An Overview of Genomics, Phylogenomics and Proteomics Approaches in Ascomycota




Abstract
:1. Introduction
1.1. Towards a Genome-Based Fungal Systematics
1.2. Proteomics Advances in Mycology
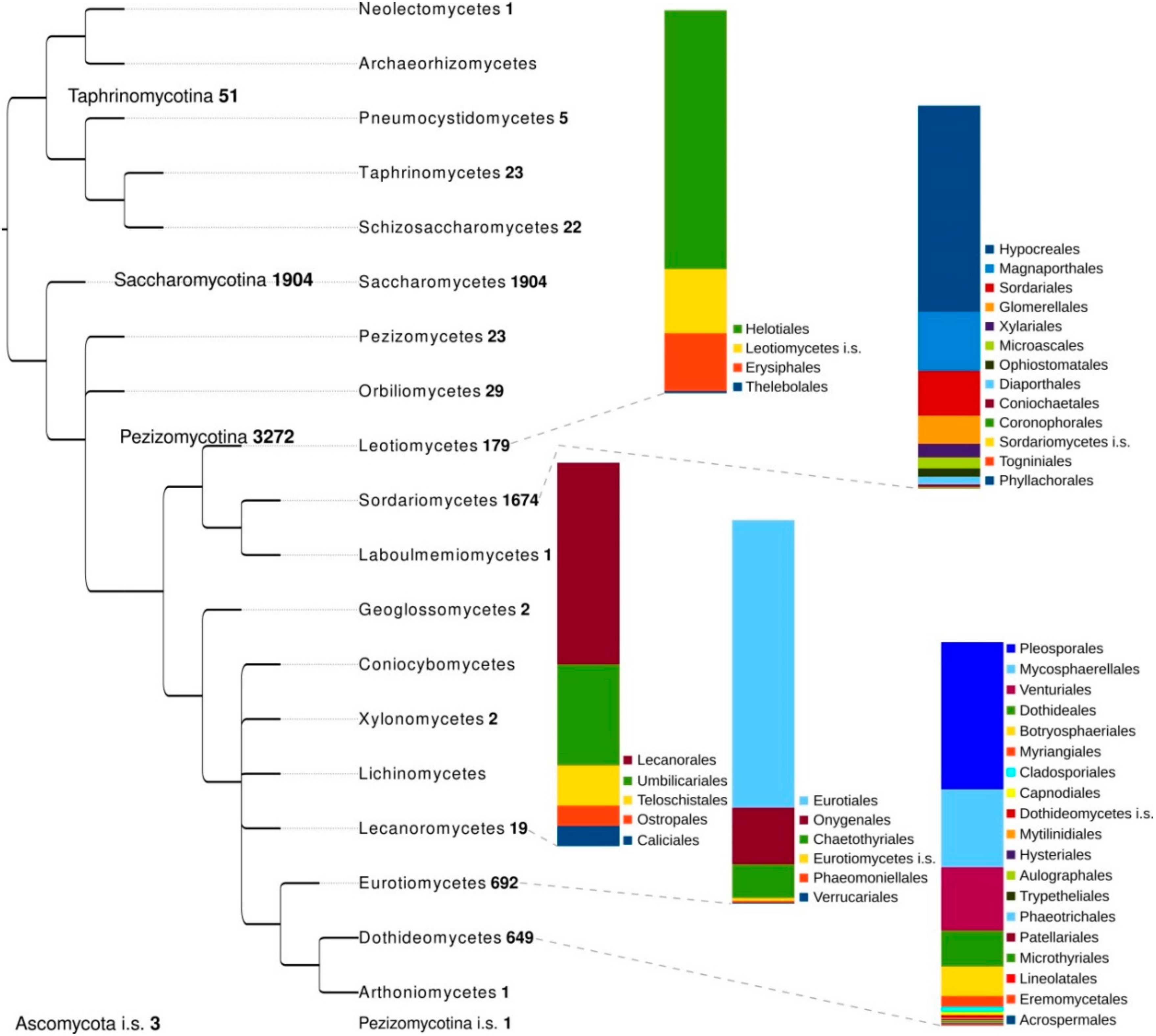
2. Genomic Advances in Ascomycota
2.1. Opportunistic and Pathogenic Fungi
2.2. Plant Pathogens
2.3. Entomopathogenic Fungi
2.4. Extremophilic/Polyextremotolerant Fungi
2.5. Lichenized Fungi
2.6. Phylogenomics and Population Genomics
3. Progress and Applications of Sequencing Technologies to Fungal Genomes
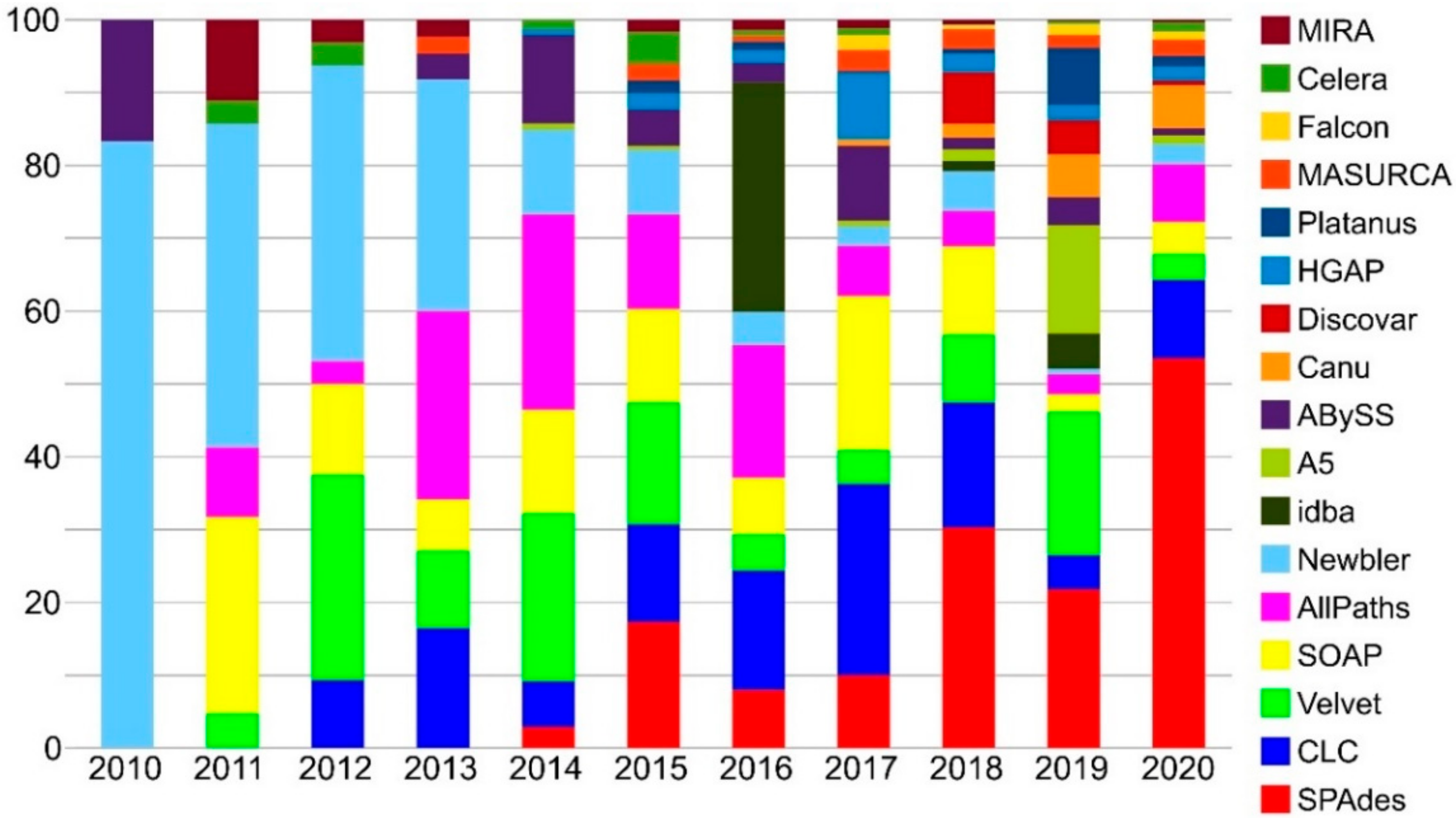
3.1. Genome Sequencing
3.2. Genome Assembly
3.3. Metagenome Assembly
3.4. Genome Annotation
3.5. Methods for Phylogenomic Analyses
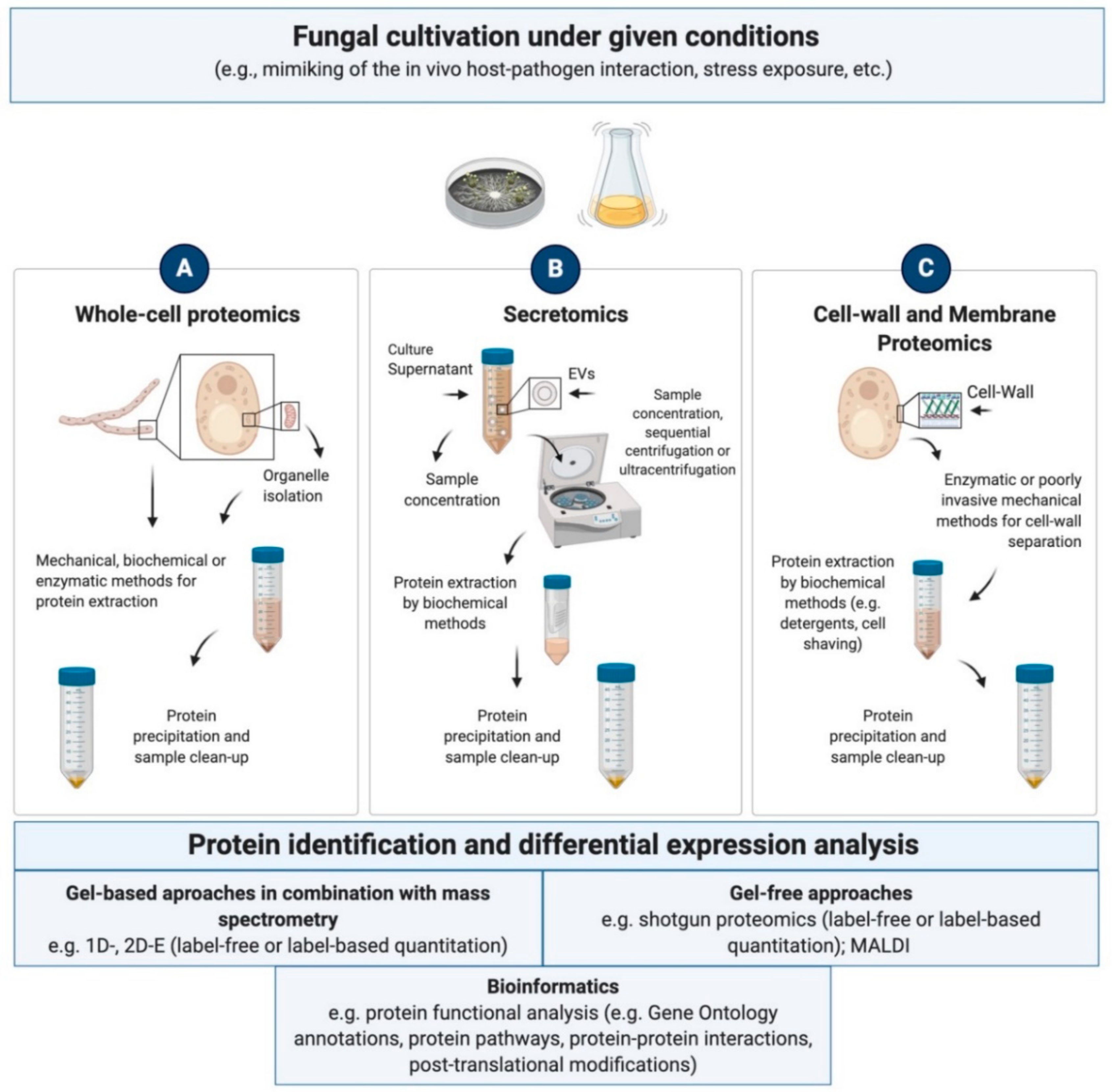
4. Proteomics Advances in Ascomycota
4.1. Opportunistic and Pathogenic Fungi
4.1.1. Whole-Cell and Subcellular Proteomics
4.1.2. Secretomics
4.1.3. Cell Wall and Membrane Proteomics
4.2. Proteomics Advances in Extremophilic and Extremotolerant Fungi
4.2.1. Whole Cell and Subcellular Proteomics
4.2.2. Secretomics
4.2.3. Membrane and Cell Wall Proteomics
4.3. Proteomics in Lichens
4.4. Use of Proteomics for Species Identification
4.5. Bioinformatics Tools for Fungal Protein Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lichen Species | Lichen Material | Applied Approach(s) | Remarks | References |
|---|---|---|---|---|
| Cetradonia linearis | whole thallus, nuclear genome | whole-genome shotgun Illumina sequencing; SNP calling | sequencing of reference genome and population genomic analysis | [116] |
| Cladonia grayi | cultured mycobiont, nuclear genome | Illumina genome sequencing; transcriptome sequencing; analyses of gene families; | identification of genes/proteins of potential symbiotic relevance | [123] |
| Arthonia radiata | cultured mycobiont, nuclear genome | Illumina genome and transcriptome sequencing; assamply with overlapping paired-end (PE) and mate pair (MP) libraries | sequencing and assembly of genome and transcriptom | [409] |
| C. apodocarpa, C. caroliniana, C. furcata, C. leporina, C. petrophila, C. peziziformis, C. robbinsii, C. stipitata, and C. subtenuis | whole thallus, mitochondrial genome | Illumina sequencing; phylogenetic analysis (Bayesian MCMC) | analysing genome size, protein coding gene content, intron-encoded retrotransposable elements, Bayesian analyses to assess efficacy of five loci | [125] |
| Lasallia hispanica | cultured mycobiont, nuclear genome | Illumina sequencing | genome asembly and annotaion, comparison with the closest related species | [131] |
| Usnea halei, U. mutabilis, U. subfusca, U. subgracilis, and U. subscabrosa | whole thallus, mitochondrial genome | Illumina sequencing | genome asembly and annotaion, phylogenetic infeence with the closest related species | [127] |
| Lasallia pustulata | whole thallus, metagenome | whole-genome shotgun (metagenome skimming), Illumina sequencing | testing the best genome ssembling method | [131,140] |
| Usnea antarctica, U. aurantiacoatra | whole thallus, metagenome | RADseq | population genomics and species distinction | [136] |
| Rhizoplaca haydenii, R. idahoensis, R. melanophthalma, R. novomexicana, R. occulta, R. parilis, R. polymorpha, R. porteri, R. shushanii | whole thallus, metagenome | RADseq | phylogenomics | [135] |
| Cladonia rangiferina | cultured mycobiont, nuclear genome | Sanger EST sequencing data | characterization of transcriptome | [124] |
| 273 lichen species | whole thallus, nuclear genome | whole-genome shotgun Illumina sequencing | comparison of WGS vs. amplicon sequencing efficiency; species identification for ecological pourposes | [139] |
| Rhizoplaca melanophthalma (species complex) | whole thallus and cultured mycobiont, nuclear and mitochondrial genomes | genome Illumina sequencing | phylogenomic; to infer evolutionary relationships and potential patterns of introgression | [133] |
| Rhizoplaca melanophthalma (species complex) | whole thallus and cultured mycobiont, nuclear genomes | genome Illumina sequencing | phylogenomic | [134] |
| Alectoria sarmentosa | whole thallus, metagenome | genome Illumina sequencing | sequencing of draft genome | [141] |
| Acarospora strigata, Arthonia rubrocinta, Dibaies baeomyces, Endocarpon pallidum, Graphis scripta, Leptogium austroamericanum, Peltula cylindrica, Physcia cf. stellaris | whole thallus and cultured mycobiont, nuclear genomes | whole-genome shotgun, Illumina sequencing | to study the evolution of the ammonium transporter/ammonia permease gene family | [118] |
