
Analysis of Differentially Expressed Genes That Aggravate Metabolic Diseases in Depression


Abstract
:
1. Introduction
2. Methods and Materials
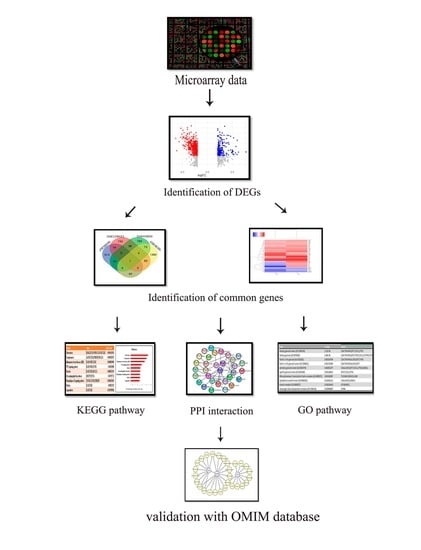
2.1. Overview of the Study
2.2. Retrieval of Microarray Datasets
2.3. Differential Gene Expression Analysis and Validation with Random Unrelated Diseases
2.4. Gene Set Enrichment Analysis
2.5. Ontology Pathway
2.6. Protein–Protein Interaction (PPI) Network and Hub-Gene Identification
2.7. Prediction of the Master Transcription Factors (TFs)
2.8. OMIM Disease and OMIM Expanded Database
2.9. Validation of the Common DEGs with other Depression-Related Datasets
2.10. Statistical Analysis
3. Results
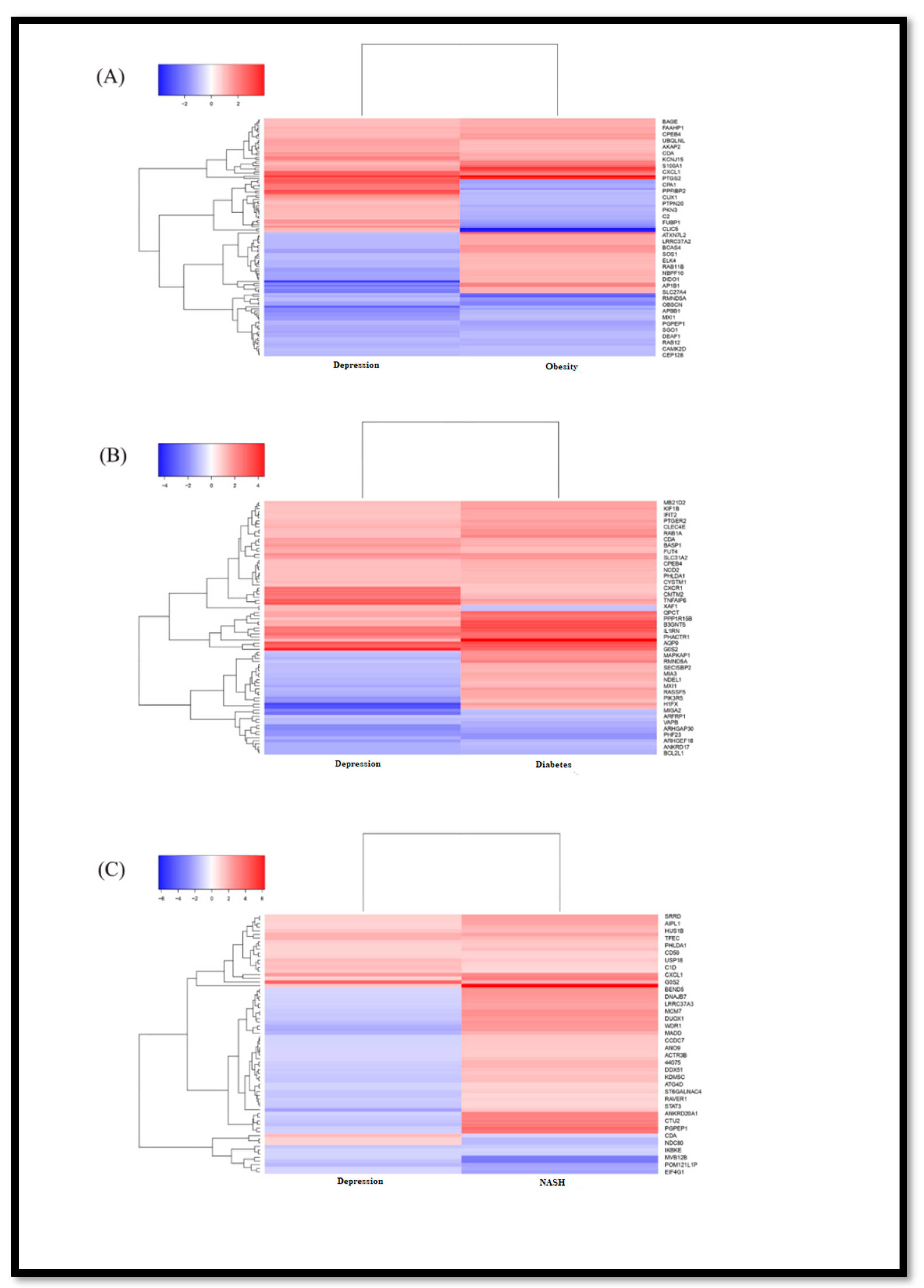
3.1. Identification of DEGs
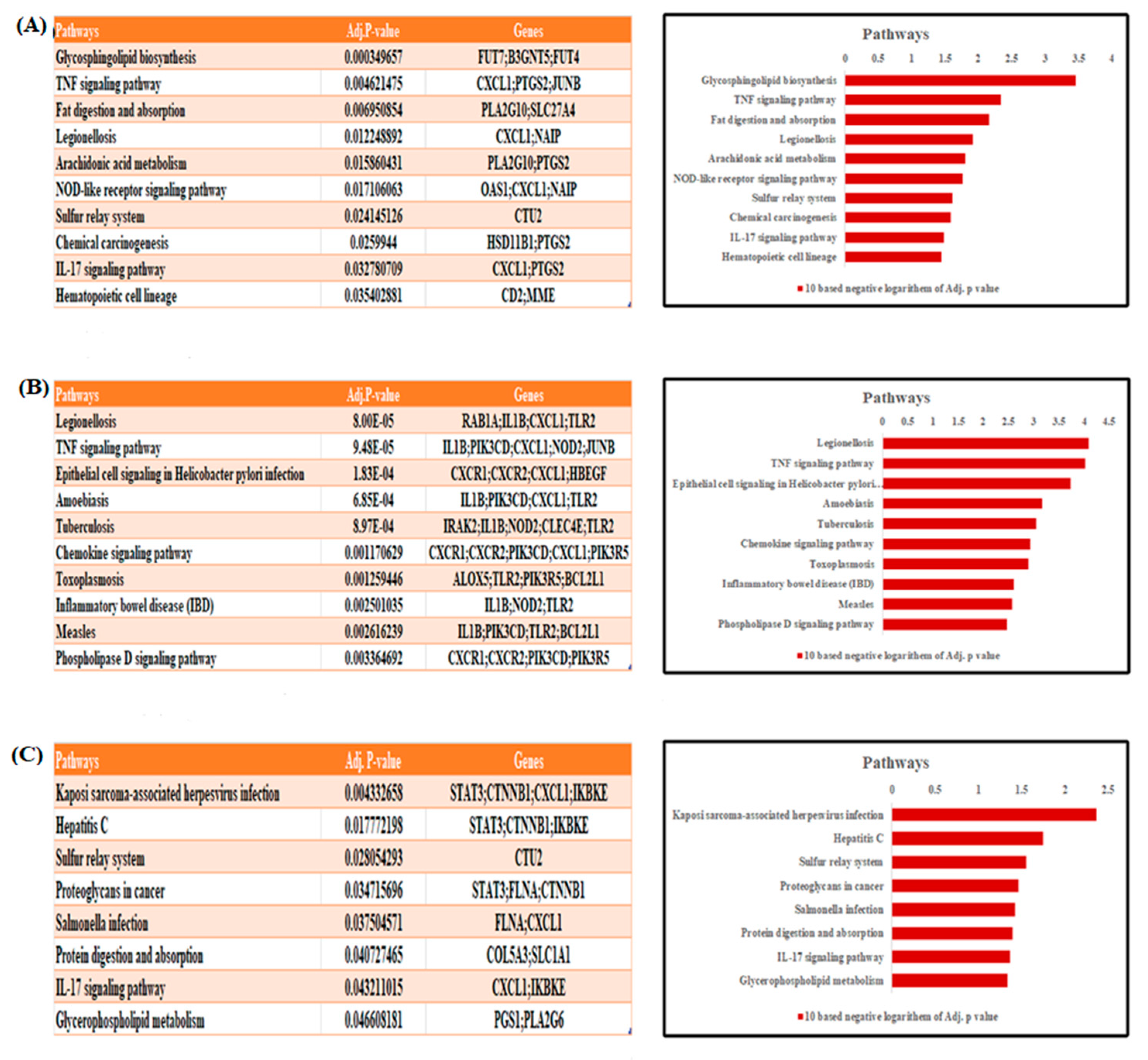
3.2. KEGG Pathway Analysis
3.3. Gene Ontological Pathway Analysis
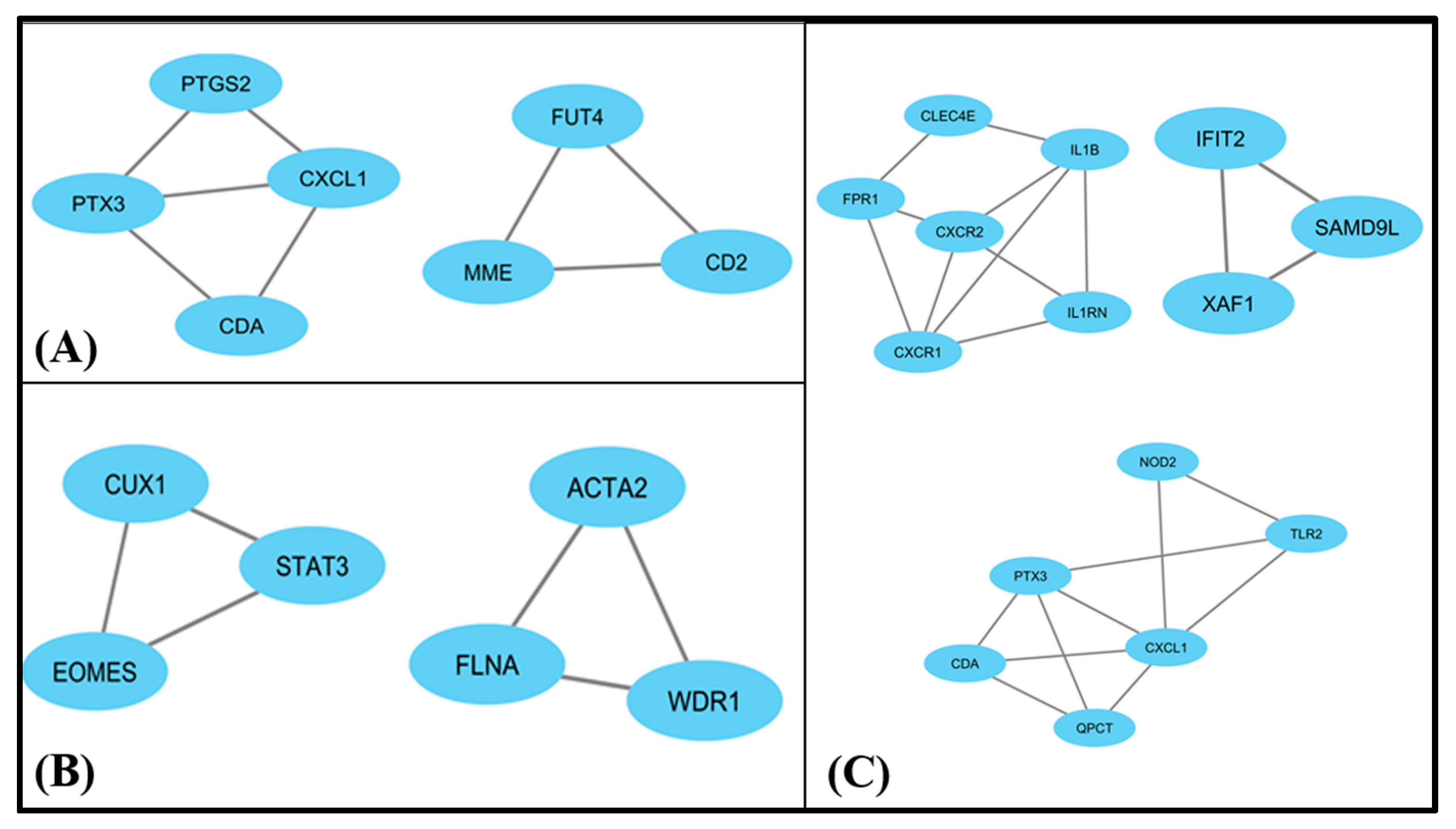
3.4. PPI Interactions
3.5. MCODE
3.6. TFs Prediction by iRegulon
3.7. Validation by OMIM Disease and dbGap
3.8. Validation of the Commonly Expressed DEGs with Other Depression-Related Datasets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DEGs | Differentially Expressed Genes |
| Log FC | Log Fold Change |
| GO | Gene Ontology |
| GEO | Gene Expression Omnibus |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| PPI | Protein–Protein Interaction |
| DAVID | Database for Annotation, Visualization, and Integrated Discovery |
| NASH | Nonalcoholic Steatohepatitis |
