genome cyclization as strategy for flavivirus RNA replication. Virus Res. 2009, 139, 230–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutard, B.; Barral, K.; Lichière, J.; Selisko, B.; Martin, B.; Aouadi, W.; Lombardia, M.; Debart, F.; Vasseur, J.; Guillemot, J.; et al. Zika Virus Methyltransferase: Structure and Functions for Drug Design Perspectives. J. Virol. 2016, 91. [Google Scholar] [CrossRef] [Green Version]
- Sadri Nahand, J.; Bokharaei-Salim, F.; Karimzadeh, M.; Moghoofei, M.; Karampoor, S.; Mirzaei, H.; Tabibzadeh, A.; Jafari, A.; Ghaderi, A.; Asemi, Z.; et al. MicroRNAs and exosomes: Key players in HIV pathogenesis. HIV Med. 2020, 21, 246–278. [Google Scholar] [CrossRef]
- Bruscella, P.; Bottini, S.; Baudesson, C.; Pawlotsky, J.; Feray, C.; Trabucchi, M. Viruses and miRNAs: More Friends than Foes. Front. Microbiol. 2017, 8, 824. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, A.; Shango, J.; Seal, A.; Shukla, D.; Nares, S. Viral miRNAs alter host cell miRNA profiles and modulate innate immune responses. Front Immunol. 2018, 9, 433. [Google Scholar] [CrossRef]
- Bernier, A.; Sagan, S. The Diverse Roles of microRNAs at the Host-Virus Interface. Viruses 2018, 10, 440. [Google Scholar] [CrossRef] [Green Version]
- Scheel, T.; Luna, J.; Liniger, M.; Nishiuchi, E.; Rozen-Gagnon, K.; Shlomai, A.; Auray, G.; Gerber, M.; Fak, J.; Keller, I.; et al. A Broad RNA Virus Survey Reveals Both miRNA Dependence and Functional Sequestration. Cell Host Microbe 2016, 19, 409–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, R.; Kumar, A.; Ingle, H.; Kumar, H. The Interplay Between Viral-Derived miRNAs and Host Immunity during Infection. Front. Immunol. 2020, 10, 3079. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Wei, Y.; Mei, M.; Tang, L.; Sun, L.; Huang, W.; Zhou, J.; Zou, C.; Zhang, S.; Qin, C.; et al. Integrative Analysis of Zika Virus Genome RNA Structure Reveals Critical Determinants of Viral Infectivity. Cell Host Microbe 2018, 24, 875–886.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Center for Biotechnology Information (NCBI). National Library of Medicine (US), National Center for Biotechnology Information: Bethesda, MD, USA. 1988. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 1 July 2020).
- Pickett, B.; Sadat, E.; Zhang, Y.; Noronha, J.; Squires, R.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2011, 40, D593–D598. [Google Scholar] [CrossRef]
- Altschul, S.; Gish, W.; Miller, W.; Myers, E.; Lipman, D. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Stajich, J. The Bioperl Toolkit: Perl Modules for the Life Sciences. Genome Res. 2002, 12, 1611–1618. [Google Scholar] [CrossRef] [Green Version]
- RStudio Team. RStudio: Integrated Development for R. RStudio; PBC: Boston, MA, USA, 2020; Available online: http://www.rstudio.com/ (accessed on 1 July 2020).
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [Green Version]
- Charif, D.; Lobry, J. SeqinR 1.0-2: A contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis. In Structural Approaches to Sequence Evolution: Molecules, Networks, Populations; Biological and Medical Physics, Bastolla, U., Porto, M., Roman, H., Vendruscolo, M., Eds.; Springer: New York, NY, USA, 2007; pp. 207–232. ISBN 978-3-540-35305-8. [Google Scholar]
- Schliep, K. Phangorn: Phylogenetic analysis in R. Bioinformatics 2010, 27, 592–593. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E.; Schliep, K. Ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2018, 35, 526–528. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Geneious Prime. 2020. Available online: https://www.geneious.com (accessed on 1 July 2020).