| Evernia prunastri, Pseudevernia furfuracea | whole thallus and cultured mycobiont, nuclear genomes | whole-genome shotgun, metagenome skimming, Illumina sequencing | to test accuracy and completeness of assemblies based on metagenomic sequences and comparison with assemblies based on pure cultured strains | [115] |
| Caloplaca flavorubescens | cultured mycobiont, nuclear genome | whole-genome shotgun, Illumina sequencing | sequencing of draft genome | [119] |
| Cladonia macilenta | cultured mycobiont, nuclear genome | whole-genome shotgun, Illumina sequencing | sequencing of draft genome | [120] |
| Umbilicaria muehlenbergii | cultured mycobiont, nuclear genome | whole-genome shotgun, Illumina sequencing | sequencing of draft genome | [121] |
| Cladonia metacorallifera | cultured mycobiont, nuclear genome | whole-genome shotgun, Illumina sequencing | sequencing of draft genome | [122] |
| 41 lichen species | whole thallus, nuclear genome | whole-genome shotgun Illumina sequencing | to characterize the MAT locus for heterothallism or homothallism | [132] |
| 58 lichen species | whole thallus, mitochondrial genome | whole-genome shotgun Illumina sequencing | to analyze mitochondrial genome evolutionby loss of introns | [142] |
| Ramalina intermedia | whole thallus, nuclear genome | whole-genome shotgun Illumina sequencing | draft genome sequence | [128] |
| Endocarpon pusillum | cultured mycobiont, nuclear genome | whole-genome shotgun Illumina sequencing | genome sequencing and characterization for symbiotic traits | [130] |
| Physcia stellaris | cultured mycobiont, nuclear genome | SpotON R9.4.1 FLO-MIN106 flowcell and Illumina sequencing | draft genome sequence | [129] |
| Peltigera malacea, P. membranacea | whole thallus, mitochondrial genome | Illumina sequencing | comparative analysis and phylogenetic infeence | [126] |
Appendix B
| Fungus | Fungal Material | Experimental Workflow | Number of Identified Proteins | Remarks | References |
|---|---|---|---|---|---|
| Aspergillus flavus | Conidia, whole cell proteome | LC-MS-QToF | Not indicated | Identification of proteins expressed under simulated deep-sea condition | [331] |
| Aspergillus fumigatus | Mycelium, whole cell proteome | iTRAQ LC-MS/MS | 471 | Quantitative proteomics of the fungus response to caspofungin to determine potential biomarkers of drug action | [261] |
| Conidia | SWATH-MS | 712 | Time course evaluation of the conidial proteome | [265] | |
| Secretome | LC-MS/MS | 484 | Secretome profiling | [302] | |
| Cell wall proteome | UHPLC-MSE | 367 | Detection of protein targets for broad-spectrum mycosis vaccines | [325] | |
| LC-MS/MS | 148 | Description of conidial surface proteins | [323] | ||
| Conidia, mycelium, whole cell proteome | TMT-LC-MS/MS | 75 | Proteomic under simulated Mars conditions | [352] | |
| Aspergillus niger | Mycelium, whole cell proteome | TMT-LC-MS/MS | 327 | Protein characterization of an isolate from the International Space Station | [347] |
| iTRAQ LC-MS/MS | 1025 | Characterization of the proteome under heat stress | [277] | ||
| Aspergillus terreus spp. | Conidia, whole cell proteome | LC-MS-QToF | Not indicated | Identification of proteins expressed under simulated deep-sea condition | [331] |
| Aspergillus sydowii | Conidia, whole cell proteome | LC-MS-QToF | Not indicated | Identification of proteins expressed under simulated deep-sea condition | [331] |
| Aspergillus versicolor | Conidia | 2D-E, IgE-immunoblotting | 20 | Identification of allergens detected in sera from patients participating in a study about indoor exposure to molds | [262] |
| Beauveria bassiana | Secretome | Label-free nano-LC-MS/MS | 50 | Secretome screening under two diet regimes | [299] |
| Botrytis cinerea | Mycelium, whole cell proteome | iTRAQ LC-MS/MS | 3816 | Differential expression study of proteins following fungal treatment with the broad-spectrum agricultural antibiotic wuyiencin | [263] |
| MALDI, LC-ESI-MS/MS | 1431 | Identification of target proteins and pathways of the antifungal peptide ETD151, homologous to insect defensin | [264] | ||
| Secretome | 2D-E, LC-MS/MS | 56, 105 | Description of the fungus secretome | [292] | |
| Nano-LC-MS/MS | 1719 | Comparative secretome analysis of wild type and non-virulent mutants | [293] | ||
