
Generation and Characterization of the Drosophila melanogaster paralytic Gene Knock-Out as a Model for Dravet Syndrome

 , and
, and
Abstract
:1. Introduction
1.1. Dravet Syndrome
1.2. The Paralytic Gene of Drosophila
2. Materials and Methods
2.1. Molecular Biology
2.2. Generation of the paraKO Allele
2.3. Drosophila Husbandry and Genetics
2.4. Seizure Phenotypes
2.5. Negative Geotaxis
2.6. Flight Assay
2.7. Locomotion Assay
2.8. Learning and Memory
2.9. Electrophysiology
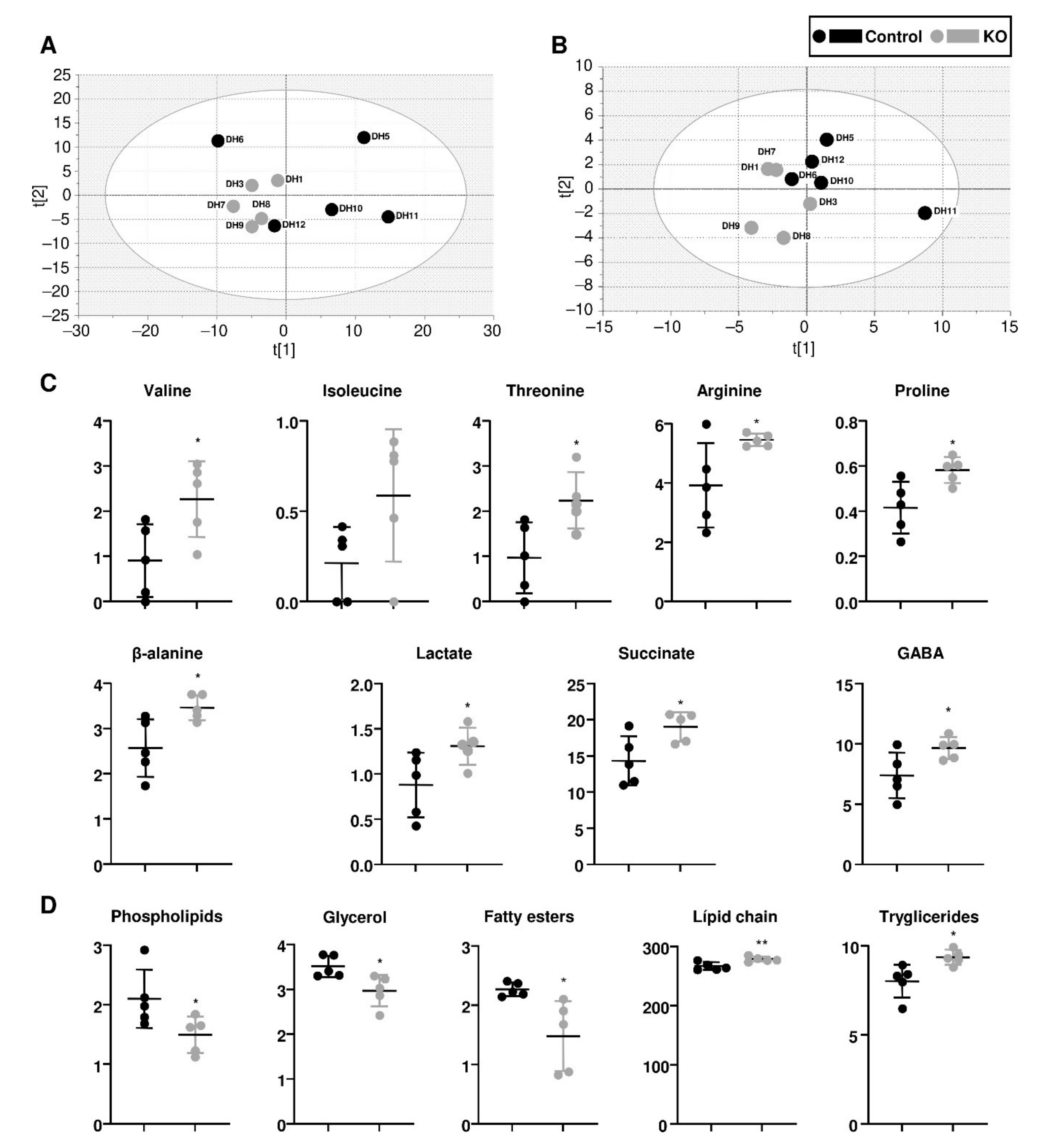
2.10. Metabolomics
2.11. Statistical Analysis
3. Results and Discussion
3.1. Generation and Validation of the para Knock-Out
3.2. The paraKO Flies Suffer Additional Neuromuscular and Cognitive Alterations
3.3. The paraKO Larvae Have Electrophysiological Defects
3.4. The Metabolome of paraKO Fly Heads
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Use | Primer Sequence | Direction |
|---|---|---|
| Generation of the A arm of homology | AGAGGCTAGCCAGTATTCGGGCAACTGCTG | Forward |
| AGAGAGGTACCTGTCTATGCGGCCAACGATGA | Reverse | |
| Generation of the B arm of homology | AGAGAACTAGTTCCGACTCGATATCTGAGGAAG | Forward |
| GTGGACATATGCTGTTCTAGT | Reverse | |
| Validation of the A arm recombination | TCGCTGGTGGTAG | Forward |
| CGAAGTTATGGTACACT | Reverse | |
| Validation of the B arm recombination | ATAGGAAGTTATCACTAGTTCCG | Forward |
| GACAGAATAGACAGTATGCAG | Reverse | |
| paraqPCR | CTGAACATGAAAAGCAGAAGG | Forward |
| CCTGTTCAAGTGTAGGATCCG | Reverse | |
| rp49qPCR (control) | CGTTTACTGCGGCGAGAT | Forward |
| GCGCTCGACAATCTCCTT | Reverse | |
| Validation of the excision by PCR | CGAAGATGAGGAAGACACAT | Forward |
| GACAGAATAGACAGTATGCAG | Reverse |
| Stock | Origin |
|---|---|
| white1118 | B#3605 |
| Canton-S | B#64349 |
| Ubiquitin-Gal43xP3-GFP | Luis Alberto Baena-Lopez |
| hs-Cre; TM2/TM6B | B#6936 |
| y1 w*/Dp(2;Y)G, P{w+mC = hs-hid}Y; P{ry+t7.2 = 70FLP}23 P{v+t1.8 = 70I-SceI}4A/TM3, P{w+mC = hs-hid}14, Sb1 | B#25679 |
| paraKO/FM7c-GFP | This work |
| Df(1)FDD-0230908, w1118/FM7c | B#23296 |
| parabss1 | Mark Tanouye |
References
- Dravet, C. The core Dravet syndrome phenotype. Epilepsia 2011, 52, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Gataullina, S.; Dulac, O. From genotype to phenotype in Dravet disease. Seizure 2017, 44, 58–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrini, R. Dravet syndrome: The main issues. Eur. J. Paediatr. Neurol. 2012, 16, S1–S4. [Google Scholar] [CrossRef] [PubMed]
- De Jonghe, P. Molecular genetics of Dravet syndrome. Dev. Med. Child Neurol. 2011, 53, 7–10. [Google Scholar] [CrossRef]
- Claes, L.; Del-Favero, J.; Ceulemans, B.; Lagae, L.; Van Broeckhoven, C.; De Jonghe, P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am. J. Hum. Genet. 2001, 68, 1327–1332. [Google Scholar] [CrossRef] [Green Version]
- Ogiwara, I.; Miyamoto, H.; Morita, N.; Atapour, N.; Mazaki, E.; Inoue, I.; Takeuchi, T.; Itohara, S.; Yanagawa, Y.; Obata, K.; et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: A circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci. 2007, 27, 5903–5914. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.H.; Mantegazza, M.; Westenbroek, R.E.; Robbins, C.A.; Kalume, F.; Burton, K.A.; Spain, W.J.; McKnight, G.S.; Scheuer, T.; Catterall, W.A. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 2006, 9, 1142–1149. [Google Scholar] [CrossRef]
