Potential for Applying Continuous Directed Evolution to Plant Enzymes: An Exploratory Study

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Directed Evolution
3. Continuous Directed Evolution
4. OrthoRep
5. EvolvR
6. Limitations of Continuous Directed Evolution Systems
7. Materials and Methods
7.1. Chemicals, Enzymes, and Oligonucleotides
7.2. OrthoRep
7.2.1. Construction of Multipurpose p1 Integration Vector
7.2.2. Transformation of the Yeast ΔTHI4 Strain with Wild Type and Error-Prone Polymerases
7.2.3. Integration of ScTHI4 into the p1 Plasmid
7.2.4. Protoplast Fusion of ΔTHI4 BY4741 and GA-Y319 Strains
7.2.5. Functional Complementation in Yeast
8. EvolvR
8.1. Bacterial Strains and Plasmid Construction
8.2. Media and Culture Condition
9. Results and Discussion
9.1. OrthoRep
9.2. EvolvR
9.3. Future Perspectives
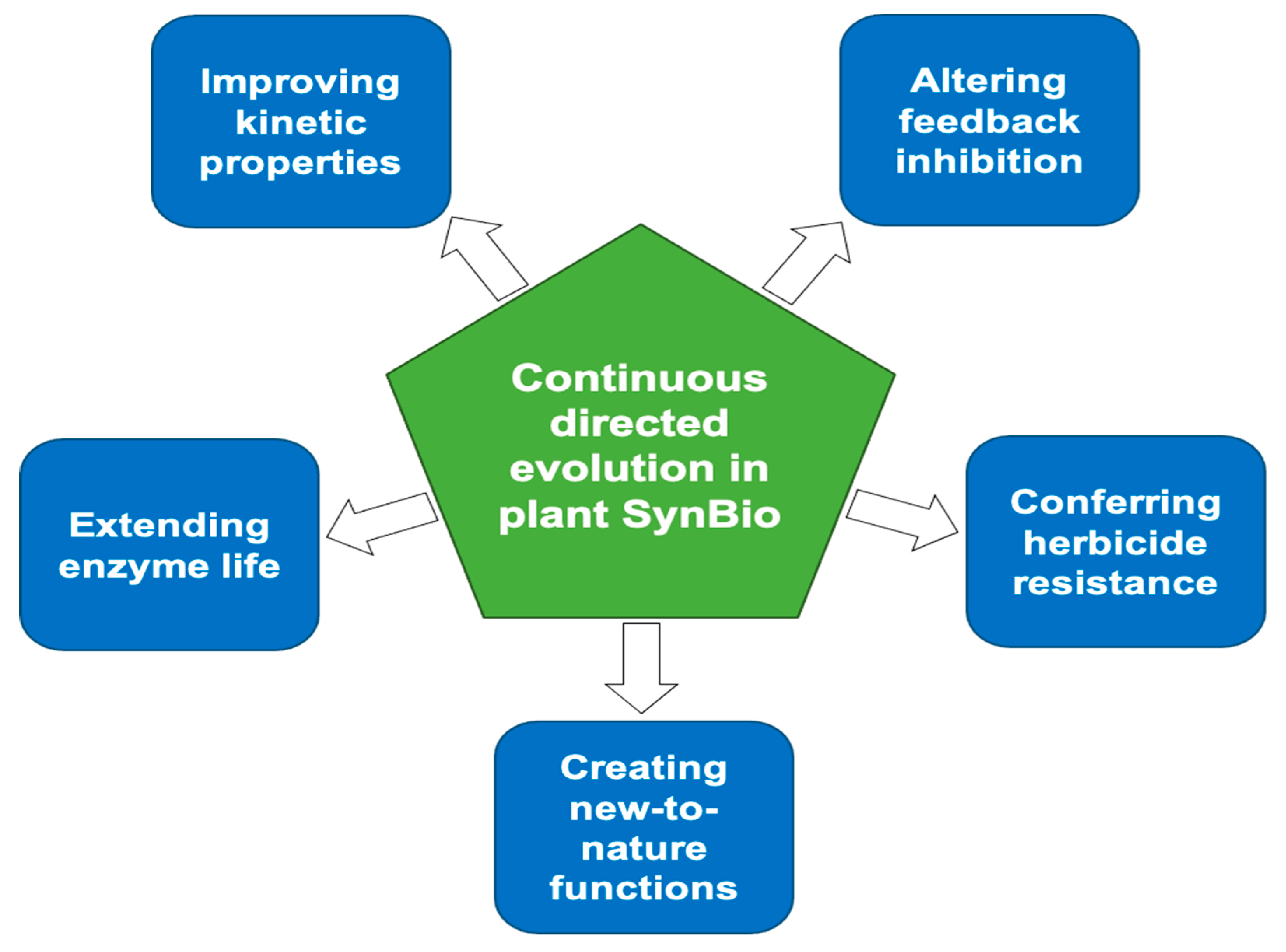
- Improving kinetic properties. Directed evolution can outdo rational design by introducing multiple, potentially beneficial changes and going beyond known or predictable key residues [22]. Because it is carried out in vivo, continuous directed evolution has the further advantage of improving enzyme function under realistic conditions (e.g., substrate and product levels and, macromolecular crowding), which in vitro screening protocols may mimic poorly [1].
- Altering feedback inhibition. Many enzymes at the head of pathways are inhibited by downstream pathway intermediates or end-products and relieving this feedback inhibition is often effective in increasing flux in such pathways [51,52]. Selecting for feedback-insensitive microbial enzymes has a long track record of success [53,54,55] and can be extended to plant enzymes in microbes engineered to express them [56,57].
- Conferring herbicide resistance. Many herbicides act by inhibiting essential metabolic enzymes, and mutations that alleviate this inhibition confer resistance [58,59]. Continuous directed evolution has already been used to evolve antibiotic resistance in microbial enzymes [27,28]; this approach could be similarly applied to evolve herbicide-resistant crop enzymes. Using genome editing to put the resistant enzyme back in the crop would avoid using transgenes, which are now banned in various countries [60].
- Creating new-to-nature functions. Because continuous directed evolution dramatically improves access to the vast protein design landscape, it can create new-to-nature features more quickly than classic directed evolution [23,61]. Such new features include altered substrate specificity [62] and catalysis of a different type of reaction [8].
