3. Literature Review of Results Relevant to Theoretical Predictions
Some of the predictions made above have already been experimentally investigated. We consider these results in terms of the hypothesis that adrenergic and opioid receptors and their ligands co-evolved by means of conserved, molecularly complementary modules.
Prediction 1. Adrenergic compounds should be molecularly complementary to opioid compounds since each molecular species alters the physiological effects of the other.
As noted in the Introduction, combining adrenergic and opioid compounds alters the effects of each on its respective receptor. As predicted, adrenergic compounds do bind to opioids and vice versa. Nuclear magnetic resonance spectroscopy, ultraviolet spectroscopy, and capillary electrophoresis have been employed to demonstrate that compounds such as epinephrine, norepinephrine, amphetamine, albuterol, propranolol, etc., bind directly to morphine, methionine- and leucine-enkephalin and endomorphin (
Table 1) [
17,
75,
76,
77]. Thus, the small-molecule complementarity predicted by Dwyer and Root-Bernstein is present in opioid–adrenergic pairs.
Prediction 2. Adrenergic receptors should have opioid-like modules within their ligand binding regions.
Since adrenergic compounds bind to opioids, primitive versions of opioid peptides such as pro-enkephalin and the endorphins may have provided the basic modules from which adrenergic receptors evolved. Molecular paleontology therefore predicts that remnants of these opioid-like sequences should be conserved in the binding regions of adrenergic receptors. Indeed, an LALIGN comparison of opioid peptides with adrenergic receptors (methods explained more fully in the
Figure 3 caption) reveal that several regions of significant homology exist between endorphins or pro-enkephalins and adrenergic receptors (
Figure 3). The location of these regions of homology map to transmembrane (TM) sequences TM5 and extracellular (EC) sequences EC3 that form part of the ligand-binding pocket of the receptors according to the UniProtKB website. The locations of these homologies within the overall receptor sequences are plotted below in Prediction 6. These homologies are statistically significant as will be discussed in greater detail in the statistical tests section that follows the final prediction below.
Prediction 3. Self-aggregation (homo-complementarity) of opioid peptides should provide the basis for the evolution of opioid receptors.
Dwyer’s theory proposes that homo-complementary peptides may evolve into ligand–receptor pairs [
59] so that it may be predicted that opioid peptides are homo-complementary. This is the case.
Enkephalins have been found to self-aggregate into homodimers arranged in an anti-parallel beta ribbon conformation [
78,
79,
80,
81,
82,
83]. Notably, the linearly extended conformation required for homodimerization is also the preferred, lowest-energy conformation and the one that best superimposes well on the pharmacophoric groups of morphine [
84]. Since endorphins include the enkephalin sequence, if follows that enkephalins can also bind to endorphins, although such aggregation has not apparently been experimentally tested.
Additionally, beta endorphin self-aggregates to form amyloid fibrils [
85,
86,
87,
88,
89], as does proenkephalin (PENKA) [
90], so that both these molecules are homo-complementary and satisfy Dwyer’s criterion for being possible precursors for opioid peptide receptors.
Following Dwyer further, opioid receptors should display evidence of opioid-like sequences associated with their ligand-binding regions. In fact, the beta endorphin and PENKA peptides have significant homologies to one another and to the mu and delta opioid receptors, and these homologies are conserved from fish through human beings (