- Pearson-Smith, J.N.; Patel, M. Metabolic Dysfunction and Oxidative Stress in Epilepsy. Int. J. Mol. Sci. 2017, 18, 2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, A.; Musto, A.E. The role of inflammation in the development of epilepsy. J. Neuroinflammation 2018, 15, 144. [Google Scholar] [CrossRef]
- Boguszewicz, L.; Jamroz, E.; Ciszek, M.; Emich-Widera, E.; Kijonka, M.; Banasik, T.; Skorupa, A.; Sokol, M. NMR-based metabolomics in pediatric drug resistant epilepsy—Preliminary results. Sci. Rep. 2019, 9, 15035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef] [PubMed]
- Auvin, S.; Avbersek, A.; Bast, T.; Chiron, C.; Guerrini, R.; Kaminski, R.M.; Lagae, L.; Muglia, P.; Cross, J.H. Drug Development for Rare Paediatric Epilepsies: Current State and Future Directions. Drugs 2019, 79, 1917–1935. [Google Scholar] [CrossRef] [PubMed]
- Devinsky, O.; Marsh, E.; Friedman, D.; Thiele, E.; Laux, L.; Sullivan, J.; Miller, I.; Flamini, R.; Wilfong, A.; Filloux, F.; et al. Cannabidiol in patients with treatment-resistant epilepsy: An open-label interventional trial. Lancet Neurol. 2016, 15, 270–278. [Google Scholar] [CrossRef]
- Parihar, R.; Ganesh, S. The SCN1A gene variants and epileptic encephalopathies. J. Hum. Genet. 2013, 58, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Cutillo, C.M.; Austin, C.P.; Groft, S.C. A Global Approach to Rare Diseases Research and Orphan Products Development: The International Rare Diseases Research Consortium (IRDiRC). Adv. Exp. Med. Biol. 2017, 1031, 349–369. [Google Scholar] [PubMed]
- Loughney, K.; Kreber, R.; Ganetzky, B. Molecular analysis of the para locus, a sodium channel gene in Drosophila. Cell 1989, 58, 1143–1154. [Google Scholar] [CrossRef]
- Parker, L.; Howlett, I.C.; Rusan, Z.M.; Tanouye, M.A. Seizure and epilepsy: Studies of seizure disorders in Drosophila. Int. Rev. Neurobiol. 2011, 99, 1–21. [Google Scholar] [PubMed] [Green Version]
- Schutte, R.J.; Schutte, S.S.; Algara, J.; Barragan, E.V.; Gilligan, J.; Staber, C.; Savva, Y.A.; Smith, M.A.; Reenan, R.; O’Dowd, D.K. Knock-in model of Dravet syndrome reveals a constitutive and conditional reduction in sodium current. J. Neurophysiol. 2014, 112, 903–912. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.H.; Giachello, C.N.; Baines, R.A. Seizure control through genetic and pharmacological manipulation of Pumilio in Drosophila: A key component of neuronal homeostasis. Dis. Model. Mech. 2017, 10, 141–150. [Google Scholar]
- Baena-Lopez, L.A.; Alexandre, C.; Mitchell, A.; Pasakarnis, L.; Vincent, J.P. Accelerated homologous recombination and subsequent genome modification in Drosophila. Development 2013, 140, 4818–4825. [Google Scholar] [CrossRef] [Green Version]