- Extending enzyme life. The unnecessarily short working lives of certain plant enzymes wastes resources [3]. Short life is likely due in part to damage to active site residues by chemically reactive substrates or products or by catalytic misfires [63]. A recently introduced metric for enzyme working life—Catalytic-Cycles-till-Replacement (CCR) = [Metabolic flux rate]/[Enzyme replacement rate]—helps identify enzymes that are candidates for life-lengthening [63]. Continuous directed evolution could potentially be applied to increase enzyme longevity in vivo, i.e., to raise CCR, e.g., by driving replacement of damage-prone residues within the active site by less vulnerable alternatives.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pettersson, G. Effect of evolution on the kinetic properties of enzymes. Eur. J. Biochem. 1989, 184, 561–566. [Google Scholar] [CrossRef]
- Bar-Even, A.; Noor, E.; Savir, Y.; Liebermeister, W.; Davidi, D. The moderately efficient enzyme: Evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry 2011, 50, 4402–4410. [Google Scholar] [CrossRef]
- Amthor, J.S.; Bar-Even, A.; Hanson, A.D.; Harvey Millar, A.; Stitt, M.; Sweetlove, L.J.; Tyerman, S.D. Engineering strategies to boost crop productivity by cutting respiratory carbon loss. Plant. Cell 2019, 31, 297–314. [Google Scholar] [CrossRef] [Green Version]
- Stitt, M. Progress in understanding and engineering primary plant metabolism. Curr. Opin. Biotechnol. 2013, 24, 229–238. [Google Scholar] [CrossRef]
- Jez, J.M.; Lee, S.S.G.; Sherp, A.M. The next green movement: Plant biology for the environment and sustainability. Science 2016, 353, 1241–1244. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.N., Jr.; Patron, N.; Hanson, A.D.; Jez, J.M. Plant metabolic engineering in the synthetic biology era: Plant chassis selection. Plant. Cell Rep. 2018, 37, 1357–1358. [Google Scholar] [CrossRef] [Green Version]
- Wurtzel, E.T.; Vickers, C.E.; Hanson, A.D.; Millar, A.H.; Cooper, M. Revolutionizing agriculture with synthetic biology. Nat. Plants 2019, 5, 1207–1210. [Google Scholar] [CrossRef]
- Jeschek, M.; Reuter, R.; Heinisch, T.; Trindler, C.; Kleur, J. Directed evolution of artificial metalloenzymes for in vivo metathesis. Nature 2016, 537, 661–665. [Google Scholar] [CrossRef]
- Trudeau, D.L.; Edlich-Muth, C.; Zarzycki, J.; Scheffen, M.; Goldsmith, M. Design and in vitro realization of carbon-conserving photorespiration. Proc. Natl. Acad. Sci. USA 2018, 115, E11455–E11464. [Google Scholar] [CrossRef] [Green Version]
- Engqvist, M.K.M.; Rabe, K.S. Applications of protein engineering and directed evolution in plant research. Plant. Physiol. 2019, 179, 907–917. [Google Scholar] [CrossRef] [Green Version]
- Peracchi, A. Enzyme catalysis: Removing chemically “essential” residues by site-directed mutagenesis. Trends Biochem. Sci. 2001, 26, 497–503. [Google Scholar] [CrossRef]
- Spreitzer, R.J.; Salvucci, M.E. Rubisco: Structure, regulatory interactions, and possibilities for a better enzyme. Annu. Rev. Plant. Biol. 2002, 53, 449–475. [Google Scholar] [CrossRef] [Green Version]
- Nishitani, Y.; Yoshida, S.; Fujihashi, M.; Kitagawa, K.; Doi, T. Structure-based catalytic optimization of a type III Rubisco from a hyperthermophile. J. Biol. Chem. 2010, 285, 39339–39347. [Google Scholar] [CrossRef] [Green Version]