| MDD | Major Depressive Disorder |
| NCBI | National Center for Biotechnology Information |
| OMIM | Online Mendelian Inheritance in Man |
| HPA | Hypothalamic–pituitary–adrenal |
| CRH | Corticotrophin-releasing hormone |
References
- Whiteford, H.A.; Ferrari, A.J.; Degenhardt, L.; Feigin, V.; Vos, T. The global burden of mental, neurological and substance use disorders: An analysis from the Global Burden of Disease Study 2010. PLoS ONE 2015, 10, e0116820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandelow, B.; Michaelis, S. Epidemiology of anxiety disorders in the 21st century. Dialogues Clin. Neurosci. 2015, 17, 327–335. [Google Scholar]
- Otte, C.; Gold, S.M.; Penninx, B.W.; Pariante, C.M.; Etkin, A.; Fava, M.; Mohr, D.C.; Schatzberg, A.F. Major depressive disorder. Nat. Rev. Dis. Primers 2016, 2, 16065. [Google Scholar] [CrossRef] [Green Version]
- Anisman, H.; Merali, Z. Understanding stress: Characteristics and caveats. Alcohol Res. Health J. Natl. Inst. Alcohol Abus. 1999, 23, 241–249. [Google Scholar]
- Varghese, F.P.; Brown, E.S. The Hypothalamic-Pituitary-Adrenal Axis in Major Depressive Disorder: A Brief Primer for Primary Care Physicians. Prim. Care Companion J. Clin. Psychiatry 2001, 3, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Nemiary, D.; Shim, R.; Mattox, G.; Holden, K. The Relationship between Obesity and Depression Among Adolescents. Psychiatr. Ann. 2012, 42, 305–308. [Google Scholar] [CrossRef] [Green Version]