- Mituzaite, J.; Petersen, R.; Claridge-Chang, A.; Baines, R.A. Characterization of Seizure Induction Methods in Drosophila. eNeuro 2021, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.O.; Escala, W.; Ruan, K.; Zhai, R.G. Assaying locomotor, learning, and memory deficits in Drosophila models of neurodegeneration. J. Vis. Exp. 2011, 49, 2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babcock, D.T.; Ganetzky, B. An improved method for accurate and rapid measurement of flight performance in Drosophila. J. Vis. Exp. 2014, 84, e51223. [Google Scholar] [CrossRef] [Green Version]
- Stone, B.; Burke, B.; Pathakamuri, J.; Coleman, J.; Kuebler, D. A low-cost method for analyzing seizure-like activity and movement in Drosophila. J. Vis. Exp. 2014, 84, e51460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marley, R.; Baines, R.A. Increased persistent Na+ current contributes to seizure in the slamdance bang-sensitive Drosophila mutant. J. Neurophysiol. 2011, 106, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez Del Amo, V.; Palomino-Schatzlein, M.; Seco-Cervera, M.; Garcia-Gimenez, J.L.; Pallardo, F.V.; Pineda-Lucena, A.; Galindo, M.I. A Drosophila model of GDAP1 function reveals the involvement of insulin signalling in the mitochondria-dependent neuromuscular degeneration. Biochim. Biophys. Acta 2017, 1863, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Ulrich, E.L.; Akutsu, H.; Doreleijers, J.F.; Harano, Y.; Ioannidis, Y.E.; Lin, J.; Livny, M.; Mading, S.; Maziuk, D.; Miller, Z.; et al. BioMagResBank. Nucleic Acids Res. 2008, 36, D402–D408. [Google Scholar] [CrossRef] [Green Version]
- Parker, L.; Padilla, M.; Du, Y.; Dong, K.; Tanouye, M.A. Drosophila as a model for epilepsy: Bss is a gain-of-function mutation in the para sodium channel gene that leads to seizures. Genetics 2011, 187, 523–534. [Google Scholar] [CrossRef] [Green Version]
- Griffin, A.; Hamling, K.R.; Hong, S.; Anvar, M.; Lee, L.P.; Baraban, S.C. Preclinical Animal Models for Dravet Syndrome: Seizure Phenotypes, Comorbidities and Drug Screening. Front. Pharmacol. 2018, 9, 573. [Google Scholar] [CrossRef] [Green Version]
- Shmuely, S.; Sisodiya, S.M.; Gunning, W.B.; Sander, J.W.; Thijs, R.D. Mortality in Dravet syndrome: A review. Epilepsy Behav. 2016, 64, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Cheah, C.S.; Yu, F.H.; Westenbroek, R.E.; Kalume, F.K.; Oakley, J.C.; Potter, G.B.; Rubenstein, J.L.; Catterall, W.A. Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 14646–14651. [Google Scholar] [CrossRef] [Green Version]
- Dutton, S.B.; Makinson, C.D.; Papale, L.A.; Shankar, A.; Balakrishnan, B.; Nakazawa, K.; Escayg, A. Preferential inactivation of Scn1a in parvalbumin interneurons increases seizure susceptibility. Neurobiol. Dis. 2013, 49, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Kalume, F.; Westenbroek, R.E.; Cheah, C.S.; Yu, F.H.; Oakley, J.C.; Scheuer, T.; Catterall, W.A. Sudden unexpected death in a mouse model of Dravet syndrome. J. Clin. Investig. 2013, 123, 1798–1808. [Google Scholar] [CrossRef] [Green Version]