Figure 4), a fact that is explored in additional vertebrate species below (zebrafish are used as an example here because they represent the most evolutionarily divergent species from
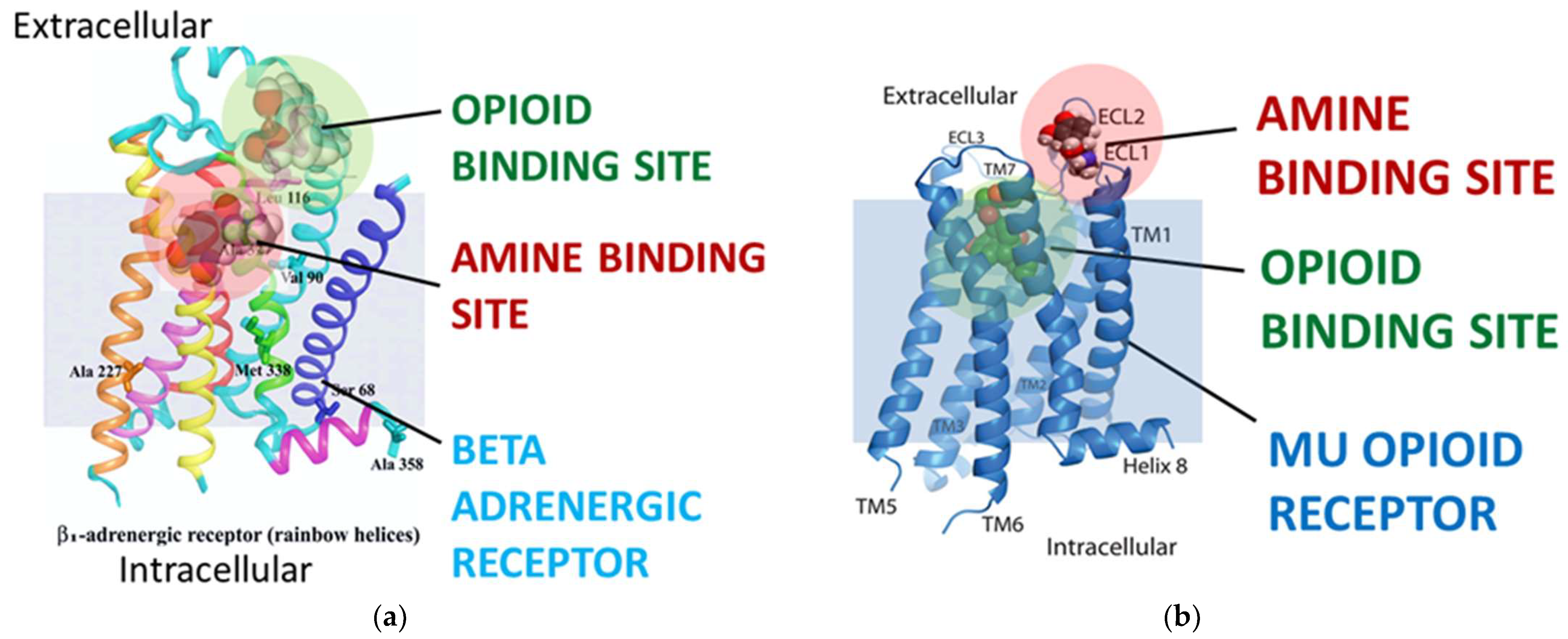
Homo sapiens for which the ranges of opioid and adrenergic receptor and ligand sequences were available). These homologies occur within the opioid ligand binding site of the opioid receptors, as will also be demonstrated in Prediction 6 below, and additional homologies are found between opioid receptors and pro-opiomelanocortin and beta endorphin in the extracellular loops. These extracellular loop homologies are significant both in terms of the fact that opioids often interact transiently with these extracellular loops prior to being drawn into their high-affinity binding site that exists in a pocket formed by the transmembrane sequences of the receptor [
2,
39,
40,
41,
42,
43,
44] (see
Figure 5). These extracellular homologies are also important with regard to adrenergic binding, as is discussed in the next prediction. Additionally, these homologies are statistically significant, as will be discussed in greater detail in the statistical tests section the follows the final prediction below.
Prediction 4. Some opioid-like regions of opioid receptors should bind adrenergic compounds acting as allosteric modifiers of opioid receptor function.
Since adrenergic agonists and antagonists bind directly to opioid peptides, and opioid-peptide-like regions are found within the opioid binding site of the opioid receptor as well as in its extracellular regions, it follows adrenergic compounds should bind to opioid receptors within one or more regions that are homologous to opioid peptides. This appears to be the case.
To begin with, adrenergic agonists and antagonists do bind directly to opioid receptors blocking access to the high-affinity opioid binding site. Among adrenergic compounds found to be effective competitors for naloxone binding to opioid receptors are: clonidine and L-phenylephrine, alpha adrenergic agonists; phentolamine, an alpha 1- and alpha-2 adrenergic antagonist; prazosin, an alpha 1-adrenergic antagonist; yohimbine, an alpha 2-adrenergic antagonist [
91]. Additionally, xylazine, an analogue of clonidine and an agonist at the α2 class of adrenergic receptor, as well as the adrenergic neurotoxin DSP4, also bind directly to opioid receptors with an IC50 competing with naloxone of about 1 µM [
92,
93]. Beta-adrenergic antagonists competitively bind to opioid receptors as well, displacing the enkephalin agonist [3H]D-Ala2-Met5-enkephalinamide and the morphine antagonist [3H]naloxone [
94]. Correspondingly, the alpha-adrenergic antagonists phentolamine and phenoxybenzamine at about 1 micromolar blocked 50% of 5 nanomolar naloxone binding to opioid receptors in brain tissue. This was ten times more potent than the IC50 of codeine and about ten times less potent than the IC50 of methadone [
95,
96,
97].
Epinephrine binds directly to purified mu opioid receptor (MOR) with about the same affinity as methadone [
16,
41,
77] and enhances binding of morphine to the receptor by a factor of about ten [
41]. Binding studies to MOR peptide fragments suggest that the epinephrine binding site on MOR is most likely located in a pocket defined by the juxtaposition of the second and third extracellular loops of the receptor (
Table 2) though, notably, the first extracellular region also binds epinephrine [
16,
41,
77]. Each of these epinephrine-binding regions corresponds to a sequence with high homology to either pro-opiomelanocortin (and, more specifically, beta endorphin) or pro-enkephalin (
Section 3, above). Thus opioid-peptide-like regions of opioid receptors appear to provide binding sites for adrenergic compounds on opioid receptors.
Prediction 5. Some of the opioid-like regions of adrenergic receptors should bind opioids and act as allosteric modulators of the receptor.