| Candida albicans | Yeast cells, whole cell proteome | 2D-E, MALDI-TOF/TOF | 51 | Identification of proteins regulated by fungal exposure to oleic acid from the curry leaf tree Murraya koenigii | [257] |
| Label-free LC-MS/MS | 1262 | Quantitative proteomics of the response to osmotic stress. | [279] | ||
| Chlamydospores | SWATH-MS | 1177 | Description of chlamydospores proteome | [280] | |
| Cell wall proteome | MALDI-TOF/TOF | 46 | Identification of surface exposed proteins. | [218] | |
| 131 | Analysis of surface exposed proteins under different growth conditions. | [315] | |||
| 50 | Proteomic analysis of a pga1 null strain | [322] | |||
| Candida glabrata | Secretome | LC-MS/MS | 119; 548 | Secretome characterization of wild type and Cgyps1-11Δ mutant strain. | [301] |
| Cladonia stellaris | Thallus proteome | IEF | n.a. | Description of enzyme polymorphism | [375] |
| Cladonia portentosa | Thallus proteome | 2D-E, MALDI-TOF, MALDI-TOF/TOF | 45 | Proteomic analysis of nitrogen stress effects | [382] |
| Cladosporium cladosporioides | Conidia, mycelium, whole cell proteome | TMT-LC-MS/MS | 269 | Proteomic under simulated Mars conditions | [352] |
| Coccidioides posadasii | Cell wall proteome | UHPLC-MSE | 314 | Detection of protein targets for broad-spectrum mycosis vaccines | [325] |
| Cryomices antarcticus | Whole cell proteome | 2D-E | n.a. | Characterization of protein patterns under simulated Mars-like conditions | [340] |
| Characterization of protein patterns under drought | [339] | ||||
| Debaryomyces hansenii | Whole cell proteome | 2D-E, MALDI-TOF/TOF | 43 | Proteomic changes under potassium starvation | [343] |
| Doratomyces stemonitis | Secretome | MALDI-TOF/TOF, Q-TOF LC-MS/MS | 44 | Secretome profiling | [364] |
| Evernia prunastri | Thallus proteome | 2D-E, LC-ESI-MS/MS | 5 | Changes of the lichen proteome during exposure to constant concentrations of mercury | [378] |
| Exophiala dermatitidis | Mycelium, whole cell proteome | 2D-DIGE, LC-ESI-MS/MS | 32 | Analysis of protein differential expression in response to temperature stress | [276] |
| Exophiala jeanselmei | Whole cell proteome | 2D-E | n.a | Characterization of protein patterns in response to sub-optimal temperature | [338] |
| Characterization of protein patterns under simulated Mars-like conditions | [340] | ||||
| Characterization of protein patterns under drought | [339] | ||||
| Fusarium oxysporum | Secretome | Label-free LC-MS/MS | 919 | Detection of effector candidates regulated under simulated in-planta status | [296] |
| Fusarium proliferatum | Secretome | 2D-E, MALDI-TOF MS | 39 | Investigation of pH-dependent changes in the fungus secretome | [295] |
| Friedmanniomyces endolithicus | Whole cell proteome | 2D-E | n.a | Characterization of protein patterns in response to sub-optimal temperature | [338] |
| Histoplasma capsulatum | Extracellular vesicles | LC-MS/MS | 206 | Proteomic analysis of extracellular vesicles | [306] |
| 2000 | Analysis of alterations in the extracellular vesicles proteome in response to different nutritional milieus | [309] | |||
| Knufia chersonesos | Secretome | LC-MS/MS | 1730 | Secretome screening for polyester degrading enzymes under exposure to PBAT. | [366] |
| Knufia perforans | Whole cell proteome | 2D-E | n.a. | Characterization of protein patterns in response to sub-optimal temperature | [338] |
| Characterization of protein patterns under simulated Mars-like conditions | [240] | ||||
| Characterization of protein patterns under drought | [339] | ||||
| Lobaria pulmonaria | Proteome of the symbiotic consortium | 1D-E, LC-ESI-MS/MS | 463 | Profiling of the metaproteome | [380] |
| 4405 | Comparative omics to explore the lichen-associated microbiome | [380] | |||
| 1D-E, GeLC-MS/MS | 6590 | Profiling of the metaproteome | [381] | ||
| Magnaporthe oryzae | Mycelium and conidia, whole cell proteome | Label-free LC-MS/MS | 355 and 559 N-glycosylation sites | Quantitative analysis of N-glycosylation regulation and elucidation of its role in development and pathogenesis of M. oryzae | [259] |
| LC-MS/MS | 5498 | Comparative proteomic analysis between nitrogen supplemented and starved conditions | [278] | ||
| Malbranchea cinnamomea | Secretome | LC-MS/MS | 53 | Secretome screening for lignocellulolytic enzymes | [361] |
| Metarhizium anisopliae | Secretome | MudPIT-MS/MS | 48 | Analysis of the secretome induced by insect cuticle | [300] |
| Metarhizium robertsii | Mycelium and conidia, whole cell proteome | iTRAQ | 2052 | Comparative quantitative analysis of conidia and mycelium proteome | [266] |
