Evaluation of GammaH2AX in Buccal Cells as a Molecular Biomarker of DNA Damage in Alzheimer’s Disease in the AIBL Study of Ageing

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Ethics and Clinical Assessment of the Participants
2.2. Buccal Cell Collection and Microscope Slide Preparation
2.3. Preparation of Buccal Cells for Immunofluorescence
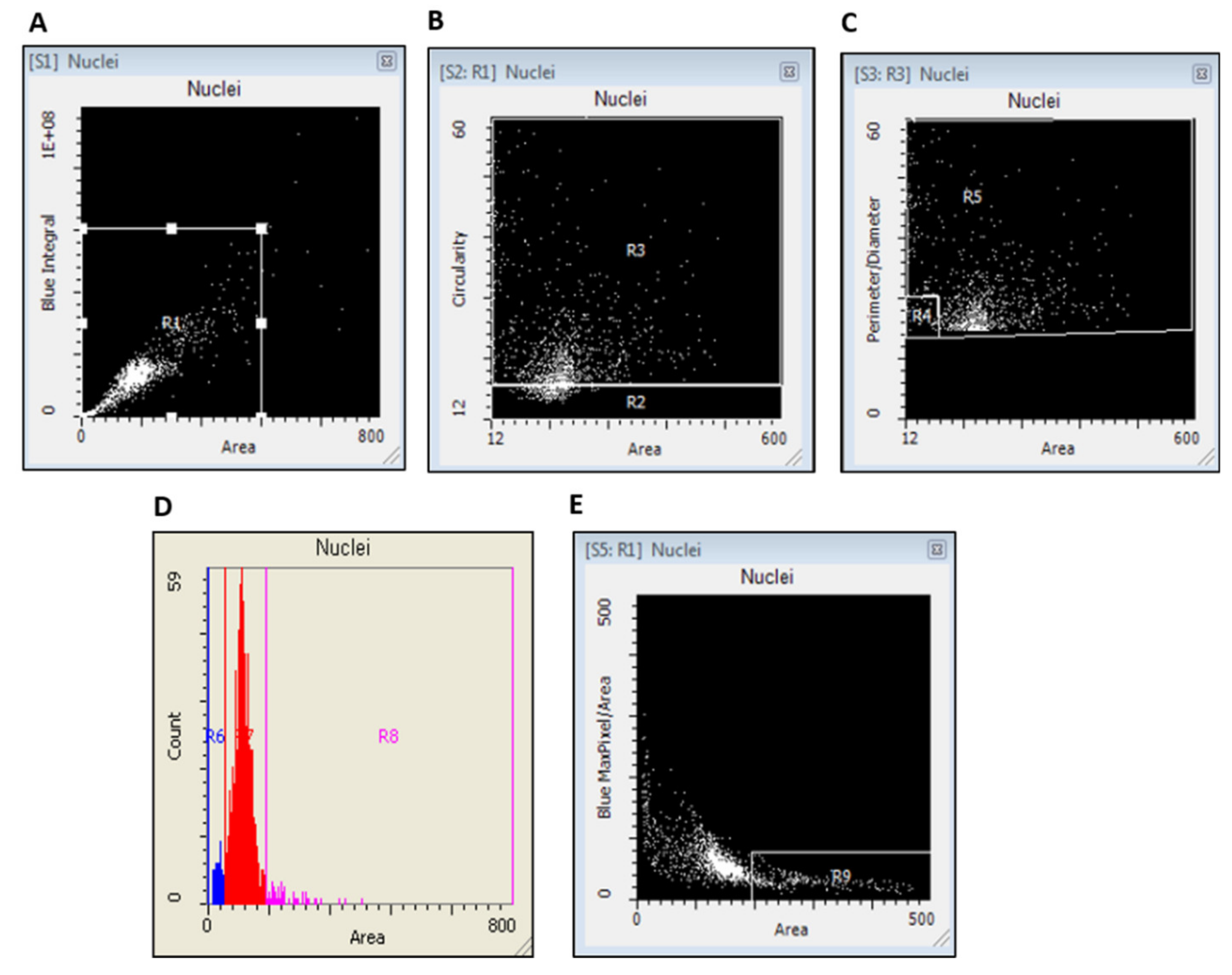
2.4. Laser Scanning Cytometry Measurements of γH2AX
2.5. Statistical Analysis
3. Results
3.1. Clinical Characteristics of Participants
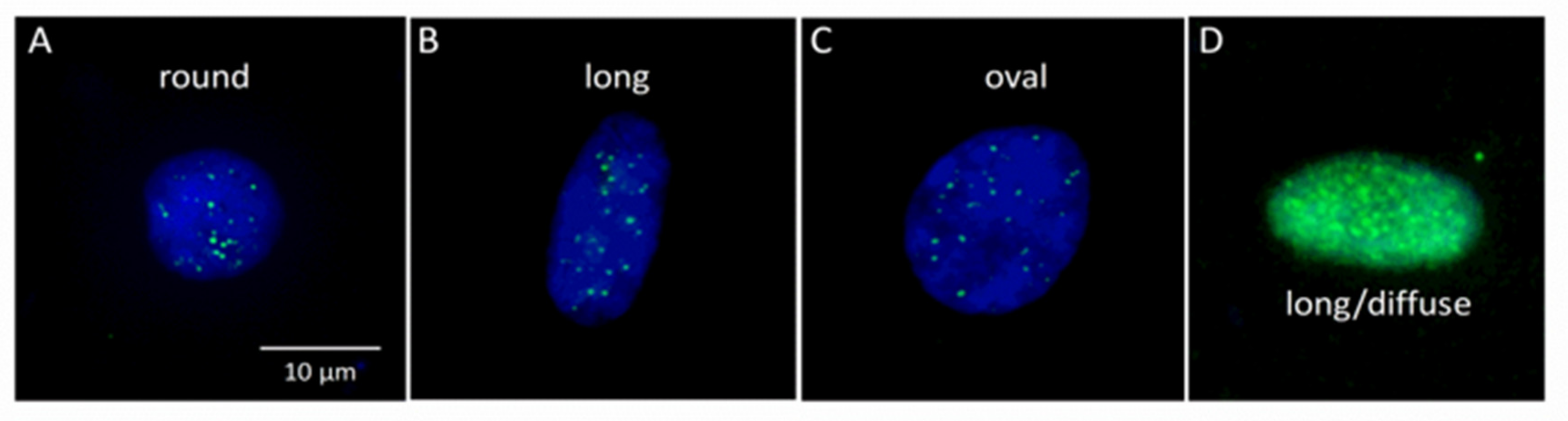
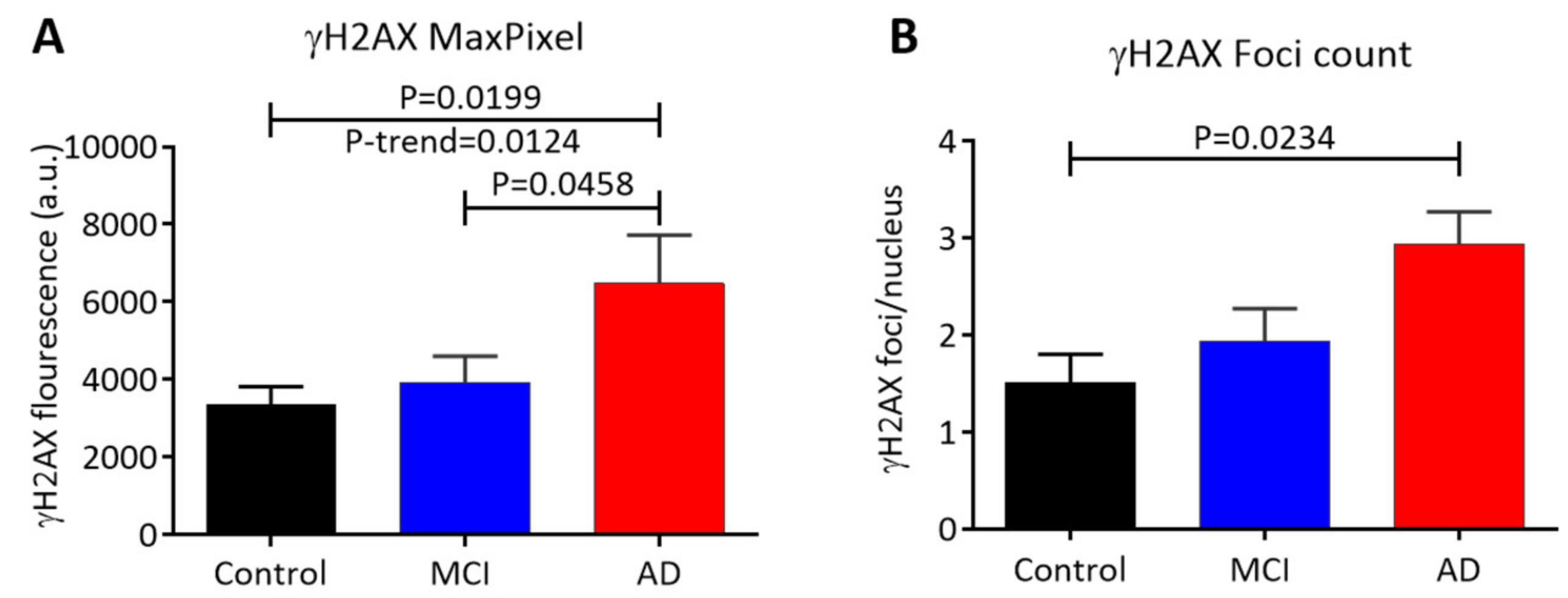
3.2. Scoring of γH2AX Signals in Buccal Cells by LSC
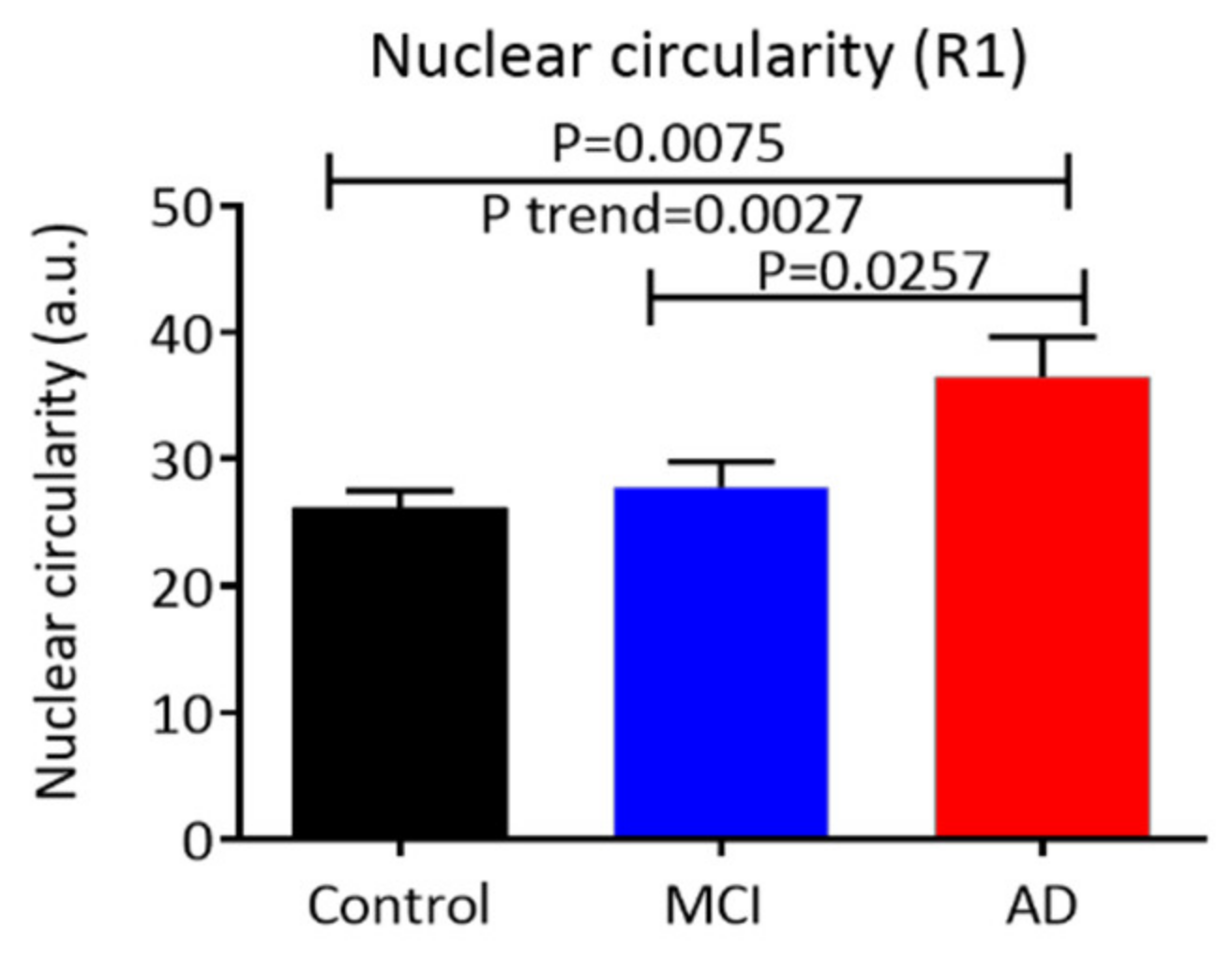
3.3. Nuclear (Morphology Characteristics) Circularity, Integral, and Area in Buccal Cells
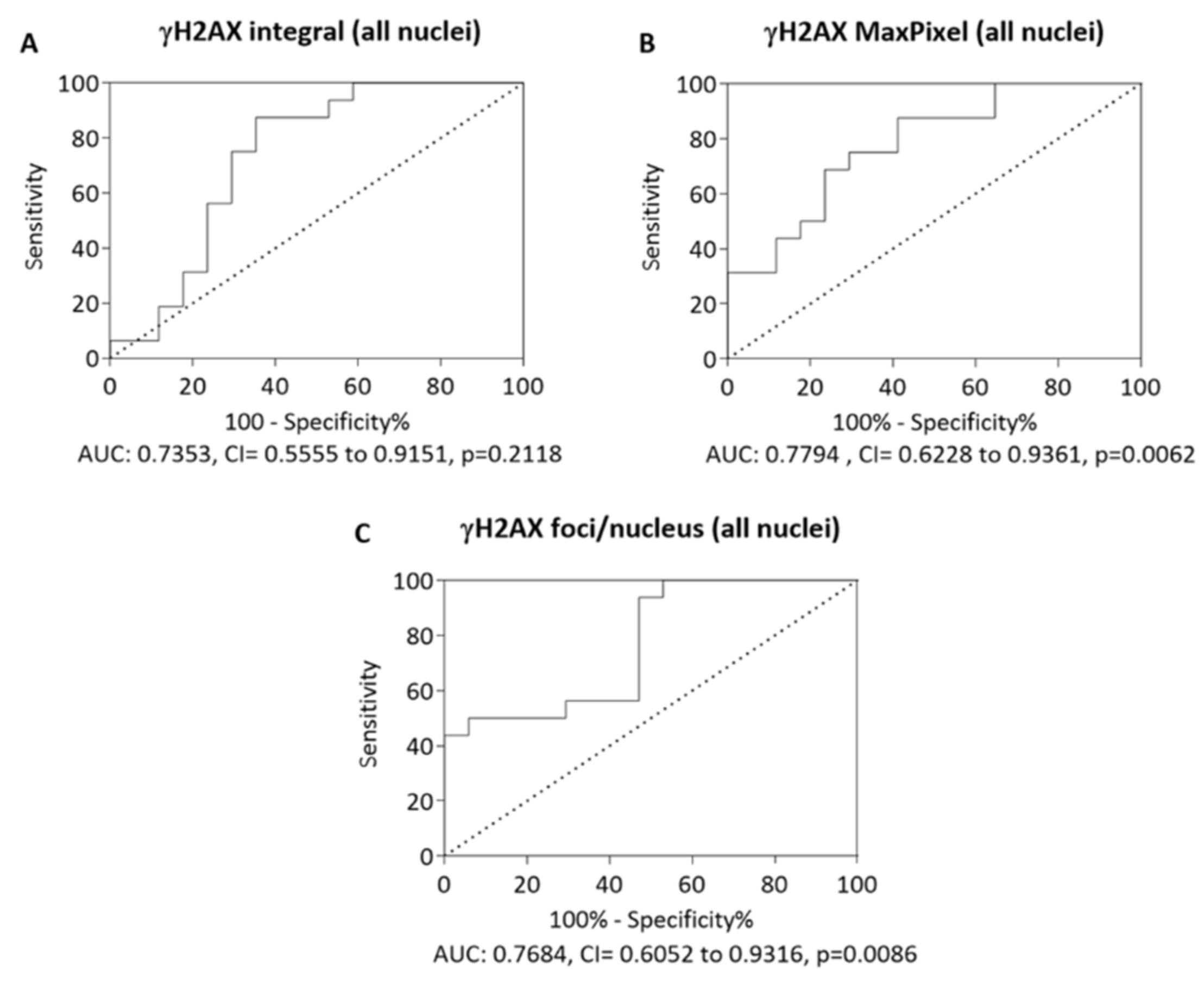
3.4. Receiver-Operating Characteristic Curve
3.5. Correlation of γH2AX Signals (Integral, MaxPixel) in Different Types of Buccal Cell Nuclei with the MMSE Score
3.6. Correlation of γH2AX Integral with Blood Parameters
3.7. Correlation of γH2AX in Control, MCI and AD Nuclei with Blood Parameters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burns, A.; Byrne, E.J.; Maurer, K. Alzheimer’s disease. Lancet 2002, 13, 163–165. [Google Scholar] [CrossRef]
- Alzheimer’s Association; Thies, W.; Bleiler, L. Alzheimer’s disease facts and figures. Alzheimers Dement 2011, 77, 208–244. [Google Scholar]
- Lobo, A.; Launer, L.J.; Fratiglioni, L.; Andersen, K.; Di Carlo, A.; Breteler, M.M.; Copeland, J.R.M.; Dartigues, J.F.; Jagger, C.; Martinez-Lage, J.; et al. Prevalence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurologic diseases in the elderly research group. Neurology 2000, 54 (Suppl. S5), S4–S9. [Google Scholar]
- Tijms, B.M.; Moller, C.; Vrenken, H.; Wink, A.M.; de Haan, W.; van der Flier, W.M.; Stam, C.J.; Scheltens, P.; Barkhof, F. Single-Subject grey matter graphs in Alzheimer’s disease. PLoS ONE 2013, 88, e58921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forlenza, O.V.; Diniz, B.S.; Stella, F.; Teixeira, A.L.; Gattaz, W.F. Mild cognitive impairment. Part 1: Clinical characteristics and predictors of dementia. Rev. Bras. Psiquiatr. 2013, 35, 178–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, M.; Wimo, A.; Guerchet, M.; Claire Ali, G.; Wu, Y.; Prina, M. The global impact of dementia. In World Alzheimer’s Disease Report 2015; Alzheimer’s Disease International: London, UK, 2015. [Google Scholar]
- Sloane, P.D.; Zimmerman, S.; Suchindran, C.; Reed, P.; Wang, L.; Boustani, M.; Sudha, S. The public health impact of Alzheimer’s disease, 2000–2050: Potential implication of treatment advances. Annu. Rev. Public Health 2002, 23, 213–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ittner, L.M.; Gotz, J. Amyloid-Beta and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef]
- Weintraub, S.; Wicklund, A.H.; Salmon, D.P. The neuropsychological profile of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 22, a006171. [Google Scholar] [CrossRef]
- Storandt, M. Neuropsychological assessment in Alzheimer’s disease. Exp. Ageing Res. 1991, 17, 100–101. [Google Scholar]
- Tombaugh, T.N.; McIntyre, N.J. The mini-mental state examination: A comprehensive review. J. Am. Geriatr. Soc. 1992, 40, 922–935. [Google Scholar] [CrossRef]