- Ott, C.M.; Smith, B.D.; Portis, A.R., Jr.; Spreitzer, R.J. Activase region on chloroplast ribulose-1,5-bisphosphate carboxylase/oxygenase: Nonconservative substitution in the large subunit alters species specificity of protein interaction. J. Biol. Chem. 2000, 275, 26241–26244. [Google Scholar] [CrossRef] [Green Version]
- Larson, E.M.; O’Brien, C.M.; Zhu, G.; Spreitzer, R.J.; Portis, A.R., Jr. Specificity for activase is changed by a Pro-89 to Arg substitution in the large subunit of ribulose-1,5-bisphosphate carboxylase oxygenase. J. Biol. Chem. 1997, 272, 17033–17037. [Google Scholar] [CrossRef] [Green Version]
- Yokota, A.; Higashioka, M.; Wadano, A. Regulation of the activity of ribulose-1,5-bisphosphate carboxylase/oxygenase through cooperative binding of 6-phosphogluconate to its regulatory sites. Eur. J. Biochem. 1992, 208, 547–557. [Google Scholar] [CrossRef]
- Parry, M.A.J.; Keys, A.J.; Madgwick, P.J.; Carmo-Silva, A.E.; Andralojc, P.J. Rubisco regulation: A role for inhibitors. J. Exp. Bot. 2008, 59, 1569–1580. [Google Scholar] [CrossRef] [Green Version]
- Silver, P.A.; Way, J.C.; Arnold, F.H.; Meyerowitz, J.T. Synthetic biology: Engineering explored. Nature 2014, 509, 166–167. [Google Scholar] [CrossRef]
- Zeymer, C.; Hilvert, D. Directed evolution of protein catalysis. Annu. Rev. Biochem. 2018, 87, 131–157. [Google Scholar] [CrossRef]
- Wilson, R.H.; Martin-Avila, E.; Conlan, C.; Whitney, S.M. An improved Escherichia coli screen for Rubisco identifies a protein-protein interface that can enhance CO2-fixation kinetics. J. Biol. Chem. 2018, 293, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Lobkovsky, A.E.; Wolf, Y.I.; Koonin, E.V. Predictability of evolutionary trajectories in fitness landscapes. PLoS Comput. Biol. 2011, 7, e1002302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldsmith, M.; Tawfik, D.S. Enzyme engineering: Reaching the maximal catalytic efficiency peak. Curr. Opin. Struct. Biol. 2017, 47, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Liu, C. Probing pathways of adapting with continuous evolution. Curr. Opin. Syst. Biol. 2019, 14, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Esvelt, K.M.; Carlson, J.C.; Liu, D.R. A system for the continuous directed evolution of biomolecules. Nature 2011, 472, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, B.; Packer, M.; Badran, A.; Liu, D.R. A system for the continuous directed evolution of proteases rapidly reveals drug-resistance mutations. Nat. Commun. 2014, 5, 5352. [Google Scholar] [CrossRef]
- Morrison, M.S.; Podracky, C.J.; Liu, D.R. The developing toolkit of continuous directed evolution. Nat. Chem. Biol. 2020, 16, 610–619. [Google Scholar] [CrossRef]
- Ravikumar, A.; Arzumanyan, G.A.; Obadi, M.K.A.; Javanpour, A.A.; Liu, C.C. Scalable, continuous evolution of genes at mutation rates above genomic error thresholds. Cell 2018, 175, 1946–1957. [Google Scholar] [CrossRef] [Green Version]