- Carreiro, A.L.; Carreiro, A.L.; Dhillon, J.; Gordon, S.; Higgins, K.A.; Jacobs, A.G.; McArthur, B.M.; Redan, B.W.; Rivera, R.L.; Schmidt, L.R.; et al. The Macronutrients, Appetite, and Energy Intake. Annu. Rev. Nutr. 2016, 36, 73–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penninx, B.W.J.H.; Lange, S.M.M. Metabolic syndrome in psychiatric patients: Overview, mechanisms, and implications. Dialogues Clin. Neurosci. 2018, 20, 63–73. [Google Scholar] [PubMed]
- Chen, S.; Zhang, Q.; Dai, G.; Hu, J.; Zhu, C.; Su, L.; Wu, X. Association of Depression with Pre-Diabetes, Undiagnosed Diabetes, and Previously Diagnosed Diabetes: A Meta-Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Roy, T.; Lloyd, C.E. Epidemiology of depression and diabetes: A systematic review. J. Affect. Disord. 2012, 142 (Suppl. S8), 21. [Google Scholar] [CrossRef]
- Silverman, M.N.; Pearce, B.D.; Biron, C.A.; Miller, A.H. Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol. 2005, 18, 41–78. [Google Scholar] [CrossRef]
- Moulton, C.D.; Pickup, J.C.; Ismail, K. The link between depression and diabetes: The search for shared mechanisms. Lancet Diabetes Endocrinol. 2015, 3, 461–471. [Google Scholar] [CrossRef]
- Mezuk, B.; Eaton, W.W.; Albrecht, S.; Golden, S.H. Depression and type 2 diabetes over the lifespan: A meta-analysis. Diabetes Care 2008, 31, 2383–2390. [Google Scholar] [CrossRef] [Green Version]