- Gruber, A.; Findeis, S.; Washielt, S.; Hofacker, I.; Stadler, P. RNAZ 2.0. Biocomputing 2010, 2009, 69–79. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; Available online: https://ggplot2.tidyverse.org (accessed on 1 September 2020)ISBN 978-3-319-24277-4.
- Kalvari, I.; Nawrocki, E.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Tof-fano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2020, 49, D192–D200. [Google Scholar] [CrossRef]
- Grubaugh, N.; Ladner, J.; Kraemer, M.; Dudas, G.; Tan, A.; Gangavarapu, K.; Wiley, M.; White, S.; Thézé, J.; Magnani, D.; et al. Genomic epidemiology reveals multiple introductions of Zika virus into the United States. Nature 2017, 546, 401–405. [Google Scholar] [CrossRef]
- Faria, N.; Quick, J.; Claro, I.; Thézé, J.; de Jesus, J.; Giovanetti, M.; Kraemer, M.; Hill, S.; Black, A.; da Costa, A.; et al. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature 2017, 546, 406–410. [Google Scholar] [CrossRef]
- Finol, E.; Ooi, E. Evolution of Subgenomic RNA Shapes Dengue Virus Adaptation and Epidemiological Fitness. SSRN Electron. J. 2018. [Google Scholar] [CrossRef] [Green Version]
- Göertz, G.; Abbo, S.; Fros, J.; Pijlman, G. Functional RNA during Zika virus infection. Virus Res. 2018, 254, 41–53. [Google Scholar] [CrossRef]
- Jerzak, G.; Bernard, K.; Kramer, L.; Ebel, G. Genetic variation in West Nile virus from naturally infected mosquitoes and birds suggests quasispecies structure and strong purifying selection. J. Gen. Virol. 2005, 86, 2175–2183. [Google Scholar] [CrossRef]
- Filomatori, C. A 5′ RNA element promotes dengue virus RNA synthesis on a circular genome. Genes Dev. 2006, 20, 2238–2249. [Google Scholar] [CrossRef] [Green Version]
- Lodeiro, M.; Filomatori, C.; Gamarnik, A. Structural and Functional Studies of the Promoter Element for Dengue Virus RNA Replication. J. Virol. 2008, 83, 993–1008. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Ray, D.; Zhao, Y.; Dong, H.; Ren, S.; Li, Z.; Guo, Y.; Bernard, K.; Shi, P.; Li, H. Structure and Function of Flavivirus NS5 Methyltransferase. J. Virol. 2007, 81, 3891–3903. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Dong, H.; Zhou, Y.; Shi, P. Genetic Interactions among the West Nile Virus Methyltransferase, the RNA-Dependent RNA Polymerase, and the 5′ Stem-Loop of Genomic RNA. J. Virol. 2008, 82, 7047–7058. [Google Scholar] [CrossRef] [Green Version]
- Bustos-Arriaga, J.; Gromowski, G.; Tsetsarkin, K.; Firestone, C.; Castro-Jiménez, T.; Pletnev, A.; Cedillo-Barrón, L.; Whitehead, S. Decreased accumulation of subgenomic RNA in human cells infected with vaccine candidate DEN4Δ30 increases viral susceptibility to type I interferon. Vaccine 2018, 36, 3460–3467. [Google Scholar] [CrossRef]
- Filomatori, C.; Carballeda, J.; Villordo, S.; Aguirre, S.; Pallarés, H.; Maestre, A.; Sánchez-Vargas, I.; Blair, C.; Fabri, C.; Morales, M.; et al. Dengue Virus Genomic Variation Associated with Mosquito Adaptation Defines the Pattern of Viral Non-Coding RNAs and Fitness in Human Cells. PLOS Pathog. 2017, 13, e1006265. [Google Scholar] [CrossRef] [PubMed]