Just as adrenergic compounds modify opioid receptor function, opioids and opioid antagonists are known to modify adrenergic receptor function (Introduction). Following the principles set out in the Introduction, it can be predicted that adrenergic receptors have allosteric opioid binding sites and that they should be composed of opioid-like regions adapted to specifically bind adrenergic compounds. This also appears to be the case. Studies have demonstrated that naloxone and morphine can enhance adrenergic receptor activity in thoracic aorta, which is a tissue notable for lacking opioid receptor, therefore suggesting direct action of opioids on adrenergic receptors. Indeed, two κ opioid antagonists are known to bind directly to adrenergic receptors: nor-binaltorphimine (nor-BNI), a kappa-selective opioid antagonist bound to the α(2C)-adrenoceptor (K(i) = 630 nM), and 6′-guanidinonaltrindole (6′-GNTI) bound to the α(1A)-adrenoceptor (EC
50 = 41 nM), enhancing calcium mobilization by noradrenaline. Neither compound directly activated the receptors [
2].
Additionally, as summarized above in
Table 2, morphine and naloxone bind directly to extracellular loops of the beta 2 adrenergic receptor (regions highly conserved in other adrenergic receptors as well) [
44,
77]. As with the opioid receptor binding site for adrenergic compounds, the adrenergic receptor binding site for opioids appears to be located at the junction of the second and third extracellular loops (
Table 2) [
44,
77]. The presence of a cysteine residue appears to be of particular importance for the formation of a disulfide bond that keeps the receptor in its highest activity conformation [
44,
98].
Finally, tramadol and tapentadol, which are generally characterized as being combined mu opioid receptor agonists/norepinephrine reuptake inhibitors, also elicit direct effects on adrenergic receptor function via binding to beta 2 and alpha 2 adrenergic receptors [
99,
100]. Significant functional effects occur at micromolar concentrations [
99,
100] and can be blocked by various alpha 2 adrenergic receptor antagonists such as yohimbine and idazoxan [
100,
101].
Together, Points 4 and 5 demonstrate that many adrenergic receptor ligands bind to and directly affect opioid receptor function while many opioid receptor ligands bind to and directly affect adrenergic receptor function. These affects appear to be mediated in both cases by binding to an allosteric pocket formed at the intersection of the second and third extracellular loops and possibly involving part of transmembrane region 2, all of which are homologous to opioid-peptide-like sequences known to bind either opioids or adrenergic compounds (
Figure 5).
Prediction 6. Opioid and adrenergic receptors will therefore share common, evolutionarily conserved modules.
The previous five predictions lead inexorably to the conclusion that opioid and adrenergic receptors share common modules, many of these associated with peptide opioid sequences. In order to explore this possibility more thoroughly, we compare the sequences of a series of opioid and adrenergic receptors from diverse species utilizing LALIGN (an alignment program for exploring similarities between pairs of proteins). The details of the methods are provided in the
Figure 6 caption.
Figure 6 illustrates the high degree of homology between the zebrafish kappa opioid receptor and the zebrafish beta 2 adrenergic receptor.
Figure 7 illustrates the high degree of homology between the African clawed toad mu opioid receptor and its alpha 1A adrenergic receptor.