| Mycothermus thermophilus | Secretome | Q-TOF LC/MS | 240 | Secretome screening for cellulases and hemicellulases | [362] |
| Paracoccidioides sp. | Yeast cells, whole cell proteome | 2D-E, MALDI-MS/MS | 135 | Comparative 2D-E to analyse protein differential expression in response to zinc deprivation. | [270] |
| 2D-E, MALDI-MS/MS | 179 | Characterization of the oxidative stress response following short- and long-term H2O2-treatment. | [271] | ||
| NanoUPLC-MSE | 421 | Investigation of the proteome response to carbon starvation. | [273] | ||
| NanoUPLC-MSE | 142 | Investigation of the proteome response to nitrosative starvation. | [274] | ||
| Paracoccidioides brasiliensis | Yeast cells, whole cell proteome | NanoUPLC-MSE | 538 | Analysis of the proteome response of P. brasiliensis Pb18following interaction with alveolar macrophages primed with interferon gamma. | [255] |
| 2D-E, LC-MS/MS | 11 | Identification of proteins commonly overexpressed in highly virulent P. brasiliensis spp. complex isolates causing disseminated disease in a murine model of PCM | [256] | ||
| Nano-ESI-UPLC-MSE | 308 | Characterization of the proteomic response to macrophage internalization. | [410] | ||
| Secretome | LC-MS/MS | 205; 260 | Vesicle and vesicle-free extracellular proteome. | [283] | |
| Paracoccidioides lutzii | Yeast cells, whole cell proteome | UPLC-MSE | 303 | Analysis of proteins differential expression during osmotic shock. | [272] |
| Cell wall proteome | NanoUPLC-MSE | 512 | Comparative analysis of yeast and mycelium cell wall proteome | [320] | |
| Penicillium sp. | Conidia, whole cell proteome | LC-MS-QToF | Not indicated | Identification of proteins expressed under simulated deep-sea condition | [331] |
| Penicillium oxalicum | Membrane and subcellular proteome | 2D-E, MALDI-TOF/TOF | 20 | Screening of membrane and cytosolic proteome in response to the polycyclic aromatic hydrocarbon (PAH) anthracene | [369] |
| Pestalotiopsis sp. | Secretome | LC-MS/MS | 209 | Characterization of saline and non-saline secretomes | [360] |
| Physcia adscendens | Thallus proteome | 2D-E, MALDI-TOF-MS | 16 | Proteomic analysis in response to cadmium stress | [372] |
| Pyrenophora tere | Secretome | LC-MS/MS | 182 | Secretome profiling | [297] |
| Pyricularia oryzae | Mycelium, whole cell proteome | LC−MS/MS combined with a high-efficiency succinyl-lysine antibody | 714 and 2109 lysine succinylation sites | Succinyl-proteome profiling of Pyricularia oryzae and role of succinylation in general metabolism and infection | [260] |
| Sclerotinia sclerotiorum | Mycelium, whole cell proteome | 1D-E, LC-MS/MS | 1471 | Proteomic profiling of S. sclerotiorum total proteome | [269] |
| Sporotrichum thermophile | Whole cell proteome | 2D-E, LC-ESI-MS/MS | 15 | Proteomic profiling under the effect of ionic liquids | [346] |
| Sporothrix brasiliensis | Extracellular vesicles | 2D-E, LC-MS/MS | 63 | Detection of EVs associated immunogenic components and virulence factors | [310] |
| Sporothrix schenckii | Extracellular vesicles | 2D-E, LC-MS/MS | 40 | Detection of EVs associated immunogenic components and virulence factors | [310] |
| Cell wall proteome | 1D-E, LC-ESI-HDMSE | 479 | Proteomic analysis of the cell wall in response to oxidative stress | [321] | |
| Talaromyces marneffei | Mycelium and conidia, whole cell proteome | 2D-DIGE, MALDI-TOF MS | 26 | Differentially expressed proteins in yeast and mycelial phases | [267] |
| Mycelium and conidia, secretome | 2D-E, MALDI-TOF MS | 12 | Proteome profiling of the extracellular proteome | [268] | |
| Thermomyces lanuginosus | Secretome | LC-MS/MS | 74 | Characterization of secretome during fungal growth on corn cobs | [363] |
| Trichoderma harzianum | Secretome | 2D-E, MALDI-TOF MS | 60 | Analysis of the fungus secretome during growth on plant cell wall | [294] |
| Trichoderma reesei | Secretome | iTRAQ | 636 | Description of secretome composition in the wild type and in a hypercellulolytic mutant | [337] |
| Umbilicaria mammulata | Thallus proteome | IEF | n.a. | Intraspecific variability of isozymes | [374] |
| Xanthoria parietina | Thallus proteome | 1D-E | n.a | Characterization of proteins involved in the lichen-algae interplay. | [373] |
| Yarrowia lipolytica | Whole cell proteome | 2D-E, MALDI-TOF/TOF | 38 | Proteomic analysis of the response to environmental pH stimuli | [342] |