- Halperin, S.O.; Tou, C.J.; Wong, E.B.; Modavi, C.; Schaffer, D.V. CRISPR-guide DNA polymerases enable diversification of all nucleotides in a tunable window. Nature 2018, 560, 248–252. [Google Scholar] [CrossRef]
- Álvarez, B.; Mencía, M.; de Lorenzo, V.; Fernández, L.A. In vivo diversification of target genomic using processive T7 RNA polymerase-base deaminase fusions blocked by RNA-guided dCas9. BioRxiv 2020, 850974. [Google Scholar]
- Zhong, Z.; Wong, B.G.; Ravikumar, A.; Arzumanyan, A.; Khalil, A.S. Automated continuous evolution of proteins in vivo. ACS Synth. Biol. 2020, 9, 1270–1276. [Google Scholar] [CrossRef]
- Gunge, N.; Sakaguchi, K. Intergeneric transfer of deoxyribonucleic acid killer plasmids, pGKl1 and pGKl2, from Kluyveromyces lactis into Saccharomyces cerevisiae by cell fusion. J. Bacteriol. 1981, 147, 155–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kämper, J.; Esser, K.; Gunge, N.; Meinhardt, F. Heterologous gene expression on the linear DNA killer plasmid from Kluyveromyces lactis. Curr. Genet. 1991, 19, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, A.; Arrieta, A.; Liu, C.C. An orthogonal DNA replication system in yeast. Nat. Chem. Biol. 2014, 10, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Abeydeera, N.D.; Bale, S.; Pai, P.J.; Dorrestein, P.C. Saccharomyces cerevisiae Thi4p is a suicide thiamine thiazole synthase. Nature 2011, 478, 542–546. [Google Scholar] [CrossRef] [Green Version]
- Hanson, A.D.; Amthor, J.S.; Sun, J.; Niehaus, T.D.; Gregory, J.F., III. Redesigning thiamin synthesis: Prospects and potential payoffs. Plant. Sci. 2018, 273, 92–99. [Google Scholar] [CrossRef]
- Joshi, J.; Beaudoin, G.A.; Patterson, J.A.; García-García, J.D.; Belisle, E. Bioinformatic and experimental evidence for suicidal and catalytic plant THI4s. Biochem. J. 2020, 477, 2055–2069. [Google Scholar] [CrossRef]
- Zhang, X.; Eser, B.E.; Chanani, P.K.; Begley, T.P.; Ealick, S.E. Structural basis for iron-mediated sulfur transfer in archael and yeast thiazole synthases. Biochemistry 2016, 55, 1826–1838. [Google Scholar] [CrossRef]
- Eser, B.E.; Zhang, X.; Chanani, P.K.; Begley, T.P.; Ealick, S.E. From suicide enzyme to catalyst: The iron-dependent sulfide transfer in Methanococcus jannaschii thiazole biosynthesis. J. Am. Chem. Soc. 2016, 138, 3639–3642. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Singler, C.L.; Beaudoin, G.A.W.; Joshi, J.; Patterson, J.A. Parts-prospecting for a high-efficiency thiamin thiazole biosynthesis pathway. Plant. Physiol. 2019, 179, 958–968. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Ravikumar, A.; Liu, C.C. Tunable expression systems for orthogonal DNA replication. ACS Synth. Biol. 2018, 7, 2930–2934. [Google Scholar] [CrossRef]
- Mount, R.C.; Jordan, B.E.; Hadfield, C. Transformation of lithium-treated yeast cells and the selection of auxotrophic and dominant markers. Methods Mol. Biol. 1996, 53, 139–145. [Google Scholar]