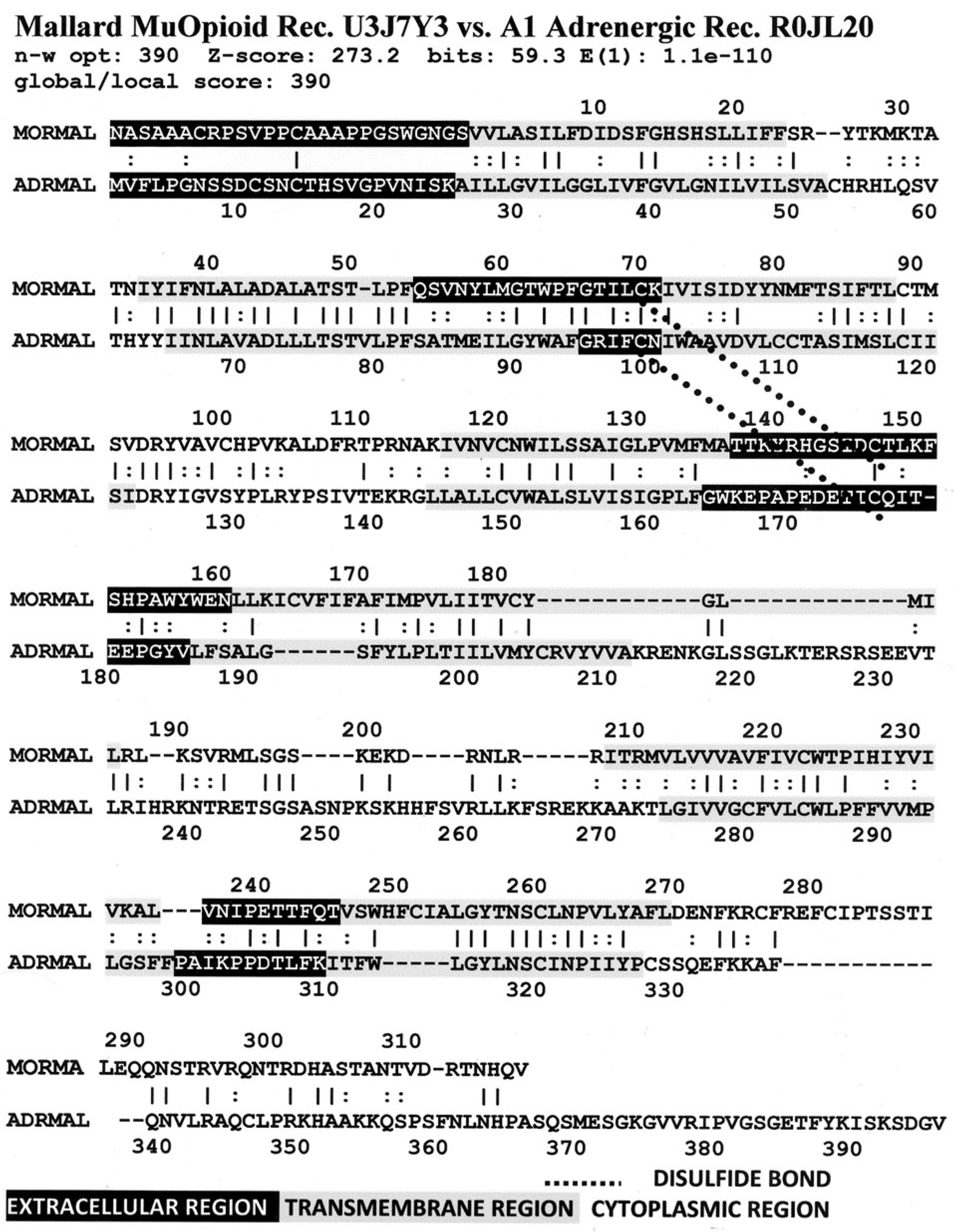
Figure 8 illustrates the high degree of homology between the mallard mu opioid receptor and its alpha 1A adrenergic receptor.
Figure 9 illustrates the high degree of homology between the mouse kappa opioid receptor and its beta 2 adrenergic receptor. Similar homology studies for the stickleback mu opioid receptor/alpha A1 adrenergic receptor and for the human kappa opioid receptor/beta 2 adrenergic receptor pairs will be discussed separately in Prediction 7 below. Additionally, we have previously published a similar study of the human mu opioid receptor and alpha 1A adrenergic receptor [
41].
A number of features are shared by all seven pairs of the homology pairs of the opioid and adrenergic receptors that we studied (zebrafish, clawed toad, and mallard, mouse (
Figure 4,
Figure 5,
Figure 6 and
Figure 7) and stickleback, human MOR/alpha ADR, and human KOR/beta ADR (which will be discussed in detail below) and these are summarized in
Table 3. Most regions of the opioid and adrenergic receptors are well-conserved across the two classes of receptors with the clear exceptions of the first, third, and fourth extracellular sequences (EC1, EC3, EC4) and the third and fourth intracellular sequences (IC3 and IC4). The rest of the receptors average 60% similarity; four regions—the second extracellular sequence (EC2, the first extracellular loop), transmembrane sequences 2 and 6 (TM2 and TM6), and the first intracellular sequence (IC1, the first intracellular loop)—average 70% similarity or greater across the entire range of species and within both adrenergic and opioid receptors.
The overall impression given by
Table 3 that the EC2, TM2, TM6, and IC1 are highly conserved is confirmed by Logoplots of these regions, generated from all fourteen receptor sequences (seven adrenergic and seven opioid receptors of the six species and seven comparisons used to generate
Table 3. The method is provided in
Figure 10 and applies to
Figure 11,
Figure 12,
Figure 13,
Figure 14 and
Figure 15 as well. The extracellular domain 2 (EC2), transmembrane region 2, or second helix, (TM2), the transmembrane region (or helix) 6 (TM6), and the first intracellular domain (IC1) Logoplots all reveal the presence of amino acids that are very highly, and in some cases completely, conserved in their positions across all seven comparisons and both types of opioid and adrenergic receptors. While the fourth transmembrane region (TM4) is also well conserved (60%:
Table 2), it displays only one completely conserved amino acid (a tryptophan) and a handful of other well-conserved residues, thus demonstrating significantly more divergence over evolutionary time than has occurred in EC2, TM2, TM6, and IC1. It is logical to conclude that shared functions, such as shared affinity for both opioids and adrenergic compounds, is preserved in these conserved regions, and that receptor dimerization is also made possible through conserved sequences in these regions—a point that will be taken up at greater length in the next part.
The second extracellular loop (EC3) provides an additional contrast: it is not well-conserved except at the crucial cysteine position that is essential for maintaining the ligand binding site integrity [
103] (
Figure 6,
Figure 7,
Figure 8 and
Figure 9). Since EC2 and EC3 make up the main part of the allosteric enhancer binding site (Predictions 4 and 5 above), it follows that the specificity of the enhancer site has evolved in these two regions.