- Elwing, J.E.; Lustman, P.J.; Wang, H.L.; Clouse, R.E. Depression, anxiety, and nonalcoholic steatohepatitis. Psychosom. Med. 2006, 68, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liu, X.; Yu, Y. Depression and Chronic Liver Diseases: Are There Shared Underlying Mechanisms? Front. Mol. Neurosci. 2017, 10, 134. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Xing, J.J.; Shan, A.Q.; Leng, L.; Liu, J.C.; Yue, S.; Yu, H.; Chen, X.; Tian, F.S.; Tang, N.J. Increased risk of nonalcoholic fatty liver disease with occupational stress in Chinese policemen: A 4-year cohort study. Medicine 2016, 95, e5359. [Google Scholar] [CrossRef] [PubMed]
- Youssef, N.A.; Abdelmalek, M.F.; Binks, M.; Guy, C.D.; Omenetti, A.; Smith, A.D.; Diehl, A.M.E.; Suzuki, A. Associations of depression, anxiety and antidepressants with histological severity of nonalcoholic fatty liver disease. Liver Int. 2013, 33, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Giuliani, F. The Role of Inflammation in Depression and Fatigue. Front. Immunol. 2019, 10, 1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Ji, Y.L.; Yang, Y.Y.; Xiong, X.Y.; Wang, I.M.; Sandford, A.J.; Liang, Z.A.; He, J.Q. Transcriptomic profiling of peripheral blood CD4+ T-cells in asthmatics with and without depression. Gene 2015, 565, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Buehrer, B.M.; Cheatham, R.B. Gene expression differences in omental mesothelial cells from lean and obese human donors. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128021 (accessed on 9 March 2019).