- Curran, B.P.; Bugeja, V.C. Protoplast fusion in Saccharomyces cerevisiae. Methods Mol. Biol. 1996, 53, 45–49. [Google Scholar] [PubMed]
- Javanpour, A.; Liu, C. Genetic compatibility and extensibility of orthogonal replication. ACS Synth. Biol. 2019, 8, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhong, Y.; Zhou, Y.; Tanaseichuk, O.; Li, L. Identification of a Xist silencing domain by Tiling CRISPR. Sci. Rep. 2019, 9, 2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, R.; Bujard, H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997, 25, 1203–1210. [Google Scholar] [CrossRef]
- Stewart, V.; Parales, J. Identification and expression of genes narL and narX of the nar (nitrate reductase) locus in Escherichia coli K-12. J. Bacteriol. 1988, 170, 1589–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korshunov, S.; Imlay, K.R.; Imlay, J.A. The cytochrome bd oxidase of Escherichia coli prevents respiratory inhibition by endogenous and exogenous hydrogen sulfide. Mol. Microbiol. 2016, 101, 62–77. [Google Scholar] [CrossRef] [Green Version]
- Paerl, R.W.; Bertrand, E.M.; Rowland, E.; Schatt, P.; Mehiri, M. Carboxythiazole is a key microbial nutrient currency and critical component of thiamin biosynthesis. Sci. Rep. 2018, 8, 5940. [Google Scholar] [CrossRef] [Green Version]
- Antoine, R.; Locht, C. Isolation and molecular characterization of a novel broad-host-range plasmid from Bordetella bronchiseptica with sequence similarities to plasmids from gram-positive organisms. Mol. Microbiol. 1992, 6, 1785–1799. [Google Scholar] [CrossRef]
- Nikel, P.I.; de Lorenzo, V. Pseudomonas putida as a functional chassis for industrial biocatalysis: From native biochemistry to trans-metabolism. Metab. Eng. 2018, 50, 142–155. [Google Scholar] [CrossRef]
- Hong, Z.; Lakkineni, K.; Zhang, Z.; Verma, D.P. Removal of feedback inhibition of delta(1)-pyrroline-5-carboxylate synthase results in increasing proline accumulation and protection of plants from osmotic stress. Plant. Physiol. 2000, 122, 1129–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitcomb, S.J.; Rakpenthai, A.; Brückner, F.; Fischer, A.; Parmar, S. Cysteine and methionine biosynthetic enzymes have distinct effects on seed nutritional quality and on molecular phenotypes associate with accumulation of a methionine-rich seed storage protein in rice. Front. Plant. Sci. 2020, 11, 1118. [Google Scholar] [CrossRef]
- Cohen, G.N.; Patte, J.C.; Truffa-Bachi, P. Parallel modifications caused by mutations in two enzymes concerned with the biosynthesis of threonine in Escherichia coli. Biochem. Biophys. Res. Commun. 1965, 19, 546–550. [Google Scholar] [CrossRef]
- Báez-Viveros, J.L.; Osuna, J.; Hernández-Chávez, G.; Soberón, X.; Bolívar, F. Metabolic engineering and protein directed evolution increasing the yield of L-phenylalanine synthesized from glucose in Escherichia coli. Biotechnol. Bioeneg. 2004, 87, 516–524. [Google Scholar] [CrossRef]