| Zymoseptoria tritici | Mycelium, whole cell proteome | 1D-E, SCX, LC-MS/MS | 6440 | Comprehensive proteomic analysis of the proteome of Z. tritici during growth in nutrient-limiting and rich media and in vivo at a late stage of wheat infection | [258] |
References
- Blackwell, M. The fungi: 1, 2, 3... 5.1 million species? Am. J. Bot. 2011, 98, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Hawksworth, D.L. The fungal dimension of biodiversity: Magnitude, significance, and conservation. Mycol. Res. 1991, 95, 641–655. [Google Scholar] [CrossRef]
- Hibbett, D.; Abarenkov, K.; Koljalg, U.; Opik, M.; Chai, B.; Cole, J.; Wang, Q.; Crous, P.; Robert, V.; Helgason, T.; et al. Sequence-based classification and identification of Fungi. Mycologia 2016, 108, 1049–1068. [Google Scholar] [PubMed]
- Onofri, S.; Selbmann, L.; De Hoog, G.S.; Grube, M.; Barreca, D.; Ruisi, S.; Zucconi, L. Evolution and adaptation of fungi at boundaries of life. Adv. Space Res. 2007, 40, 1657–1664. [Google Scholar] [CrossRef]
- Zhang, N.; Luo, J.; Bhattacharya, D. Advances in fungal phylogenomics and their impact on fungal systematics. Adv. Genet. 2017, 100, 309–328. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.; Van Kan, J.; Pretorius, Z.A.; Hammond, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Khaman, R.; Ellis, J.; et al. The top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [Green Version]
- Köhler, J.R.; Casadevall, A.; Perfect, J. The spectrum of fungi that infects humans. Cold SpringHarb. Perspect. Med. 2014, 5, a019273. [Google Scholar] [CrossRef] [Green Version]
- Sterflinger, K. Fungi: Their role in deterioration of cultural heritage. Fungal Biol. Rev. 2010, 24, 47–55. [Google Scholar] [CrossRef]
- Perez-Nadales, E.; Nogueira, M.F.; Baldin, C.; Castanheira, S.; El Ghalid, M.; Grund, E.; Lengeler, K.; Marchegiani, E.; Mehrotra, P.V.; Moretti, M.; et al. Fungal model systems and the elucidation of pathogenicity determinants. Fungal Gen. Biol. 2014, 70, 42–67. [Google Scholar] [CrossRef] [Green Version]
- Stajich, J.E. Fungal genomes and insights into the evolution of the kingdom. Microbiol Spectrum. 2017, 5. [Google Scholar] [CrossRef]
- Aylward, J.; Steenkamp, E.T.; Dreyer, L.L.; Roets, F.; Wingfeld, B.D.; Wingfeld, M.J. A plant pathology perspective of fungal genome sequencing. IMA Fungus 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Queiroz, K. Species Concepts and Species Delimitation. Syst. Biol. 2007, 56, 879–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutzoni, F.; Kauff, F.; Cox, C.J.; McLaughlin, D.; Celio, G.; Dentinger, B.; Padamsee, M.; Hibbett, D.; James, T.Y.; Baloch, E.; et al. Assembling the fungal tree of life: Progress, classification, and evolution of subcellular traits. Am. J. Bot. 2004, 91, 1446–1480. [Google Scholar] [CrossRef] [PubMed]
- Spatafora, J.W.; Sung, G.H.; Johnson, D.; Hesse, C.; O’Rourke, B.; Serdani, M.; Spotts, R.; Lutzoni, F.; Hofstetter, V.; Miadlikowska, J.; et al. A five-gene phylogeny of Pezizomycotina. Mycologia 2006, 98, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Sung, G.H.; Lopez-Giraldez, F.; Townsend, J.P.; Miadlikowska, J.; Hofstetter, V.; Robbertse, B.; Matheny, P.B.; Kauff, F.; Wang, Z.; et al. The Ascomycota tree of life: A phylum-wide phylogeny clarifies the origin and evolution of fundamental reproductive and ecological traits. Syst. Biol. 2009, 58, 224–239. [Google Scholar] [CrossRef]
- Miadlikowska, J.; Kauff, F.; Högnabba, F.; Oliver, J.C.; Molnar, K.; Fraker, E.; Gaya, E.; Hafellner, J.; Hofstetter, V.; Gueidan, C.; et al. A multigene phylogenetic synthesis for the class Lecanoromycetes (Ascomycota): 1307 fungi representing 1139 infrageneric taxa, 317 genera and 66 families. Mol. Phylogen. Evol. 2014, 79, 132–168. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.J.; Kim, S.H. A genome tree of life for the Fungi kingdom. Proc. Natl. Acad. Sci. USA 2017, 114, 9391–9396. [Google Scholar] [CrossRef] [Green Version]
- Ebersberger, I.; de Matos Simoes, R.; Kupczok, A.; Gube, M.; Kothe, E.; Voigt, K.; von Haeseler, A. A consistent phylogenetic backbone for the fungi. Mol. Biol. Evol. 2012, 29, 1319–1334. [Google Scholar] [CrossRef] [Green Version]
- Rokas, A.; Williams, B.L.; King, N.; Carroll, S.B. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 2003, 425, 798–804. [Google Scholar] [CrossRef]
- Matute, D.R.; Sepulveda, V.E. Fungal species boundaries in the genomic era. Fungal Gen. Biol. 2019, 131, 103249. [Google Scholar] [CrossRef]