- Hazra, S.S.V.; Bhatwadekar, A.D.; Boulton, M.E.; Higgins, P.J.; Nikonova, E.V.; Pepine, C.J.; Thut, C.; Finney, E.M.; Sitnicka, E.; Iversen, P.; et al. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi (accessed on 1 February 2013).
- Wruck, W.; Kashofer, K.; Rehman, S.; Daskalaki, A.; Berg, D.; Gralka, E.; Jozefczuk, J.; Drews, K.; Pandey, V.; Regenbrecht, C.; et al. Multi-omic profiles of human non-alcoholic fatty liver disease tissue highlight heterogenic phenotypes. Sci. Data 2015, 2, 150068. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janky, R.; Verfaillie, A.; Imrichova, H.; Van de Sande, B.; Standaert, L.; Christiaens, V.; Hulselmans, G.; Herten, K.; Naval Sanchez, M.; Potier, D.; et al. iRegulon: From a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol. 2014, 10, e1003731. [Google Scholar] [CrossRef] [Green Version]
- Hamosh, A.; Scott, A.F.; Amberger, J.S.; Bocchini, C.A.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005, 33, D514–D517. [Google Scholar] [CrossRef] [PubMed]
- Tryka, K.A.; Hao, L.; Sturcke, A.; Jin, Y.; Wang, Z.Y.; Ziyabari, L.; Lee, M.; Popova, N.; Sharopova, N.; Kimura, M.; et al. NCBI’s Database of Genotypes and Phenotypes: dbGaP. Nucleic Acids Res. 2014, 42, D975–D979. [Google Scholar] [CrossRef]
- Tömböl, Z.; Szabo, P.M.; Molnar, V.; Wiener, Z.; Tölgyesi, G.; Horanyi, J.; Riesz, P.; Reismann, P.; Patocs, A.; Liko, I.; et al. Integrative molecular bioinformatics study of human adrenocortical tumors: microRNA, tissue-specific target prediction, and pathway analysis. Endocr. Relat. Cancer 2009, 16, 895–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Wang, Y.; Li, H.; Feng, L.; Li, W.; Cheng, S. Identification of Prognostic Biomarkers for Multiple Solid Tumors Using a Human Villi Development Model. Front. Cell Dev. Biol. 2020, 8, 492. [Google Scholar] [CrossRef]
- Breen, M.S.; Wingo, A.P.; Koen, N.; Donald, K.A.; Nicol, M.; Zar, H.J.; Ressler, K.J.; Buxbaum, J.D.; Stein, D.J. Gene expression in cord blood links genetic risk for neurodevelopmental disorders with maternal psychological distress and adverse childhood outcomes. Brain Behav. Immun. 2018, 73, 320–330. [Google Scholar] [CrossRef]
- Rana, H.K.; Akhtar, M.R.; Islam, M.B.; Ahmed, M.B.; Lió, P.; Huq, F.; Quinn, J.M.; Moni, M.A. Machine Learning and Bioinformatics Models to Identify Pathways that Mediate Influences of Welding Fumes on Cancer Progression. Sci. Rep. 2020, 10, 2795. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef]
- Foley, D.L.; Morley, K.I.; Madden, P.A.; Heath, A.C.; Whitfield, J.B.; Martin, N.G. Major depression and the metabolic syndrome. Twin Res. Hum. Genet. 2010, 13, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Wang, F.; Lan, X.; Li, C.; Feng, J.; Bai, J.; Cao, L.; Gui, S.; Hong, L.; Zhang, Y. Lower PRDM2 expression is associated with dopamine-agonist resistance and tumor recurrence in prolactinomas. BMC Cancer 2015, 15, 272. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.; Piao, Z.; Simon, D.; Sheu, J.C.; Huang, S. Mapping of a minimal deleted region in human hepatocellular carcinoma to 1p36.13-p36.23 and mutational analysis of the RIZ (PRDM2) gene localized to the region. Genes Chromosomes Cancer 2000, 28, 269–275. [Google Scholar] [CrossRef]