- Mitchell, A.J. A meta-analysis of the accuracy of the mini-mental state examination in the detection of dementia and mild cognitive impairment. J. Psychiatr. Res. 2009, 43, 411–431. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A. Plaques and tangles and the pathogenesis of Alzheimer’s disease. Folia Neuropathol. 2006, 44, 1–11. [Google Scholar] [PubMed]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Tredici, K.D.; et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Humpel, C. Identifying and validating biomarkers for Alzheimer’s disease. Trends Biotechnol. 2011, 29, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, M.S.; Passmore, A.P.; Todd, S.; McGuinness, B.; Craig, D.; Johnston, J.A. The development of effective biomarkers for Alzheimer’s disease: A review. Int. J. Geriatr. Psychiatry 2013, 28, 331–340. [Google Scholar] [CrossRef]
- Gauthier, S.; Reisberg, B.; Zaudig, M.; Petersen, R.C.; Ritchie, K.; Broich, K.; Belleville, S.; Brodaty, H.; Bennett, D.; Chertkow, H.; et al. Mild cognitive impairment. Lancet 2006, 367, 1262–1270. [Google Scholar] [CrossRef]
- Fischer, P.; Jungwirth, S.; Zehetmayer, S.; Weissgram, S.; Hoenigschnabl, S.; Gelpi, E.; Broich, K.; Belleville, S.; Brodaty, H.; Bennett, D.; et al. Conversion from subtypes of mild cognitive impairment to Alzheimer dementia. Neurology 2007, 68, 288–291. [Google Scholar] [CrossRef]
- Farias, S.T.; Mungas, D.; Reed, B.R.; Harvey, D.; DeCarli, C. Progression of mild cognitive impairment to dementia in clinic- vs. community-based cohorts. Arch. Neurol. 2009, 66, 1151–1157. [Google Scholar] [CrossRef] [Green Version]
- Petersen, R.C.; Roberts, R.O.; Knopman, D.S.; Boeve, B.F.; Geda, Y.E.; Ivnik, R.J.; Smith, G.E.; Jack, C.R. Mild cognitive impairment: Ten years later. Arch. Neurol. 2009, 66, 1447–1455. [Google Scholar] [CrossRef] [Green Version]
- Gasparini, L.; Racchi, M.; Binetti, G.; Trabucchi, M.; Solerte, S.B.; Alkon, D.; Etcheberrigaray, R.; Gibson, G.; Blass, J.; Paoletti, R.; et al. Peripheral markers in testing pathophysiological hypotheses and diagnosing Alzheimer’s disease. FASEB J. 1998, 12, 17–34. [Google Scholar] [CrossRef] [Green Version]
- Joachim, C.L.; Mori, H.; Selkoe, D.J. Amyloid beta-protein deposition in tissues other than brain in Alzheimer’s disease. Nature 1989, 341, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Soininen, H.; Syrjanen, S.; Heinonen, O.; Neittaanmaki, H.; Miettinen, R.; Paljarvi, L. Amyloid beta-protein deposition in skin of patients with dementia. Lancet 1992, 339, 245. [Google Scholar] [CrossRef]
- Khan, T.K.; Alkon, D.L. Peripheral biomarkers of Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 44, 729–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, L.E.; Muffat, J.A.; Cherny, R.A.; Moir, R.D.; Ericsson, M.H.; Huang, X.; Mavros, C.; Coccia, J.A.; Faget, K.Y.; Fitch, K.A.; et al. Cytosolic beta-amyloid deposition and supranuclear cataracts in lenses from people with Alzheimer’s disease. Lancet 2003, 361, 1258–1265. [Google Scholar] [CrossRef]
- Lee, M.; Guo, J.P.; Kennedy, K.; McGeer, E.G.; McGeer, P.L. A method for diagnosing Alzheimer’s disease based on salivary amyloid-beta protein 42 levels. J. Alzheimer’s Dis. 2017, 55, 1175–1182. [Google Scholar] [CrossRef]
- Francois, M.; Leifert, W.; Martins, R.; Thomas, P.; Fenech, M. Biomarkers of Alzheimer’s disease risk in peripheral tissues; focus on buccal cells. Curr. Alzheimer Res. 2014, 11, 519–531. [Google Scholar] [CrossRef] [Green Version]