- Rustagi, A.; Singh, G.; Agrawal, S.; Gupta, P.K. Proteomic studies revealing enigma of plant-pathogen interaction. In Molecular Aspects of Plant-Pathogen Interactions; Singh, A., Singh, I., Eds.; Springer: Singapore, 2018; pp. 239–264. [Google Scholar] [CrossRef]
- Pandey, A.; Mann, M. Proteomics to study genes and genomes. Nature 2000, 405, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Tesei, D.; Sterflinger, K.; Marzban, G. Global Proteomics of Extremophilic Fungi: Mission Accomplished? Tiquia-Arashiro, M.G., Ed.; Springer Nature Switzerland AG 2019: Cham, Switzerland, 2019; ISBN 9783030190309. [Google Scholar]
- Griss, J.; Perez-Riverol, Y.; Hermjakob, H.; Vizcaíno, J.A. Identifying novel biomarkers through data mining—A realistic scenario? Proteomics Clin. Appl. 2015, 9, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Shiny, M.C.; Madhusudan, I.; Gaurav Isola, R.; Shanthi, C. Potential of proteomics to probe microbes. J. Basic Microbiol. 2020, 60, 471–483. [Google Scholar] [CrossRef]
- Archer, D.B.; Dyer, P.S. From genomics to post-genomics in Aspergillus. Curr. Opin. Microbiol. 2004, 7, 499–504. [Google Scholar] [CrossRef]
- Doyle, S. Fungal proteomics: From identification to function. FEMS Microbiol. Lett. 2011, 321, 1–9. [Google Scholar] [CrossRef]
- Uranga, C.C.; Ghassemian, M.; Hernández-Martínez, R. Novel proteins from proteomic analysis of the trunk disease fungus Lasiodiplodia theobromae (Botryosphaeriaceae). Biochim. Open 2017, 4, 88–98. [Google Scholar] [CrossRef]
- Özhak-Baysan, B.; Ögünc, D.; Dögen, A.; Ilkit, M.; De Hoog, G.S. MALDI-TOF MS-based identification of black yeasts of the genus Exophiala. Med. Mycol. 2015, 53, 347–352. [Google Scholar] [CrossRef] [Green Version]
- Bhadauria, V.; Banniza, S.; Wang, L.-X.; Wei, Y.-D.; Peng, Y.-L. Proteomic studies of phytopathogenic fungi, oomycetes and their interactions with hosts. Eur. J. Plant Pathol. 2009, 126, 81–95. [Google Scholar] [CrossRef]
- Loginov, D.; Šebela, M. Proteomics of survival structures of fungal pathogens. N. Biotechnol. 2016, 33, 655–665. [Google Scholar] [CrossRef]
- Karányi, Z.; Holb, I.; Hornok, L.; Pócsi, I.; Miskei, M. FSRD: Fungal stress response database. Database 2013, 2013, bat0037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Park, J.; Kim, D.; Jung, K.; Kang, S. Fungal Secretome Database: Integrated platform for annotation of fungal secretomes. BMC Genom. 2010, 11, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudimella, R.; Nallapeta, S.; Varadwaj, P.; Suravajhala, P. Fungome: Annotating proteins implicated in fungal pathogenesis. Bioinformation 2010, 5, 202–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egbuta, A.M.; Mwanza, M.; Oluranti Babalola, O. A Review of the ubiquity of ascomycetes filamentous fungi in relation to their economic and medical importance. Adv. Microbiol. 2016, 6, 1140–1158. [Google Scholar] [CrossRef] [Green Version]
- Greco, T.M.; Cristea, I.M. Proteomics tracing the footsteps of infectious disease. Mol. Cell. Proteomics 2017, 16, S5–S14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grinyer, J.; Hunt, S.; McKay, M.; Herbert, B.R.; Nevalainen, H. Proteomic response of the biological control fungus Trichoderma atroviride to growth on the cell walls of Rhizoctonia solani. Curr. Genet. 2005, 47, 381–388. [Google Scholar] [CrossRef]
- Ibrar, M.; Ullah, M.W.; Manan, S.; Farooq, U.; Rafiq, M.; Hasan, F. Fungi from the extremes of life: An untapped treasure for bioactive compounds. Appl. Microbiol. Biotechnol. 2020, 104, 2777–2801. [Google Scholar] [CrossRef]
- Sharma Ghimire, P.; Jin, C. Genetics, molecular, and proteomics advances in filamentous fungi. Curr. Microbiol. 2017, 74, 1226–1236. [Google Scholar] [CrossRef]
- Kroll, K.; Pähtz, V.; Kniemeyer, O. Elucidating the fungal stress response by proteomics. J. Proteomics 2014, 97, 151–163. [Google Scholar] [CrossRef]
- Muñoz, J.F.; Gauthier, G.M.; Desjardins, C.A.; Gallo, J.E.; Holder, J.; Sullivan, T.D.; Marty, A.J.; Carmen, J.C.; Chen, Z.; Ding, L.; et al. The dynamic genome and transcriptome of the human fungal pathogen Blastomyces and close relative Emmonsia. PLoS Genet. 2015, 11, e1005493. [Google Scholar] [CrossRef] [Green Version]
- De Hoog, G.S.; Guarro, J.; Gené, S.; Ahmed, A.M.S. Atlas of Clinical Fungi, 3rd ed.; Cold Spring Harbor Laboratory Press: Utrech, The Netherlands, 2019; Available online: http://www.clinicalfungi.org/. (accessed on 12 November 2020).