- Xie, X.; Man, X.; Zhu, Z.; Yuan, D.; Huang, S. Tumor suppressor RIZ1 in obesity and the PI3K/AKT/mTOR pathway. Obesity 2016, 24, 389–397. [Google Scholar] [CrossRef] [Green Version]
- Chai, H.H.; Fu, X.C.; Ma, L.; Sun, H.T.; Chen, G.Z.; Song, M.Y.; Chen, W.X.; Chen, Y.S.; Tan, M.X.; Guo, Y.W.; et al. The chemokine CXCL1 and its receptor CXCR2 contribute to chronic stress-induced depression in mice. FASEB J. 2019, 33, 8853–8864. [Google Scholar] [CrossRef]
- Ślusarczyk, J.; Chwastek, J.; Basta-Kaim, A. A Potential Contribution of Chemokine Network Dysfunction to the Depressive Disorders. Curr. Neuropharmacol. 2016, 14, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Tseng, C.; Zhang, Y.; Sirin, O.; Corn, P.G.; Li-Ning-Tapia, E.M.; Troncoso, P.; Davis, J.; Pettaway, C.; Ward, J.; et al. CXCL1 mediates obesity-associated adipose stromal cell trafficking and function in the tumour microenvironment. Nat. Commun. 2016, 7, 11674. [Google Scholar] [CrossRef]
- Nunemaker, C.S.; Chung, H.G.; Verrilli, G.M.; Corbin, K.L.; Upadhye, A.; Sharma, P.R. Increased serum CXCL1 and CXCL5 are linked to obesity, hyperglycemia, and impaired islet function. J. Endocrinol. 2014, 222, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.; He, Y.; Xiang, X.; Seo, W.; Kim, S.J.; Ma, J.; Ren, T.; Park, S.H.; Zhou, Z.; Feng, D.; et al. Interleukin-22 Ameliorates Neutrophil-Driven Nonalcoholic Steatohepatitis Through Multiple Targets. Hepathology 2020, 72, 412–429. [Google Scholar] [CrossRef]
- Yun, H.; Park, E.S.; Choi, S.; Shin, B.; Yu, J.; Yu, J.; Amarasekara, D.S.; Kim, S.; Lee, N.; Choi, J.S.; et al. TDAG51 is a crucial regulator of maternal care and depressive-like behavior after parturition. PLoS Genet. 2019, 15, e1008214. [Google Scholar] [CrossRef] [PubMed]
- Basseri, S.; Lhoták, Š.; Fullerton, M.D.; Palanivel, R.; Jiang, H.; Lynn, E.G.; Ford, R.J.; Maclean, K.N.; Steinberg, G.R.; Austin, R.C. Loss of TDAG51 results in mature-onset obesity, hepatic steatosis, and insulin resistance by regulating lipogenesis. Diabetes 2013, 62, 158–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gałecka, E.; Talarowska, M.; Maes, M.; Su, K.P.; Górski, P.; Szemraj, J. Polymorphisms of iodothyronine deiodinases (DIO1, DIO3) genes are not associated with recurrent depressive disorder. Pharmacol. Rep. 2016, 68, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Frances, A.; Cordelier, P. The emerging role of cytidine deaminase in human diseases: A new opportunity for therapy? Mol. Ther. 2020, 28, 357–366. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SL | Disease Name | GEO Accession | Sample | Platform | |
|---|---|---|---|---|---|
| Healthy | Disease | ||||
| 1 | Depression | GSE58430 | 6 | 6 | GPL14550 |
| 3 | Obesity | GSE128021 | 3 | 3 | GPL10558 |
| 2 | Diabetes | GSE43950 | 5 | 4 | GPL10379 |
| 4 | NASH | GSE43600 | 8 | 10 | GPL10558 |
| Disease Name | GEO Accession | Disease Name | GEO Accession | % of Upregulated Common DEGs | % of Down Regulated Common DEGs |
|---|---|---|---|---|---|
| Depression | GSE58430 | Obesity | GSE128021 | 3.5 | 4.6 |