- Francois, M.; Leifert, W.; Hecker, J.; Faunt, J.; Martins, R.; Thomas, P.; Fenech, M. Altered cytological parameters in buccal cells from individuals with mild cognitive impairment and Alzheimer’s disease. Cytom. Part A 2014, 85, 698–708. [Google Scholar] [CrossRef] [Green Version]
- Leifert, W.R.; Francois, M.; Thomas, P.; Luther, E.; Holden, E.; Fenech, M. Automation of the buccal micronucleus cytome assay using laser scanning cytometry. Methods Cell Biol. 2011, 102, 321–339. [Google Scholar]
- Kummer, C.; Wehner, S.; Quast, T.; Werner, S.; Herzog, V. Expression and potential function of beta-amyloid precursor proteins during cutaneous wound repair. Exp. Cell Res. 2002, 280, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991, 11, 213–216. [Google Scholar] [CrossRef]
- Khan, S.S.; Bloom, G.S. Tau: The center of a signaling nexus in Alzheimer’s disease. Front. Neurosci. 2016, 10, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, H.; Matsumoto, M.; Iwai, K.; Tsuchiya, H.; Miyauchi, E.; Takasaki, M.; Kamino, K.; Munehira, J.; Kimura, Y.; Kawanishi, K.; et al. The tau protein of oral epithelium increases in Alzheimer’s disease. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2002, 57, M64–M70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, P.; Fenech, M. Chromosome 17 and 21 aneuploidy in buccal cells is increased with ageing and in Alzheimer’s disease. Mutagenesis 2008, 23, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Fraga, C.G.; Shigenaga, M.K.; Park, J.W.; Degan, P.; Ames, B.N. Oxidative damage to DNA during aging: 8-hydroxy-2’-deoxyguanosine in rat organ DNA and urine. Proc. Natl. Acad. Sci. USA 1990, 87, 4533–4537. [Google Scholar] [CrossRef] [Green Version]
- Goukassian, D.; Gad, F.; Yaar, M.; Eller, M.S.; Nehal, U.S.; Gilchrest, B.A. Mechanisms and implications of the age-associated decrease in DNA repair capacity. FASEB J. 2000, 14, 1325–1334. [Google Scholar]
- Wilson, D.M., Ш; Bohr, V.A.; McKinnon, P.J. DNA damage, DNA repair, ageing and age-related disease. Mech. Aging Dev. 2008, 129, 349–352. [Google Scholar] [CrossRef] [Green Version]
- Bucholtz, N.; Demuth, I. DNA-Repair in mild cognitive impairment and Alzheimer’s disease. DNA Repair (Amst.) 2013, 12, 811–816. [Google Scholar] [CrossRef]
- Lovell, M.A.; Xie, C.; Markesbery, W.R. Decreased base excision repair and increased helicase activity in Alzheimer’s disease brain. Brain Res. 2000, 855, 116–123. [Google Scholar] [CrossRef]
- Thomas, P.; Holland, N.; Bolognesi, C.; Kirsch-Volders, M.; Bonassi, S.; Zeiger, E.; Knasmueller, S.; Fenech, M. Buccal micronucleus cytome assay. Nat. Protoc. 2009, 44, 825–837. [Google Scholar] [CrossRef]
- Iqbal, K.; Grundke-Iqbal, I.; Smith, A.J.; George, L.; Tung, Y.C.; Zaidi, T. Identification and localization of a tau peptide to paired helical filaments of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 5646–5650. [Google Scholar] [CrossRef] [Green Version]
- Koo, E.H. The beta-amyloid precursor protein (APP) and Alzheimer’s disease: Does the tail wag the dog? Traffic 2002, 33, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; O’Callaghan, N.J.; Fenech, M. Telomere length in white blood cells, buccal cells and brain tissue and its variation with ageing and Alzheimer’s disease. Mech. Ageing Dev. 2008, 129, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Migliore, L.; Fontana, I.; Trippi, F.; Colognato, R.; Coppede, F.; Tognoni, G.; Nucciarone, B.; Siciliano, G. Oxidative DNA damage in peripheral leukocytes of mild cognitive impairment and AD patients. Neurobiol. Aging 2005, 26, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [Green Version]