- Nobrega, J.P.; Rosemberg, S.; Adami, A.M.; Heins-Vaccari, E.M.; da Silva Lacaz, C.; de Brito, T. Fonsecaea pedrosoi cerebral phaeohyphomycosis (chromoblastomycosis): First human culture-proven case reported in Brazil. Rev. Inst. Med. Trop Sao Paulo 2003, 45, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Surash, S.; Tyagi, A.; De Hoog, G.S.; Zeng, J.S.; Barton, R.C.; Hobson, R.P. Cerebral phaeohyphomycosis caused by Fonsecaea monophora. Med. Mycol. 2005, 43, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, S.; Klompas, M.; Marty, F.M. Fonsecaea monophora cerebral phaeohyphomycosis: Case report of successful surgical excision and voriconazole treatment and review. Med. Mycol. 2010, 48, 769–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalar, P.; Novak, M.; de Hoog, G.S.; Gunde-Cimerman, N. Dishwashers—A man-made ecological niche accommodating human opportunistic fungal pathogens. Fungal Biol. 2010, 115, 997–1007. [Google Scholar] [CrossRef]
- Gostinčar, C.; Grube, M.; Gunde-Cimerman, N. Evolution of fungal pathogens in domestic environments? Fungal Biol. 2011, 115, 1008–1018. [Google Scholar] [CrossRef]
- Teixeira, M.M.; Moreno, L.F.; Stielow, B.; Muszewska, A.; Hainaut, M.; Gonzaga, L.; Abouelleil, A.; Patané, J.S.L.; Priest, M.; Souza, R.; et al. Exploring the genomic diversity of black yeasts and relatives (Chaetothyriales, Ascomycota). Stud. Mycol. 2017, 86, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Gostinčar, C.; Muggia, L.; Grube, M. Polyextremotolerant black fungi: Oligotrophism, adaptive potential, and a link to lichen symbioses. Front. Microbiol. 2012, 3, 390. [Google Scholar] [CrossRef] [Green Version]
- Moreno, L.F.; Vicente, V.A.; de Hoog, S. Black yeasts in the omics era: Achievements and challenges. Med. Mycol. 2018, 56, 32–41. [Google Scholar] [CrossRef]
- Gostinčar, C.; Zajc, J.; Lenassi, M.; Plemenitas, A.; de Hoog, S.; Al-Hatmi, A.M.S.; Gunde-Cimerman, N. Fungi between extremotolerance and opportunistic pathogenicity on humans. Fungal Div. 2018, 93, 195–213. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Martinez, D.A.; Gujja, S.; Sykes, S.M.; Zeng, Q.; Szaniszlo, P.J.; Wang, Z.; Cuomo, C.A. Comparative genomic and transcriptomic analysis of Wangiella dermatitidis, a major cause of phaeohyphomycosis and a model black yeast human pathogen. G3 Genes Genomes Genet. 2014, 4, 561–578. [Google Scholar]
- Moreno, L.F.; Menezes da Silva, N.; Weiss, V.A.; de Fátima Costa, F.; Bittencourt, J.V.; Medina Macedo, L.; Gomes, R.R.; Souza, E.M.; Vicente, V.A.; Stielow, B.; et al. Genome sequence of the human opportunistic fungus Arthrocladium fulminans (CBS 136243). G3 Genes Genomes Genet. 2020, 10, 1817–1821. [Google Scholar] [CrossRef] [Green Version]
- Chomnunti, P.D.J.; Bhat, E.B.G.; Jones, E.; Chukeatirote, A.H.; Bahkali, A.H.; Hyde, K.D. Trichomeriaceae, a new sooty mould family of Chaetothyriales. Fungal Div. 2012, 56, 63–76. [Google Scholar] [CrossRef]
- Vicente, V.A.; Weiss, V.A.; Bombassaro, A.; Moreno, L.F.; Costa, F.F.; Raittz, R.T.; Leão, A.C.; Gomes, R.R.; Bocca, A.L.; Fornari, G.; et al. Comparative genomics of sibling species of Fonsecaea associated with human chromoblastomycosis. Front. Microbiol. 2017, 8, 1924. [Google Scholar] [CrossRef] [PubMed]
- Möller, M.; Stukenbrock, E. Evolution and genome architecture in fungal plant pathogens. Nat. Rev. Microbiol. 2017, 15, 756–771. [Google Scholar] [CrossRef]
- Covo, S. Genomic Instability in Fungal Plant Pathogens. Genes 2020, 11, 421. [Google Scholar] [CrossRef] [Green Version]
- Pedro, H.; Maheswari, U.; Urban, M.; Irvine, A.G.; Cuzick, A.; McDowall, M.D.; Staines, D.M.; Kulesha, E.; Hammond-Kosack, K.E.; Kersey, P.J. PhytoPath: An integrative resource for plant pathogen genomics. Nuc. Ac. Res. 2016, 44, D688–D693. [Google Scholar] [CrossRef] [Green Version]
- Plissonneau, C.; Benevenuto, J.; Mohd-Assaad, N.; Fouché, S.; Hartmann, F.E.; Croll, D. Using population and comparative genomics to understand the genetic basis of effector-driven fungal pathogen evolution. Front. Plant Sci. 2017, 8, 119. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.J.; van der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef]
- Eschenbrenner, C.J.; Feurtey, A.; Stukenbrock, E.H. Population genomics of fungal plant pathogens and the analyses of rapidly evolving genome compartments. In Statistical Population Genomics. Methods in Molecular Biology; Dutheil, J., Ed.; Humana: New York, NY, USA, 2020; Volume 2090. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-T.; Jeon, J.; Choi, J.; Cheong, K.; Song, H.; Choi, G.; Kang, S.; Lee, Y.H. Kingdom- wide analysis of fungal small secreted proteins (SSPs) reveals their potential role in host association. Front. Plant Sci. 2016, 7, 186. [Google Scholar] [CrossRef] [Green Version]
- Lo Presti, L.; Lanver, D.; Schweizer, G.; Tanaka, S.; Liang, L.; Tollot, M.; Zuccar, A.; Reissmann, S.; Kahmann, R. Fungal effectors and plant susceptibility. Annu. Rev. Plant Biol. 2015, 66, 513–545. [Google Scholar] [CrossRef]
- Potgieter, L.; Feurtey, A.; Dutheil, J.Y.; Stukenbrock, E.H. On variant discovery in genomes of fungal plant pathogens. Front. Microbiol. 2020, 11, 626. [Google Scholar] [CrossRef] [Green Version]
- Milgroom, M.G.; Sotirovski, K.; Risteski, M.; Brewer, M.T. Heterokaryons and parasexual recombinants of Cryphonectria parasitica in two clonal populations in southeastern Europe. Fungal Genet. Biol. 2009, 46, 849–854. [Google Scholar] [CrossRef]