| Diabetes | GSE43950 | 5.1 | 3.9 | ||
| NASH | GSE43600 | 3.4 | 2.8 | ||
| Influenza | GSE111449 | Obesity | GSE128021 | 1.2 | 1.0 |
| Diabetes | GSE43950 | 0.7 | 0.6 | ||
| NASH | GSE46300 | 1.6 | 0.9 |
| GO Pathways | Adj p-Value | Genes | ||
|---|---|---|---|---|
| Molecular functions | RNA polymerase II regulatory-region sequence-specific DNA binding | (GO:0000977) | 0.000059725 | ELK4;CUX1;MAX;DEAF1;MAZ;TFEC;HSF1;MXI1;PRDM2;NKX2-5;JUNB |
| alpha-(1->3)-fucosyltransferase activity | (GO:0046920) | 0.000866475 | FUT7;FUT4 | |
| G-protein-activated inward rectifier potassium channel activity | (GO:0015467) | 0.001382294 | KCNJ15;KCNJ2 | |
| fucosyltransferase activity | (GO:0008417) | 0.002369586 | FUT7;FUT4 | |
| titin binding | (GO:0031432) | 0.002754338 | CAMK2D;OBSCN | |
| Biological pathways | cellular response to type I interferon | (GO:0071357) | 0.000033205 | RSAD2;OAS1;IFIT1;XAF1;IFIT3 |
| type I interferon signaling pathway | (GO:0060337) | 0.000033205 | RSAD2;OAS1;IFIT1;XAF1;IFIT3 | |
| cytokine-mediated signaling pathway | (GO:0019221) | 0.000244271 | CAMK2D;RSAD2;OAS1;IRAK2;TNFRSF10C;CXCL1;IFIT1;XAF1;SOS1;PTGS2;JUNB;IFIT3 | |
| insulin secretion involved in cellular response to glucose stimulus | (GO:0035773) | 0.000467629 | PTPRN;RAB11B | |
| L-fucose metabolic process | (GO:0042354) | 0.001109929 | FUT7;FUT4 | |
| Cellular components | Golgi subcompartment | (GO:0098791) | 0.001591532 | SLC35A2;FUT7;CUX1;CDH1;B3GNT5;RAB12;AP1B1;MAN1C1;FUT4 |
| spindle pole | (GO:0000922) | 0.003189301 | SGO1;STAG2;HSF1;CEP128 | |
| tertiary granule lumen | (GO:1904724) | 0.003721186 | CDA;CXCL1;PTX3 | |
| nuclear transcription-factor complex | (GO:0044798) | 0.007595372 | MAX;MXI1;NKX2-5 | |
| mitotic spindle pole | (GO:0097431) | 0.008720051 | STAG2;HSF1 | |
| GO Pathways | Adj p-Value | Genes | ||
|---|---|---|---|---|
| Molecular functions | interleukin-1 receptor binding | (GO:0005149) | 0.001725028 | IL1RN;IL1B |
| peptidoglycan binding | (GO:0042834) | 0.001966159 | NOD2;TLR2 | |
| phosphatidylinositol-4,5-bisphosphate 3-kinase activity | (GO:0046934) | 0.002844067 | PIK3CD;HBEGF;PIK3R5 | |
| phosphatidylinositol bisphosphate kinase activity | (GO:0052813) | 0.00321474 | PIK3CD;HBEGF;PIK3R5 | |
| chemokine receptor activity | (GO:0004950) | 0.003394807 | CXCR1;CXCR2 | |
| Biological pathways | neutrophil degranulation | (GO:0043312) | 0.0000000834 | CDA;TNFAIP6;MME;FPR1;CXCL1;CXCR1;ALOX5;QPCT;CXCR2;CYSTM1;PTX3;S100A11;TLR2 |
| neutrophil activation involved in immune response | (GO:0002283) | 0.0000000918 | CDA;TNFAIP6;MME;FPR1;CXCL1;CXCR1;ALOX5;QPCT;CXCR2;CYSTM1;PTX3;S100A11;TLR2 | |
| neutrophil-mediated immunity | (GO:0002446) | 0.0000001009 | CDA;TNFAIP6;MME;FPR1;CXCL1;CXCR1;ALOX5;QPCT;CXCR2;CYSTM1;PTX3;S100A11;TLR2 | |
| inflammatory response | (GO:0006954) | 0.000001012 | TNFAIP6;IL1B;PTGER2;CXCR2;FPR1;PIK3CD;CXCL1;PTX3;NOD2 | |
| cytokine-mediated signaling pathway | (GO:0019221) | 0.0000113203 | IL1RN;IRAK2;IL1B;ALOX5;FPR1;PIK3CD;CXCL1;NOD2;XAF1;JUNB;IFIT2;BCL2L1 | |
| Cellular components | tertiary granule lumen | (GO:1904724) | 0.00000322 | CDA;TNFAIP6;QPCT;CXCL1;PTX3 |