- Myung, N.H.; Zhu, X.; Kruman, I.I.; Castellani, R.J.; Petersen, R.B.; Siedlak, S.L.; Perry, G.; Smith, M.A.; Lee, H.G. Evidence of DNA damage in Alzheimer disease: Phosphorylation of histone H2AX in astrocytes. Age 2008, 30, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.R.; Santos, A.C.F.; Farfel, J.M.; Grinberg, L.T.; Ferretti, R.E.; Campos, A.H.J.F.M.; Cunha, I.W.; Begnami, M.D.; Rocha, R.M.; Carraro, D.M.; et al. Repair of oxidative DNA damage, cell-cycle regulation and neuronal death may influence the clinical manifestation of Alzheimer’s disease. PLoS ONE 2014, 9, e99897. [Google Scholar] [CrossRef]
- Schurman, S.H.; Dunn, C.A.; Greaves, R.; Yu, B.; Ferrucci, L.; Croteau, D.L.; Seidman, M.M.; Bohr, V.A. Age-Related disease association of endogenous gamma-H2AX foci in mononuclear cells derived from leukapheresis. PLoS ONE 2012, 7, e45728. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef] [Green Version]
- Savic, V.; Yin, B.; Maas, N.L.; Bredemeyer, A.L.; Carpenter, A.C.; Helmink, B.A.; Yang-Iott, K.S.; Sleckman, B.P.; Bassing, C.H. Formation of dynamic gamma-H2AX domains along broken DNA strands is distinctly regulated by ATM and MDC1 and dependent upon H2AX densities in chromatin. Mol. Cell 2009, 34, 298–310. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, M.S.; Francois, M.; Fenech, M.F.; Leifert, W.R. GammaH2AX responses in human buccal cells exposed to ionizing radiation. Cytom. Part A 2015, 87, 296–308. [Google Scholar] [CrossRef]
- Gonzalez, J.E.; Roch-Lefevre, S.H.; Mandina, T.; Garcia, O.; Roy, L. Induction of gamma-H2AX foci in human exfoliated buccal cells after in vitro exposure to ionising radiation. Int. J. Radiat. Biol. 2010, 86, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Ellis, K.A.; Bush, A.I.; Darby, D.; De Fazio, D.; Foster, J.; Hudson, P.; Lautenschlager, N.T.; Lenzo, N.; Martins, R.N.; Maruff, P.; et al. The Australian imaging, biomarkers and lifestyle (AIBL) study of aging: Methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer’s disease. Int. Psychogeriatr. 2009, 21, 672–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doecke, J.D.; Laws, S.M.; Faux, N.G.; Wilson, W.; Burnham, S.C.; Lam, C.P.; Mondal, A.; Bedo, J.; Bush, A.I.; Brown, B.; et al. Blood-Based protein biomarkers for diagnosis of Alzheimer disease. Arch. Neurol. 2012, 69, 1318–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faux, N.G.; Ellis, K.A.; Porter, L.; Fowler, C.J.; Laws, S.M.; Martins, R.N.; Pertile, K.K.; Rembach, A.; Rowe, C.C.; Rumble, R.L.; et al. Homocysteine, vitamin B12, and folic acid levels in Alzheimer’s disease, mild cognitive impairment, and healthy elderly: Baseline characteristics in subjects of the Australian imaging biomarker lifestyle study. J. Alzheimer’s Dis. 2011, 27, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Shukkur, E.A.; Shimohata, A.; Akagi, T.; Yu, W.; Yamaguchi, M.; Murayama, M.; Chui, D.; Takeuchi, T.; Amano, K.; Subramhanya, K.H.; et al. Mitochondrial dysfunction and tau hyperphosphorylation in Ts1Cje, a mouse model for Down syndrome. Hum. Mol. Genet. 2006, 15, 2752–2762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader Lange, M.L.; St Clair, D.; Markesbery, W.R.; Studzinski, C.M.; Murphy, M.P.; Butterfield, D.A. Age-Related loss of phospholipid asymmetry in APP(NLh)/APP(NLh) x PS-1(P264L)/PS-1(P264L) human double mutant knock-in mice: Relevance to Alzheimer disease. Neurobiol. Dis. 2010, 38, 104–115. [Google Scholar] [CrossRef] [Green Version]