In order to validate the utility of the Logoplot results, the highest-scoring consensus sequences were used to perform a BLAST search against the human proteome and the vertebrate proteome. Since the results of the human and vertebrate searches were virtually identical, only the results of the human proteome search are presented below in
Table 4, which demonstrates that the three consensus sequences tested all elicited matches to both OPR and ADR, confirming their shared nature. Notably, other receptor classes were also reliably identified as sharing these consensus sequences as well: among the amines, the dopamine, histamine, and serotonin receptors; and, among the peptide receptors, somatostatin, chemokine, angiotensin II, and neuropeptide B/W and Y. Thus, these consensus sequences are more broadly shared than among just the ADR and OPR. On the other hand, the absence of other classes of GPCR from the homology list in
Table 4 also suggests that these ADR-OPR consensus sequences are far from being universally conserved in GPCR generally. The special relationships of the receptors found by this BLAST search of the most highly conserved regions of the ADR and OPR to other GPCR will be investigated further in the Discussion section below.
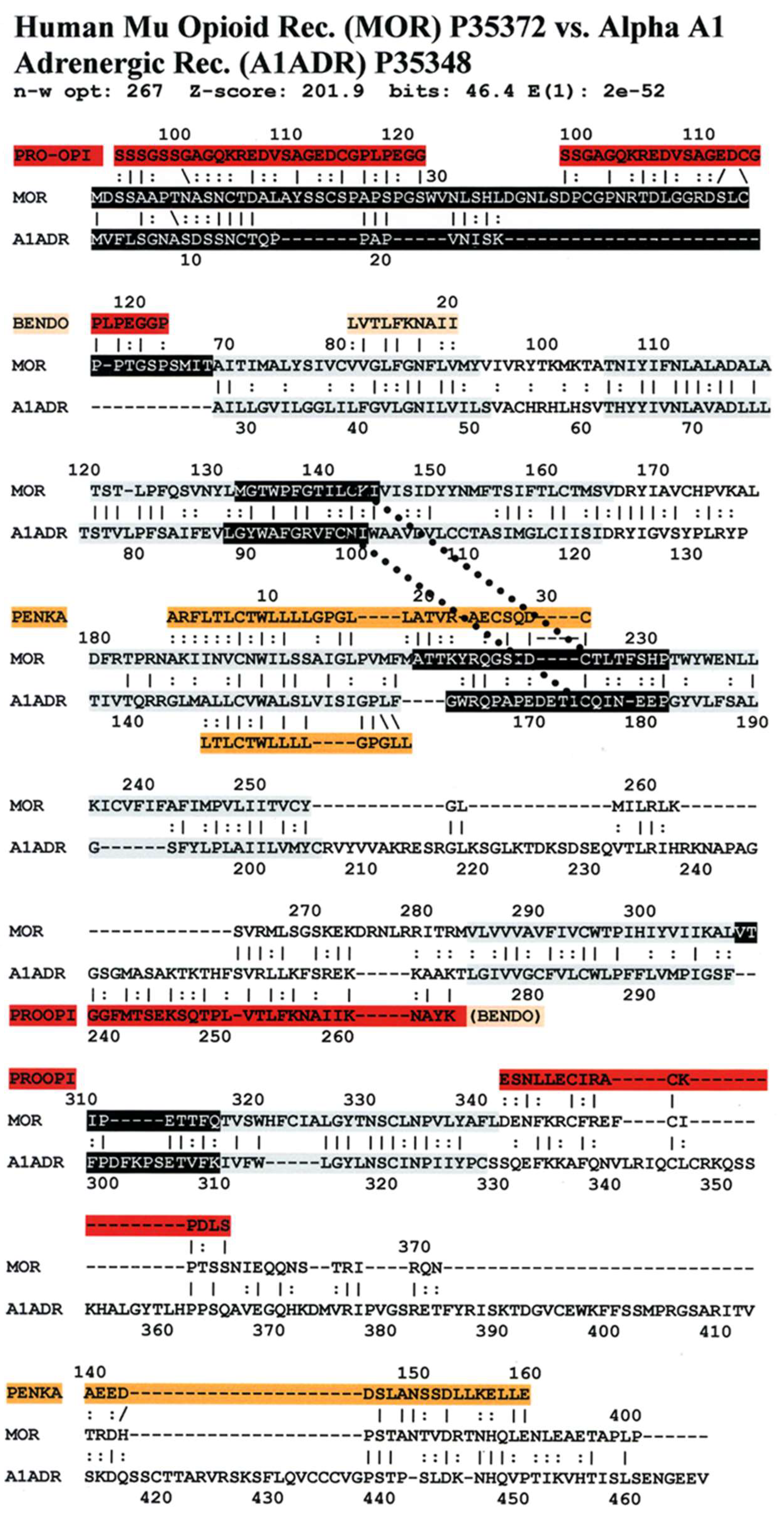
An equally important observation is that several of the sequences of the highly conserved regions are homologous to the sequences of the opioid precursor proteins pro-enkephalin (PENKA) or pro-opiomelanocortin (PROOPI) (which contains the beta endorphin sequence (BENDO)), as demonstrated in
Figure 16,
Figure 17 and
Figure 18 for stickleback mu opioid receptor versus alpha A1 adrenergic receptor, human mu opioid receptor versus alpha A1 adrenergic receptor, and human kappa opioid receptor versus beta 2 adrenergic receptor. The conservation of these opioid-peptide-like regions across both opioid and adrenergic receptors and across species that span a large portion of the evolutionary tree suggest that these regions represent important functional modules that are resistant to mutation.