| tertiary granule | (GO:0070820) | 0.00000540 | CDA;TNFAIP6;QPCT;FPR1;CXCL1;CYSTM1;PTX3 | |
| ficolin-1-rich granule | (GO:0101002) | 0.001014794 | CDA;TNFAIP6;ALOX5;QPCT;FPR1 | |
| ficolin-1-rich granule lumen | (GO:1904813) | 0.001721089 | CDA;TNFAIP6;ALOX5;QPCT | |
| secretory granule lumen | (GO:0034774) | 0.002052377 | CDA;ALOX5;QPCT;CXCL1;PTX3;S100A11 | |
| GO Pathways | Adj p-Value | Genes | ||
|---|---|---|---|---|
| Molecular functions | ATP-dependent helicase activity | (GO:0008026) | 0.003438361 | MCM7;DDX51;DDX52 |
| RNA polymerase II transcription-factor binding | (GO:0001085) | 0.009156092 | EOMES;STAT3;CTNNB1 | |
| RNA polymerase II activating transcription-factor binding | (GO:0001102) | 0.012116708 | EOMES;CTNNB1 | |
| inorganic anion transmembrane transporter activity | (GO:0015103) | 0.012614773 | SLC22A4;SLC1A1 | |
| small GTPase binding | (GO:0031267) | 0.021109349 | ACAP1;FLNA | |
| Biological Pathways | cardiolipin biosynthetic process | (GO:0032049) | 3.43 × 10−4 0.000343 | PGS1;PLA2G6 |
| phosphatidylglycerol biosynthetic process | (GO:0006655) | 5.49 × 10−4 0.000549 | PGS1;PLA2G6 | |
| thyroid-hormone generation | (GO:0006590) | 5.49 × 10−4 0.000549 | DUOX1;DIDO1 | |
| cardiolipin metabolic process | (GO:0032048) | 9.45 × 10−4 0.000945 | PGS1;PLA2G6 | |
| cytoplasmic sequestering of protein | (GO:0051220) | 0.00126624 | MXI1;FLNA | |
| Cellular functions | nucleolus | (GO:0005730) | 0.010004394 | C1D;MXI1;FLNA;DDX51;RGS12;DDX52;PHLDA1 |
| RNA polymerase II transcription-factor complex | (GO:0090575) | 0.015449845 | MXI1;STAT3;CTNNB1 | |
| tertiary granule lumen | (GO:1904724) | 0.016344963 | CDA;CXCL1 | |
| tertiary granule | (GO:0070820) | 0.020605435 | CDA;CXCL1;CD59 | |
| THO complex part of transcription export complex | (GO:0000445) | 0.021114315 | THOC3 | |
| GEO Accession | Sample Type | PRDM2 | CXCL1 | PHLDA1 | DIDO1 | CDA |
|---|---|---|---|---|---|---|
| GSE14922 | Control vs. cortisol secreting adenoma | Down regulated | Down regulated | Down regulated | Up regulated | Up regulated |
| GSE109857 | Control vs. glioma | Up regulated | Down regulated | Down regulated | Down regulated | Down regulated |
| GSE114852 | Control vs. depression | absent | Down regulated | Down regulated | Up regulated | Down regulated |
| GSE32280 | Control vs. depression | Up regulated | Down regulated | Down regulated | Down regulated | Up regulated |
| GSE58430 | Control vs. depression | Down regulated | Down regulated | Up regulated | Down regulated | Up regulated |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhadra, S.; Chen, S.; Liu, C. Analysis of Differentially Expressed Genes That Aggravate Metabolic Diseases in Depression. Life 2021, 11, 1203. https://doi.org/10.3390/life11111203
Bhadra S, Chen S, Liu C. Analysis of Differentially Expressed Genes That Aggravate Metabolic Diseases in Depression. Life. 2021; 11(11):1203. https://doi.org/10.3390/life11111203
Chicago/Turabian StyleBhadra, Sukanta, Siyu Chen, and Chang Liu. 2021. "Analysis of Differentially Expressed Genes That Aggravate Metabolic Diseases in Depression" Life 11, no. 11: 1203. https://doi.org/10.3390/life11111203