- DelleDonne, A.; Kouri, N.; Reinstatler, L.; Sahara, T.; Li, L.; Zhao, J.; Dickson, D.W.; Ertekin-Taner, N.; Leissring, M.A. Development of monoclonal antibodies and quantitative ELISAs targeting insulin-degrading enzyme. Mol. Neurodegener. 2009, 4, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Fagan, A.M.; Roe, C.M.; Xiong, C.; Mintun, M.A.; Morris, J.C.; Holtzman, D.M. Cerebrospinal fluid tau/beta-amyloid (42) ratio as a prediction of cognitive decline in nondemented older adults. Arch. Neurol. 2007, 64, 343–349. [Google Scholar] [CrossRef]
- Ingber, A.P.; Hassenstab, J.; Fagan, A.M.; Benzinger, T.L.; Grant, E.A.; Holtzman, D.M.; Morris, J.C.; Roe, C.M. Cerebrospinal fluid biomarkers and reserve variables as predictors of future “non-cognitive” outcomes of Alzheimer’s disease. J. Alzheimer’s Dis. 2016, 52, 1055–1164. [Google Scholar] [CrossRef] [Green Version]
- Zetterberg, H. Cerebrospinal fluid biomarkers for Alzheimer’s disease: Current limitations and recent developments. Curr. Opin. Psychiatry 2015, 28, 402–409. [Google Scholar] [CrossRef]
- Donald, P.M.; George, R.; Sriram, G.; Kavitha, B.; Sivapathasundharam, B. Hormonal changes in exfoliated normal buccal mucosal cells. J. Cytol. 2013, 30, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.M.; Cruchley, A.T. Structural aspects of ageing in the oral mucosa. In The Effect of Ageing in the Oral Mucosa and Skin; Squier, C.A., Hill, M.W., Eds.; CRC Press: London, UK; Bocca Raton, FL, USA, 1994; pp. 65–74. [Google Scholar]
- Thomas, P.; Hecker, J.; Faunt, J.; Fenech, M. Buccal micronucleus cytome biomarkers may be associated with Alzheimer’s disease. Mutagenesis 2007, 22, 371–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, Y.K.; Tsai, M.H.; Huang, D.C.; Zheng, Z.H.; Hung, K.D. Leukocyte nucleus segmentation and nucleus lobe counting. BMC Bioinform. 2010, 11, 558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, R.M.; Lia, E.N.; Guimaraes, R.M.; Bocca, A.L.; Cavalcante Neto, F.F.; da Silva, T.A. Cytologic and cytometric analysis of oral mucosa in Alzheimer’s disease. Anal. Quant. Cytol. Histol. 2008, 30, 113–118. [Google Scholar] [PubMed]
- Thomas, P.; Harvey, S.; Gruner, T.; Fenech, M. The buccal cytome and micronucleus frequency is substantially altered in Down’s syndrome and normal ageing compared to young healthy controls. Mutat. Res. 2008, 638, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Hozak, P.; Sasseville, A.M.; Raymond, Y.; Cook, P.R. Lamin proteins form an internal nucleoskeleton as well as a peripheral lamina in human cells. J. Cell Sci. 1995, 108, 635–644. [Google Scholar]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef] [Green Version]
- Gonzalo, S. DNA damage and lamins. Adv. Exp. Med. Biol. 2014, 773, 377–399. [Google Scholar]
- Gonzalez-Suarez, I.; Redwood, A.B.; Perkins, S.M.; Vermolen, B.; Lichtensztejin, D.; Grotsky, D.A.; Morgado-Palacin, L.; Gapud, E.J.; Sleckman, B.P.; Sullivan, T.; et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J. 2009, 28, 2414–2427. [Google Scholar] [CrossRef]