Comparing
Figure 16,
Figure 17 and
Figure 18 with each other and with
Figure 6,
Figure 7,
Figure 8 and
Figure 9 reveals several important characteristics of the co-evolution of opioid and adrenergic receptors. One is that highly conserved regions within the opioid-adrenergic pairs of one species are highly conserved in all of the other species, a point that is emphasized by the results of the Logoplots for EC2, EC3, TM2, TM4, TM6, and IC1 (
Figure 10,
Figure 11,
Figure 12,
Figure 13,
Figure 14 and
Figure 15). Thus, rather than diverging from one another over evolutionary time, opioid–adrenergic pairs have maintained a high degree of similarity. This conservation argues for the critical importance of these sequences to both OPR and ADR function. Equally important from the perspective of modular complementarity, the similarities between opioid peptides and specific regions of both opioid and adrenergic receptors are also conserved across evolutionarily divergent species (
Figure 16,
Figure 17 and
Figure 18). Regions of the stickleback receptors that mimic opioid peptides are regions in the human opioid and adrenergic receptors that also mimic opioid peptides. Thus, these highly conserved functional regions also tend to be related to the opioid–peptide modules identified above. Additionally,
Figure 16,
Figure 17 and
Figure 18 clearly emphasize the point made by
Figure 3,
Figure 4 and
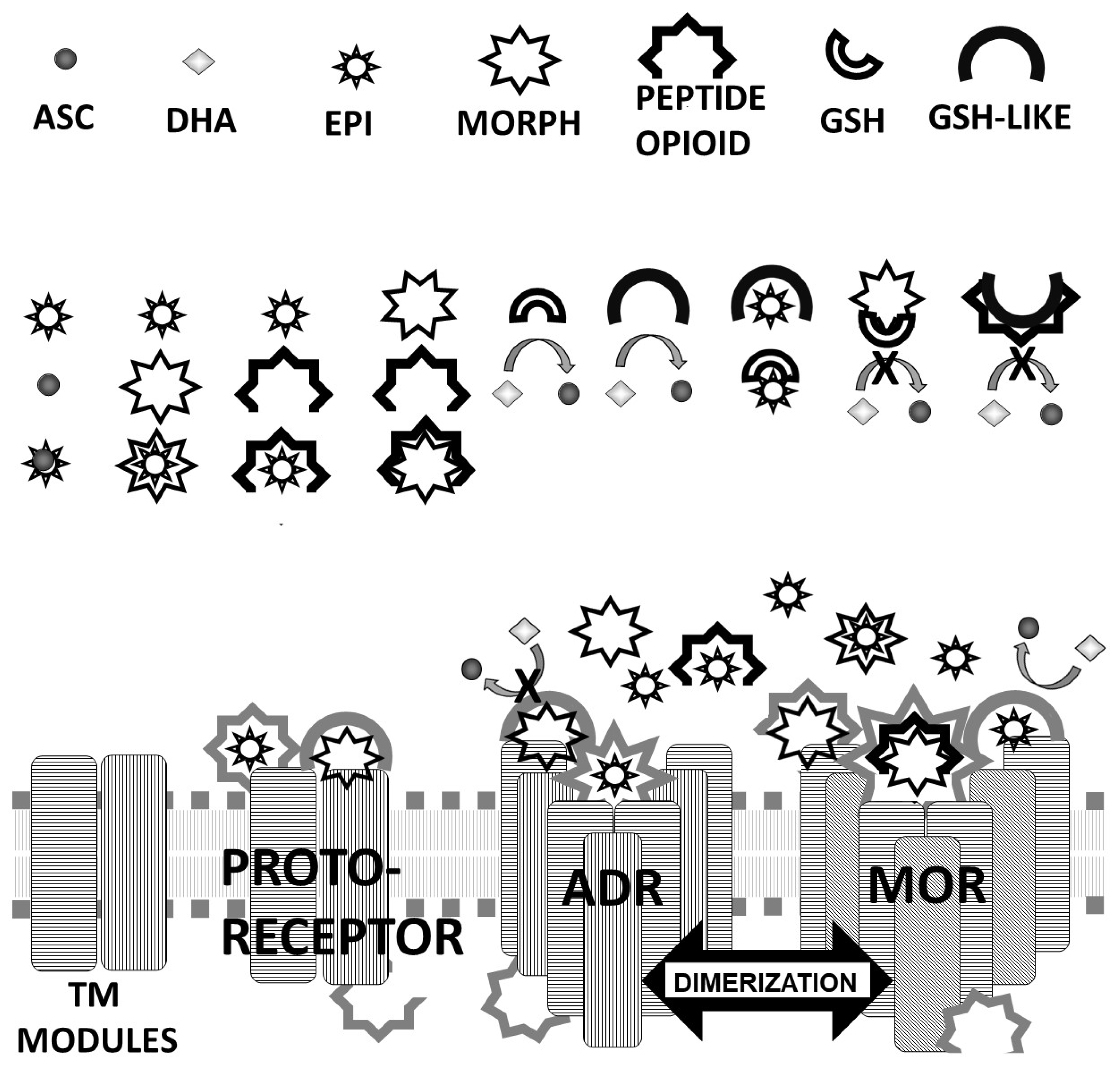
Figure 5 that these opioid peptide–receptor similarities occur at multiple sites in the receptors and that these similar regions occur both in the extracellular (including the first extracellular sequence, EC1, as well as EC3) and the fourth intracellular region (IC4) of the opioid receptors but only in one extracellular loop (EC3) and the third intracellular region (IC3) of the adrenergic receptors. Thus, these modules appear to have been rearranged in adrenergic receptors as compared with opioid receptors, providing a mechanism for explaining their divergent ligand specificities. This rearrangement is easily observed in the abstracted diagram below (
Figure 19) where it is clear that the opioid receptors are characterized by copies of opioid peptides in both their first extracellular and fourth intracellular regions that are not present in the adrenergic receptors, which display opioid receptors in regions that share little sequence similarity to the opioid receptors. The presence of opioid peptide-like regions on the extracellular and intracellular regions of the opioid receptor has possible importance in understanding the evolution of receptor signaling that will be taken up further below.
Prediction 7. Conserved modules include the transmembrane regions of the receptors.
Our seventh prediction is that homo- and hetero-dimerization domains should be highly conserved between the adrenergic and opioid receptors, thus representing yet another class of evolutionarily stable, homo-complementary modules. Adrenergic receptors can homodimerize and heterodimerize with each other (reviewed briefly below); opioid receptors can homo- and heterodimerize with each other (reviewed briefly below); and adrenergic receptors can heterodimerize with opioid receptors [
47,
48,
49,
50,
51,
52,
53,
54,