- Oakley, J.C.; Kalume, F.; Yu, F.H.; Scheuer, T.; Catterall, W.A. Temperature- and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc. Natl. Acad. Sci. USA 2009, 106, 3994–3999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, A.D.; Kalume, F.; Westenbroek, R.E.; Catterall, W.A. A more efficient conditional mouse model of Dravet syndrome: Implications for epigenetic selection and sex-dependent behaviors. J. Neurosci. Methods 2019, 325, 108315. [Google Scholar] [CrossRef] [PubMed]
- Ricobaraza, A.; Mora-Jimenez, L.; Puerta, E.; Sanchez-Carpintero, R.; Mingorance, A.; Artieda, J.; Nicolas, M.J.; Besne, G.; Bunuales, M.; Gonzalez-Aparicio, M.; et al. Epilepsy and neuropsychiatric comorbidities in mice carrying a recurrent Dravet syndrome SCN1A missense mutation. Sci. Rep. 2019, 9, 14172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gitiaux, C.; Chemaly, N.; Quijano-Roy, S.; Barnerias, C.; Desguerre, I.; Hully, M.; Chiron, C.; Dulac, O.; Nabbout, R. Motor neuropathy contributes to crouching in patients with Dravet syndrome. Neurology 2016, 87, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Nolan, K.; Camfield, C.S.; Camfield, P.R. Coping with a child with Dravet syndrome: Insights from families. J. Child Neurol. 2008, 23, 690–694. [Google Scholar] [CrossRef]
- Satta, V.; Alonso, C.; Diez, P.; Martin-Suarez, S.; Rubio, M.; Encinas, J.M.; Fernandez-Ruiz, J.; Sagredo, O. Neuropathological Characterization of a Dravet Syndrome Knock-In Mouse Model Useful for Investigating Cannabinoid Treatments. Front. Mol. Neurosci. 2021, 13, 602801. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Dravet Syndrome: A Sodium Channel Interneuronopathy. Curr. Opin. Physiol. 2018, 2, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Sinoo, C.; de Lange, I.M.; Westers, P.; Gunning, W.B.; Jongmans, M.J.; Brilstra, E.H. Behavior problems and health-related quality of life in Dravet syndrome. Epilepsy Behav. 2019, 90, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.H.; He, M.; Baines, R.A. Seizure suppression through manipulating splicing of a voltage-gated sodium channel. Brain 2015, 138, 891–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wengert, E.R.; Patel, M.K. The Role of the Persistent Sodium Current in Epilepsy. Epilepsy Curr. 2021, 21, 40–47. [Google Scholar] [CrossRef]
- Rhodes, T.H.; Lossin, C.; Vanoye, C.G.; Wang, D.W.; George, A.L., Jr. Noninactivating voltage-gated sodium channels in severe myoclonic epilepsy of infancy. Proc. Natl. Acad. Sci. USA 2004, 101, 11147–11152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasuya, J.; Iyengar, A.; Chen, H.L.; Lansdon, P.; Wu, C.F.; Kitamoto, T. Milk-whey diet substantially suppresses seizure-like phenotypes of para(Shu), a Drosophila voltage-gated sodium channel mutant. J. Neurogenet. 2019, 33, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Radlicz, C.; Chambers, A.; Olis, E.; Kuebler, D. The addition of a lipid-rich dietary supplement eliminates seizure-like activity and paralysis in the Drosophila bang sensitive mutants. Epilepsy Res. 2019, 155, 106153. [Google Scholar] [CrossRef]
- Honda, T. Amino acid metabolism in the brain with convulsive disorders. Part 3: Free amino acid patterns in cerebrospinal fluid in infants and children with convulsive disorders. Brain Dev. 1984, 6, 27–32. [Google Scholar] [CrossRef]