- Smogorzewska, A.; Karlseder, J.; Holtgreve-Grez, H.; Jauch, A.; de Lange, T. DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr. Biol. 2002, 12, 1635–1644. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikora, E.; Arendt, T.; Bennett, M.; Narita, M. Impact of cellular senescence signature on ageing research. Ageing Res. Rev. 2011, 10, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Berkenkamp, B.; Susnik, N.; Baisantry, A.; Kuznetsova, I.; Jacobi, C.; Sörensen-Zender, I.; Broecker, V.; Haller, H.; Melk, A.; Schmitt, R. In vivo and in vitro analysis of age-associated changes and somatic cellular senescence in renal epithelial cells. PLoS ONE 2014, 9, e88071. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Jurk, D.; Maddick, M.; Nelson, G.; Martin-Ruiz, C.; von Zglinicki, T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009, 8, 311–323. [Google Scholar] [CrossRef]
- Zhao, H.; Darzynkiewicz, Z. Biomarkers of cell senescence assessed by imaging cytometry. Methods Mol. Biol. 2013, 965, 83–92. [Google Scholar]
- Zhao, H.; Halicka, H.D.; Traganos, F.; Jorgensen, E.; Darzynkiewicz, Z. New biomarkers probing depth of cell senescence assessed by laser scanning cytometry. Cytom. Part A 2010, 77, 999–1007. [Google Scholar] [CrossRef]
- Faux, N.G.; Rembach, A.; Wiley, J.; Ellis, K.A.; Ames, D.; Fowler, C.J.; Martins, R.N.; Pertile, K.K.; Rumble, R.L.; Trounson, B.; et al. An anemia of Alzheimer’s disease. Mol. Psychiatry 2014, 19, 1227–1234. [Google Scholar] [CrossRef] [Green Version]
- Fischer, P.; Gotz, M.E.; Danielczyk, W.; Gsell, W.; Riederer, P. Blood transferrin and ferritin in Alzheimer’s disease. Life Sci. 1997, 60, 2273–2278. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control n = 18 | MCI n = 17 | AD n = 16 | |
|---|---|---|---|
| Sex (M:F) | 12:6 | 11:6 | 9:7 |
| Age (years) | 72.2 ± 1.5 | 78.7 ± 1.9 | 81.0 ± 1.8 ** |
| BMI | 27.0 ± 1.3 | 23.4 ± 1.3 | 24.8 ± 1.1 |
| MMSE score | 29.1 ± 0.2 | 26.0 ± 0.8 * | 12.8 ± 1.8 *** |
| Parameters | Correlation (r) | CI | p-Value | |
|---|---|---|---|---|
| All nuclei | γH2AX integral | −0.1899 | −0.4014–0.0408 | 0.0959 |
| γH2AX MaxPixel | −0.2266 | −0.4331–0.0024 | 0.0460 | |
| Round | γH2AX integral | −0.3535 | −0.5816 to −0.0737 | 0.0148 |
| γH2AX MaxPixel | −0.4550 | −0.6565 to −0.1930 | 0.0013 | |
| Long | γH2AX integral | −0.3039 | −0.5437 to −0.0183 | 0.0378 |
| γH2AX MaxPixel | −0.4141 | −0.6268 to −0.1440 | 0.0038 | |
| Oval | γH2AX integral | −0.3534 | −0.5816 to −0.0736 | 0.0148 |
| γH2AX MaxPixel | −0.4678 | −0.6656 to −0.2086 | 0.0009 | |
| Senescent | γH2AX integral | −0.5229 | −0.7044 to −0.2773 | 0.0002 |
| γH2AX MaxPixel | −0.5156 | −0.6993 to −0.2680 | 0.0002 |
| Parameters | Correlation (r) | 95% Confidence Interval | p-Value |
|---|---|---|---|
| Homocysteine | 0.0092 | −0.1537–0.4472 | 0.9541 |
| Serum folate | 0.1617 | −0.377–0.198 | 0.3125 |
| Vitamin B12 | −0.1295 | −0.4205–0.1856 | 0.4195 |
| Red cell folate | 0.0005 | −0.3151–0.3161 | 0.9975 |
| Calcium | 0.0422 | −0.2770–0.3531 | 0.7985 |
| Cholesterol | −0.0270 | −0.1924–0.4290 | 0.4261 |
| Triglycerides | −0.118 | −0.3397–0.2911 | 0.8704 |
| HDL | −0.1846 | −0.4726–0.1391 | 0.2606 |
| LDL | 0.2371 | −0.08484–0.5142 | 0.1461 |
| Albumin | 0.0305 | −0.2879–0.3428 | 0.8539 |
| Bilirubin | −0.2013 | −0.4860–0.1220 | 0.2191 |