- Petit, C.; Kim, Y.; Lee, S.; Brown, J.; Larsen, E. Reduction of feedback inhibition in homoserine kinase (ThrB) of Corynebacterium glutamicum enhances l-threonine biosynthesis. ACS Omega 2018, 3, 1178–1186. [Google Scholar] [CrossRef] [Green Version]
- Alberstein, M.; Eisenstein, M.; Abeliovich, H. Removing allosteric feedback inhibition of tomato 4-coumarate: CoA ligase by directed evolution. Plant. J. 2012, 69, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.; Moghe, G.D.; Leong, B.; Kim, J.; Ofner, I. A feedback-insensitive isopropylmalate synthase affects acylsugar composition in cultivated and wild tomato. Plant. Physiol. 2015, 169, 1821–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangani, G.; Salas-Perez, R.A.; Aponte, R.A.; Knapp, M.; Craig, I.R. A novel single-site mutation in the catalytic domain of protoporphyrinogen oxidase IX (PPO) confers resistance to PPO-inhibiting herbicides. Front. Plant. Sci. 2019, 15, 568. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, T.R.; Langford, M.P.; Viner, R.; Blain, R.E.; Callaghan, F.M. Characterization of 4-hydroxyphenylpyruvate dioxygenases, inhibition by herbicides and engineering for herbicide tolerance in crops. Pestic. Biochem. Physiol. 2019, 156, 9–28. [Google Scholar] [CrossRef]
- Mackelprang, R.; Lemaux, P.G. Genetic engineering and editing of plants: An analysis of new and persisting questions. Annu. Rev. Plant. Biol. 2020, 71, 659–687. [Google Scholar] [CrossRef] [Green Version]
- Badran, A.H.; Liu, D.R. In vivo continuous directed evolution. Curr. Opin. Chem. Biol. 2015, 24, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.G.; Domann, S.; Nelson, A.; Berry, A. Modifying the stereochemistry of an enzyme-catalyzed reaction by directed evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 3143–3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tivendale, N.D.; Hanson, A.D.; Henry, C.S.; Hegeman, A.D.; Millar, A.H. Enzymes as parts in need of replacement–and how to extend their working life. Trends Plant. Sci. 2020, 25, 661–669. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-García, J.D.; Joshi, J.; Patterson, J.A.; Trujillo-Rodriguez, L.; Reisch, C.R.; Javanpour, A.A.; Liu, C.C.; Hanson, A.D. Potential for Applying Continuous Directed Evolution to Plant Enzymes: An Exploratory Study. Life 2020, 10, 179. https://doi.org/10.3390/life10090179
García-García JD, Joshi J, Patterson JA, Trujillo-Rodriguez L, Reisch CR, Javanpour AA, Liu CC, Hanson AD. Potential for Applying Continuous Directed Evolution to Plant Enzymes: An Exploratory Study. Life. 2020; 10(9):179. https://doi.org/10.3390/life10090179
Chicago/Turabian StyleGarcía-García, Jorge D., Jaya Joshi, Jenelle A. Patterson, Lidimarie Trujillo-Rodriguez, Christopher R. Reisch, Alex A. Javanpour, Chang C. Liu, and Andrew D. Hanson. 2020. "Potential for Applying Continuous Directed Evolution to Plant Enzymes: An Exploratory Study" Life 10, no. 9: 179. https://doi.org/10.3390/life10090179