- Guedes, D.; Paiva, M.; Donato, M.; Barbosa, P.; Krokovsky, L.; Rocha, S.; Saraiva, K.; Crespo, M.; Rezende, T.; Wallau, G.; et al. Zika virus replication in the mosquito Culex quinquefasciatus in Brazil. Emerg. Microbes Infect. 2017, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kawai, Y.; Nakayama, E.; Takahashi, K.; Taniguchi, S.; Shibasaki, K.; Kato, F.; Maeki, T.; Suzuki, T.; Tajima, S.; Saijo, M.; et al. Increased growth ability and pathogenicity of American- and Pacific-subtype Zika virus (ZIKV) strains compared with a Southeast Asian-subtype ZIKV strain. PLoS Negl. Trop. Dis. 2019, 13, e0007387. [Google Scholar] [CrossRef] [Green Version]
- Schnettler, E.; Sterken, M.; Leung, J.; Metz, S.; Geertsema, C.; Goldbach, R.; Vlak, J.; Kohl, A.; Khromykh, A.; Pijlman, G. Noncoding Flavivirus RNA Displays RNA Interference Suppressor Activity in Insect and Mammalian Cells. J. Virol. 2012, 86, 13486–13500. [Google Scholar] [CrossRef] [Green Version]






| Length | Identical Sites | Mean Pairwise Identity | |||||
|---|---|---|---|---|---|---|---|
| Region | Sequences(n) | Median | Range | N° Columns | % | % | SD |
| Global | 410 | 10,729 | 10,368–11,119 | 7205 | 64.8 | 98.29 | 0.031 |
| Africa | 24 | 10,782 | 10,617–11,119 | 8917 | 80.2 | 94.11 | 0.037 |
| Asia | 106 | 10,762 | 10,415–10,808 | 8970 | 83.0 | 99.01 | 0.009 |
| Oceania | 14 | 10,644 | 10,585–11,155 | 11,021 | 98.8 | 99.86 | 0.001 |
| America | 266 | 10,692 | 10,368–10,864 | 8973 | 82.6 | 99.59 | 0.001 |
| Brazil_Cl | 58 | 10,752 | 10,455–10,864 | 10,288 | 94.7 | 99.65 | 0.001 |
| Colombia_Cl | 53 | 10,659 | 10,385–10,808 | 10,375 | 96.0 | 99.80 | 0.002 |
| Mexico_Cl | 66 | 10,696 | 10,398–10,807 | 10,191 | 94.3 | 99.75 | 0.001 |
| Caribbean_CL | 89 | 10,727 | 10,368–10,808 | 9986 | 92.4 | 99.55 | 0.002 |
| Annotated Structure | Window | p.ident | Length | Mismatch | gap.open | q.start | q.end | s.start | s.end | E-Value | Bit-Score |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Flavivirus DB | 16 | 100 | 29 | 0 | 0 | 122 | 150 | 1 | 29 | 3.29 × 10−11 | 49.6 |
| Flavi_SLA | 1 | 100 | 57 | 0 | 0 | 1 | 57 | 17 | 73 | 4.48 × 10−25 | 95.7 |
| Flavi_CRE | no hits | / | / | / | / | / | / | / | / | / | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicolas Calderon, K.; Fabian Galindo, J.; Bermudez-Santana, C.I. Evaluation of Conserved RNA Secondary Structures within and between Geographic Lineages of Zika Virus. Life 2021, 11, 344. https://doi.org/10.3390/life11040344
Nicolas Calderon K, Fabian Galindo J, Bermudez-Santana CI. Evaluation of Conserved RNA Secondary Structures within and between Geographic Lineages of Zika Virus. Life. 2021; 11(4):344. https://doi.org/10.3390/life11040344
Chicago/Turabian StyleNicolas Calderon, Kevin, Johan Fabian Galindo, and Clara Isabel Bermudez-Santana. 2021. "Evaluation of Conserved RNA Secondary Structures within and between Geographic Lineages of Zika Virus" Life 11, no. 4: 344. https://doi.org/10.3390/life11040344