| Urea | −0.0181 | −0.3318–0.2992 | 0.9131 |
| Creatinine | 0.0134 | −0.3035–0.3276 | 0.9354 |
| eGFR | 0.0427 | −0.2766–0.3535 | 0.7964 |
| Glucose | −0.2302 | −0.5088–0.09207 | 0.1586 |
| Total protein | 0.332 | 0.01837–0.5862 | 0.0389 |
| ALT | 0.0088 | −0.3077–0.3234 | 0.9579 |
| AP | 0.0101 | −0.3065–0.3247 | 0.9514 |
| GGT | 0.0708 | −0.2504–0.3779 | 0.6684 |
| Ceruloplasmin | −0.2476 | −0.5224–0.07374 | 0.1286 |
| Fe | −0.2834 | −0.5498–0.03533 | 0.0804 |
| Transferrin | 0.170 | −0.1539–0.4608 | 0.3009 |
| Trsat | −0.2688 | −0.5387–0.05111 | 0.0980 |
| Ferritin | −0.0201 | −0.3336–0.2973 | 0.9031 |
| Insulin | −0.1066 | −0.4084–0.2163 | 0.5185 |
| Testosterone | 0.1546 | −0.1692–0.4483 | 0.3472 |
| LH | 0.0245 | −0.2933–0.3375 | 0.8822 |
| FT4 | 0.1808 | −0.1429–0.4696 | 0.2707 |
| TSH | 0.1425 | −0.1812–0.4384 | 0.3868 |
| FT3 | 0.1999 | −0.1234–0.4849 | 0.2223 |
| Cl | 0.04746 | −0.2722–0.3577 | 0.7742 |
| AST | −0.1123 | −0.4132–0.2108 | 0.4961 |
| PCV | −0.0888 | −0.3933–0.2334 | 0.5911 |
| Mg | 0.1919 | −0.1317–0.4785 | 0.2418 |
| RCC | −0.0009 | −0.3165–0.3147 | 0.9952 |
| MCV | −0.226 | −0.5055–0.09647 | 0.1665 |
| MCH | −0.2427 | −0.5185–0.07897 | 0.1366 |
| MCHC | −0.1327 | −0.4303–0.1909 | 0.4206 |
| RDW | −0.208 | −0.4913–0.1152 | 0.2039 |
| ESR | −0.1164 | −0.4167–0.2068 | 0.4803 |
| Platelets | −0.05805 | −0.3669–0.2623 | 0.7255 |
| MPV | −0.1251 | −0.4239–0.1983 | 0.4481 |
| WCC | −0.2584 | −0.5307–0.06222 | 0.1122 |
| Neutrophils | −0.2226 | −0.5028–0.1001 | 0.1733 |
| Lymphocytes | −0.1001 | −0.4030–0.2225 | 0.5442 |
| Monocytes | −0.2631 | −0.5343–0.05722 | 0.1056 |
| Eosinophils | −0.1277 | −0.4261–0.1958 | 0.4386 |
| Basophils | −0.2012 | −0.4859–0.1222 | 0.2194 |
| Control | MCI | AD | |
|---|---|---|---|
| Homocysteine | r = −0.070, p = 0.804 | r = 0.514, p = 0.106 | r = −0.175, p = 0.518 |
| Serum folate | r = 0.193, p = 0.491 | r = 0.256, p = 0.448 | r = 0.134, p = 0.635 |
| Vitamin B12 | r = −0.041, p = 0.883 | r = −0.293, p = 0.382 | r = −0.243, p = 0.383 |
| Red cell folate | r = 0.288, p = 0.299 | r = 0.003, p = 0.993 | r = −0.149, p = 0.595 |
| Calcium | r = −0.041, p = 0.884 | r = −0.433, p = 0.244 | r = 0.065, p = 0.817 |
| Cholesterol | r = 0.467, p = 0.079 | r = −0.279, p = 0.467 | r = −0.072, p = 0.799 |
| Triglycerides | r = 0.114, p = 0.685 | r = −0.516, p = 0.155 | r = −0.033, p = 0.906 |
| HDL | r = 0.194, p = 0.489 | r = −0.266, p = 0.488 | r = −0.292, p = 0.292 |
| LDL | r = 0.465, p = 0.080 | r = −0.016, p = 0.968 | r = 0.292, p = 0.802 |
| Albumin | r = 0.209, p = 0.454 | r= 0.724, p = 0.027 | r = −0.018, p = 0.951 |
| Bilirubin | r = −0.286, p = 0.300 | r = −0.173, p = 0.656 | r = −0.187, p = 0.504 |
| Urea | r = 0.500, p = 0.058 | r = −0.181, p = 0.640 | r = −0.326, p = 0.236 |
| Creatinine | r = −0.276, p = 0.320 | r = 0.407, p = 0.277 | r = −0.038, p = 0.893 |
| eGFR | r = 0.186, p = 0.508 | r = −0.259, p = 0.502 | r = 0.092, p = 0.745 |
| Glucose | r = −0.457, p = 0.087 | r = 0.112, p = 0.775 | r = −0.175, p = 0.534 |
| Total protein | r= 0.557, p = 0.031 | r = 0.127, p = 0.745 | r = 0.133, p = 0.636 |
| ALT | r = −0.224, p = 0.421 | r = 0.109, p = 0.779 | r = −0.035, p = 0.901 |
| AP | r = −0.189, p = 0.498 | r= 0.681, p = 0.043 | r = −0.046, p = 0.870 |
| GGT | r = −0.108, p = 0.700 | r = −0.087, p = 0.824 | r = 0.025, p = 0.931 |
| Ceruloplasmin | r = −0.133, p = 0.638 | r = −0.149, p = 0.703 | r = −0.294, p = 0.287 |