- Patel, M. A Metabolic Paradigm for Epilepsy. Epilepsy Curr. 2018, 18, 318–322. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, N.; Curatolo, N.; Benoist, J.F.; Auvin, S. Ketogenic diet exhibits anti-inflammatory properties. Epilepsia 2015, 56, e95–e98. [Google Scholar] [CrossRef]
- Gietzen, D.W.; Lindstrom, S.H.; Sharp, J.W.; Teh, P.S.; Donovan, M.J. Indispensable Amino Acid-Deficient Diets Induce Seizures in Ketogenic Diet-Fed Rodents, Demonstrating a Role for Amino Acid Balance in Dietary Treatments for Epilepsy. J. Nutr. 2018, 148, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.G.; Rowley, S.; Fulton, R.; Dinday, M.T.; Baraban, S.C.; Patel, M. Altered Glycolysis and Mitochondrial Respiration in a Zebrafish Model of Dravet Syndrome. eNeuro 2016, 3, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrogini, P.; Torquato, P.; Bartolini, D.; Albertini, M.C.; Lattanzi, D.; Di Palma, M.; Marinelli, R.; Betti, M.; Minelli, A.; Cuppini, R.; et al. Excitotoxicity, neuroinflammation and oxidant stress as molecular bases of epileptogenesis and epilepsy-derived neurodegeneration: The role of vitamin E. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- Tiraboschi, E.; Martina, S.; van der Ent, W.; Grzyb, K.; Gawel, K.; Cordero-Maldonado, M.L.; Poovathingal, S.K.; Heintz, S.; Satheesh, S.V.; Brattespe, J.; et al. New insights into the early mechanisms of epileptogenesis in a zebrafish model of Dravet syndrome. Epilepsia 2020, 61, 549–560. [Google Scholar] [CrossRef]
- Farmer, B.C.; Walsh, A.E.; Kluemper, J.C.; Johnson, L.A. Lipid Droplets in Neurodegenerative Disorders. Front. Neurosci. 2020, 14, 742. [Google Scholar] [CrossRef]
- Bozza, P.T.; Viola, J.P. Lipid droplets in inflammation and cancer. Prostaglandins Leukot. Essent. Fatty Acids 2010, 82, 243–250. [Google Scholar] [CrossRef]
- Jin, Y.; Tan, Y.; Chen, L.; Liu, Y.; Ren, Z. Reactive Oxygen Species Induces Lipid Droplet Accumulation in HepG2 Cells by Increasing Perilipin 2 Expression. Int. J. Mol. Sci. 2018, 19, 3445. [Google Scholar] [CrossRef] [Green Version]
- Ralhan, I.; Chang, C.L.; Lippincott-Schwartz, J.; Ioannou, M.S. Lipid droplets in the nervous system. J. Cell. Biol. 2021, 220, e202102136. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tapia, A.; Giachello, C.N.; Palomino-Schätzlein, M.; Baines, R.A.; Galindo, M.I. Generation and Characterization of the Drosophila melanogaster paralytic Gene Knock-Out as a Model for Dravet Syndrome. Life 2021, 11, 1261. https://doi.org/10.3390/life11111261
Tapia A, Giachello CN, Palomino-Schätzlein M, Baines RA, Galindo MI. Generation and Characterization of the Drosophila melanogaster paralytic Gene Knock-Out as a Model for Dravet Syndrome. Life. 2021; 11(11):1261. https://doi.org/10.3390/life11111261
Chicago/Turabian StyleTapia, Andrea, Carlo N. Giachello, Martina Palomino-Schätzlein, Richard A. Baines, and Máximo Ibo Galindo. 2021. "Generation and Characterization of the Drosophila melanogaster paralytic Gene Knock-Out as a Model for Dravet Syndrome" Life 11, no. 11: 1261. https://doi.org/10.3390/life11111261