55]. Logically, this conservation of dimerization potential requires that the dimerization determinants in adrenergic and opioid receptors be highly conserved as well. Since dimerization regions generally involve transmembrane sequences (discussed in detail below), transmembrane regions involved in dimerization might therefore be expected to be highly conserved, as is summarized in
Table 3 and
Figure 19. As with the previous predictions, this appears to be the case. As noted in the previous section, all of the transmembrane (TM) regions are conserved to at least 60% across all fourteen of the receptor sequences investigated with the exception of TM5 (57% homology); TM2 and TM6 are particularly well-conserved across all of the opioid and adrenergic receptors (72% and 76%, respectively) (
Table 3;
Figure 11,
Figure 12 and
Figure 14). This conservation is also apparent in
Figure 6,
Figure 7,
Figure 8,
Figure 9,
Figure 16,
Figure 17 and
Figure 18 and is independent of the opioid–peptide modules.
The question now becomes whether these highly conserved TM regions are particularly important determinants of receptor dimerization. This appears to be the case, though much uncertainty still attains to the specific interactions governing dimerization and even which transmembrane regions of the receptors are involved in both ADR and OPR.
Three very different models have been proposed for homodimerization in beta-2 adrenergic receptors (B2AR), one involving helices V and VI (TM5 and TM6) [
104], another involving helix VI (TM6) binding to a complementary helix [
105], and the third involving binding between either helices 1 and VII (TM1 and TM7) [
106] or between helices I/II with III/IV (TM1/TM2 with TM3/TM4) [
107]. Further studies determined that helix I (TM1) was unlikely to be involved in homodimerization since mutations of helix 1 had little effect on dimerization [
108]. However, studies of the crystal structure of B1AR also suggested that oligomerization may involve TM1/TM2 interactions as well as TM4/TM5 interactions [
109], though the extent to which crystal interactions predict membrane interactions is open to question since the receptors in a crystal are packed in ways that are not evident when they are embedded in a cell membrane. Thus, evidence from studies of alpha-adrenergic receptor homodimerization have demonstrated that neither helix 1 (TM1) nor helix 7 (TM7) are involved, but isolated helices III, IV, V, and VI could each, independently, bind to full-length receptor [
110]. Again, the relevance of these studies to membrane-embedded receptor is questionable since these studies were done in solution. Additionally, a peptide derived from helix VI (TM6) was able to inhibit B2AR homodimerization, indicating that TM6 plays an essential role [
105]. Finally, TM4 is able to self-associate but not at a sufficiently high affinity to account for observed dimerization interactions [
110,
111]. In sum, for adrenergic receptors, helix II (TM2) may interact with helices III or IV (TM3 or TM4), while helices V or VI (TM5 or TM6) may also participate in adrenergic receptor dimerization. There is no evidence that helix VII (TM7) is involved in dimerization, and evidence for the participation of TM1 is contradictory.