| Fe | r = −0.298, p = 0.280 | r = −0.385, p = 0.306 | r = −0.309, p = 0.261 |
| Transferrin | r= 0.628, p = 0.012 | r = −0.225, p = 0.560 | r = −0.034, p = 0.904 |
| Trsat | r = −0.344, p = 0.209 | r = −0.294, p = 0.442 | r = −0.282, p = 0.308 |
| Ferritin | r = −0.252, p = 0.366 | r = 0.025, p = 0.949 | r = −0.100, p = 0.721 |
| Insulin | r = −0.162, p = 0.565 | r = 0.013, p = 0.975 | r = 0.280, p = 0.310 |
| Testosterone | r = −0.162, p = 0.565 | r= 0.684, p = 0.042 | r = 0.175, p = 0.532 |
| LH | r= 0.522, p = 0.046 | r = −0.235, p = 0.542 | r = −0.177, p = 0.527 |
| FT4 | r= 0.648, p = 0.009 | r = −0.078, p = 0.842 | r = 0.155, p = 0.582 |
| TSH | r = 0.228, p = 0.411 | r = 0.056, p = 0.887 | r = 0.146, p = 0.603 |
| FT3 | r = 0.431, p = 0.109 | r = −0.014, p = 0.972 | r = 0.115, p = 0.684 |
| Cl | r = −0.173, p = 0.650 | r = −0.269, p = 0.485 | r = 0.173, p = 0.538 |
| AST | r = −0.173, p = 0.536 | r = 0.032, p = 0.935 | r = −0.185, p = 0.508 |
| PCV | r = −0.267, p = 0.335 | r = 0.074, p = 0.850 | r = −0.061, p = 0.829 |
| Mg | r = −0.016, p = 0.954 | r = 0.263, p = 0.495 | r = 0.255, p = 0.359 |
| RCC | r = −0.081, p = 0.773 | r = 0.279, p = 0.467 | r = −0.071, p = 0.799 |
| MCV | r = −0.425, p = 0.115 | r= −0.678, p = 0.045 | r = −0.045, p = 0.871 |
| MCH | r= −0.658, p = 0.008 | r = −0.657, p = 0.055 | r = 0.054, p = 0.848 |
| MCHC | r= −0.689, p = 0.005 | r = −0.479, p = 0.193 | r = 0.307, p = 0.265 |
| RDW | r = −0.197, p = 0.481 | r = 0.213, p = 0.582 | r = −0.378, p = 0.165 |
| ESR | r = −0.157, p = 0.577 | r = −0.209, p = 0.589 | r = −0.186, p = 0.507 |
| Platelets | r = 0.049, p = 0.861 | r = 0.265, p = 0.490 | r = −0.158, p = 0.576 |
| MPV | r = 0.057, p = 0.844 | r = −0.143, p = 0.713 | r = −0.438, p = 0.103 |
| WCC | r = −0.163, p = 0.563 | r = 0.369, p = 0.327 | r = −0.473, p = 0.075 |
| Neutrophils | r = −0.292, p = 0.291 | r = 0.588, p = 0.096 | r = −0.496, p = 0.059 |
| Lymphocytes | r = 0.412, p = 0.127 | r = −0.356, p = 0.347 | r = −0.206, p = 0.460 |
| Monocytes | r = −0.420, p = 0.119 | r = 0.091, p = 0.815 | r = −0.335, p = 0.223 |
| Eosinophils | r = 0.015, p = 0.958 | r = −0.517, p = 0.154 | r = −0.218, p = 0.435 |
| Basophils | r = −0.171, p = 0.542 | r = 0.408, p = 0.275 | r = −0.331, p = 0.226 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siddiqui, M.S.; Francois, M.; Rainey-Smith, S.; Martins, R.; Masters, C.L.; Ames, D.; Rowe, C.C.; Macaulay, L.S.; Fenech, M.F.; Leifert, W.R. Evaluation of GammaH2AX in Buccal Cells as a Molecular Biomarker of DNA Damage in Alzheimer’s Disease in the AIBL Study of Ageing. Life 2020, 10, 141. https://doi.org/10.3390/life10080141
Siddiqui MS, Francois M, Rainey-Smith S, Martins R, Masters CL, Ames D, Rowe CC, Macaulay LS, Fenech MF, Leifert WR. Evaluation of GammaH2AX in Buccal Cells as a Molecular Biomarker of DNA Damage in Alzheimer’s Disease in the AIBL Study of Ageing. Life. 2020; 10(8):141. https://doi.org/10.3390/life10080141
Chicago/Turabian StyleSiddiqui, Mohammad Sabbir, Maxime Francois, Stephanie Rainey-Smith, Ralph Martins, Colin L. Masters, David Ames, Christopher C. Rowe, Lance S. Macaulay, Michael F. Fenech, and Wayne R. Leifert. 2020. "Evaluation of GammaH2AX in Buccal Cells as a Molecular Biomarker of DNA Damage in Alzheimer’s Disease in the AIBL Study of Ageing" Life 10, no. 8: 141. https://doi.org/10.3390/life10080141