The situation regarding opioid receptors is equally complex. Crystal structures of the mu opioid receptor (MOR) and kappa opioid receptor (KOR) show closely packed interfaces between TM5 and TM6 as well as a less compact interface involving TM1, TM2, and a helix found in the intracellular domain 4 (IC4) [
112]. Again, it is questionable whether crystal interactions are a good indicator of membrane interactions. Notably, MOR has a functional truncated splice version that lacks the first helix (TM1) but which still forms homodimers [
113] as well as heterodimers with B2AR [
114]. Thus, while helix 1 (TM1) may normally be involved in dimerization, it cannot be necessary for homo- or heterodimerization of MOR. As in adrenergic receptors, TM4 self-aggregates and may also bind to TM5 in DOR homodimerization [
115]. Crystal studies demonstrate that MOR oligomerization is mediated by TM5/TM6 binding [
116]. Modeling studies of opioid receptor homo- and heterodimerization tend to confirm that TM1/TM2 and TM5/TM6 participate in dimerization. One study revealed high probabilities of interactions between TM1,2 and TM5,6, but also TM4 and TM5 (as well as TM4,5/TM1,2 for heterodimers); TM3 and TM7 were never involved [
117]. In opioid receptor heterodimers, additional computer simulations identified TM1/TM2/IC4 complexes and TM3/TM6 complexes as the most likely means of dimerization [
118]. So, the consensus of the existing data is that TM1/TM2 may interact with TM5/TM6, while TM 3 and TM7 are never implicated in dimerization, and TM1 and TM4 seem to be dispensable.
In sum, the question of how homo- and heterodimerization are mediated is still open for both adrenergic and opioid receptors. The only firm conclusions that can be reached are that TM1 may participate, but is not necessary, and there is little or no evidence at present for the participation of TM3 or TM7. Notably, TM2, TM4, and TM6 are the best-conserved helical (or transmembrane) regions of the opioid and adrenergic receptors across species (
Table 3), suggesting that dimerization may be mediated mostly through interactions involving these helices. Treating each of these sequences as a conserved module may permit novel experiments to be performed to determine which modules are complementary to the others (or to themselves). Additionally, since receptors are often localized in lipid rafts with specific lipid and protein compositions, the possibility that conserved TM regions may have evolved in part due to raft composition should also be considered.
Statistical Significance of the Results Reported Above
The statistical significance of the homology results reported in the previous four sections can be evaluated in three ways. One is to compare the present results with a previous study of the probability of any given peptide ligand mimicking its own, or any other, receptor [
63]. In the study just cited, 13 peptide ligands were tested against 25 peptide receptors (for a total of 325 permutations) using LALIGN to determine how often sequence matches occurred at any given statistical value. Two criteria were employed for significance: one was whether the match included a sequence of 10 amino acids in which at least 5 were identical, and the second was a Waterman–Eggert score of 35 or greater. Only 3.6% of the matches satisfied those simultaneous criteria (i.e.,
p = 0.036), indicating that the probability of any given peptide ligand having a homologous region in a random peptide receptor was extremely small. Of the 17 homologies illustrated in
Figure 3 and
Figure 4, comparisons of opioid peptide sequences with adrenergic and opioid receptor sequences using LALIGN yielded Waterman–Eggert (WE) scores above 35 (and as high as 57) with corresponding E values (which correspond to internal probability values) of less than one (and as small as 0.001). The higher the Waterman–Eggert score, the less probable the match is to have occurred by chance; conversely, the smaller the E value, the less the probability [
119]. The probability that all seventeen homologies illustrated in
Figure 3 and
Figure 4 would satisfy both WE and E value criteria simultaneously is 0.036/17 or 0.0021.
However, Waterman and Eggert caution that their probability scores should always be complemented with a statistical approach that specifically tests the hypothesis generating the homology search [
119], so a second statistical approach involves interpreting the results in terms of a random search of endorphins and proenkephalin sequences with each other and with a range of opioid and adrenergic receptors from a variety of species (
Appendix A). These results confirm that the types of high Waterman–Eggert score/low E-values/high identity sequences found in
Figure 3 and
Figure 4 occur almost solely between evolutionarily related molecules such as endorphins from different species or endorphins with pro-enkephalins. The fact that endorphin- and proenkephalin-like sequences occur within both opioid receptors and adrenergic receptors at equally high Waterman–Eggert scores and E values is therefore indicative of an evolutionary relationship.
Finally, the Waterman–Eggert scores and E values provided for receptor homologies (
Figure 6,
Figure 7,
Figure 8,
Figure 9,
Figure 16,
Figure 17 and
Figure 18) as a function of the LALIGN search algorithm also provide internal tests of probability. Comparisons of opioid with adrenergic receptors universally displayed Waterman–Eggert scores or their equivalent Z scores above 200, while the E values range from 10
−24 to 10
−110, which are extraordinarily unlikely to occur by chance and are generally found only for evolutionarily related proteins, as is evident from
Appendix B.
In short, whatever statistical measure is used, the results reported here are extremely unlikely to have occurred by chance and strongly suggest the non-random conservation of key structural and functional modules across all of the adrenergic and opioid receptors tested.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



