- Wang, C.S.; St Leger, R.J.; Wang, C. Advances in genomics of entomopathogenic fungi. Adv. Genet. 2016, 94, 67–105. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.S.; St Leger, R.J. Genomics of entomopathogenic fungi. In The Ecological Genomics of Fungi; Martin, F., Ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2014; pp. 243–260. [Google Scholar] [CrossRef]
- Shu, R.; Zhang, J.; Meng, Q.; Zhang, H.; Zhou, G.; Li, M.; Wu, P.; Zhao, Y.; Chen, C.; Qin, Q. A new high-quality draft genome assembly of the Chinese Cordyceps Ophiocordyceps sinensis. Genome Biol. Evol. 2020, 12, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Ying, S.; Zheng, P.; Wang, Z.L.; Zhang, S.; Xie, X.Q.; Shang, Y.; Leger, R.J.S.; Zhao, G.P.; Wang, C.; et al. Genomic perspectives on the evolution of fungal entomopathogenicity in Beauveria bassiana. Sci. Rep. 2012, 2, 483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattemore, J.A.; Hane, J.K.; Williams, A.H.; Wilson, B.A.; Stodart, B.J.; Ash, G.J. The genome sequence of the biocontrol fungus Metarhizium anisopliae and comparative genomics of Metarhizium species. BMC Genom. 2014, 15, 660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Wang, S. Insect pathogenic fungi: Genomics, molecular interactions, and genetic improvements. Ann. Rev. Entom. 2017, 62, 73–90. [Google Scholar] [CrossRef]
- Staats, C.C.; Junges, Â.; Guedes, R.L.M.; Thompson, C.E.; de Morais, G.L.; Boldo, J.T.; de Almeida, L.G.P.; Andreis, F.C.; Gerber, A.L.; Sbaraini, N.; et al. Comparative genome analysis of entomopathogenic fungi reveals a complex set of secreted proteins. BMC Genom. 2014, 15, 822. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.Q.; Xu, Z.W.; Zhang, B.; Yi, M.; Weng, C.Y.; Lin, S.; Wu, H.; Qin, X.T.; Xu, F.; Teng, Y.; et al. Genome sequencing and analysis of fungus Hirsutella sinensis isolated from Ophiocordyceps sinensis. AMB Express 2020, 10, 105. [Google Scholar] [CrossRef]
- Maheshwar, R.; Bharadwaj, G.; Bhat, M.K. Thermophilic fungi: Their physiology and enzymes. Microbiol. Mol. Biol. Rev. 2000, 64, 461–488. [Google Scholar] [CrossRef] [Green Version]
- Morgenstern, I.; Powlowski, J.; Ishmael, N.; Darmond, C.; Marqueteau, S.; Moisan, M.C.; Quenneville, G.; Tsang, A. A molecular phylogeny of thermophilic fungi. Fungal Biol. 2012, 116, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Amlacher, S.; Sarges, P.; Flemming, D.; van Noort, V.; Kunze, R.; Devos, D.P.; Arumugam, M.; Bork, P.; Hurt, E. Insight into structure and assembly of the nuclear pore complex by utilizing the genome of a eukaryotic thermophile. Cell 2011, 146, 277–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, T.; Chen, W.H.; Ori, A.; Malik, N.; Silva-Martin, N.; Huerta-Cepas, J.; Powell, S.T.; Kastritis, P.L.; Smyshlyaev, G.; Vonkova, I.; et al. An integrated approach for genome annotation of the eukaryotic thermophile Chaetomium thermophilum. Nucleic Acids Res. 2014, 42, 13525–13533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berka, R.M.; Grigoriev, I.V.; Otillar, R.; Salamov, A.; Grimwood, J.; Reid, I.; Ishmael, N.; John, T.; Darmond, C.; Moisan, M.C.; et al. Comparative genomic analysis of the thermophilic biomass-degrading fungi Myceliophthora thermophila and Thielavia terrestris. Nat. Biotechnol. 2011, 29, 922–929. [Google Scholar] [CrossRef]
- Mchunu, N.P.; Permaul, K.; Abdul Rahman, A.Y.; Saito, J.A.; Singh, S.; Alam, M. Xylanase superproducer: Genome sequence of a compost-loving thermophilic fungus, Thermomyces lanuginosus strain SSBP. Genome Announc. 2013, 1, e00388-13. [Google Scholar] [CrossRef] [Green Version]
- Staszczak, M. The role of the ubiquitin-proteasome system in the response of the ligninolytic fungus Trametes versicolor to nitrogen deprivation. Fungal Genet. Biol. 2008, 45, 328–337. [Google Scholar] [CrossRef]
- De Oliveira, T.B.; Gostinčar, C.; Gunde-Cimerman, N.; Rodrigueset, A. Genome mining for peptidases in heat-tolerant and mesophilic fungi and putative adaptations for thermostability. BMC Genom. 2018, 19, 152. [Google Scholar] [CrossRef] [Green Version]
- Hassan, N.; Rafiq, M.; Hayat, M.; Shah, A.A.; Hasan, F. Psychrophilic and psychrotrophic fungi: A comprehensive review. Rev. Environ. Sci. Biotechnol. 2016, 15, 147–172. [Google Scholar] [CrossRef]
- Voets, I.K. From ice-binding proteins to bio-inspired antifreeze materials. Soft Matter 2017, 13, 4808–4823. [Google Scholar] [CrossRef] [Green Version]
- Leushkin, E.V.; Logacheva, M.D.; Penin, A.A.; Sutormin, R.A.; Gerasimov, E.S.; Kochkina, G.A.; Ivanushkina, N.E.; Vasilenko, O.V.; Kondrashov, A.S.; Ozerskaya, S.M. Comparative genome analysis of Pseudogymnoascus spp. reveals primarily clonal evolution with small genome fragments exchanged between lineages. BMC Genom. 2015, 16, 400. [Google Scholar] [CrossRef] [Green Version]
- Forsythe, A.; Xu, J. The complete mitochondrial genome of the White-Nose Syndrome pathogen, Pseudogymnoascus destructans. Mitochondrial DNA Part B 2017, 2, 48–49. [Google Scholar] [CrossRef]
- Su, Y.; Jiang, X.; Wu, W.; Wang, M.; Hamid, M.I.; Xiang, M.; Liu, X. Genomic, transcriptomic, and proteomic analysis provide insights into the cold adaptation mechanism of the obligate psychrophilic fungus Mrakia psychrophila. G3 Genes Genomes Genet 2016, 6, 3603–3613. [Google Scholar] [CrossRef] [Green Version]
- De Menezes, G.C.A.; Godinho, V.M.; Porto, B.A.; Goncalves, V.N.; Rosa, L.H. Antarctomyces pellizariae sp. nov., a new, endemic, blue, snow resident psychrophilic ascomycete fungus from Antarctica. Extremophiles 2017, 21, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Batista, T.M.; Hilario, H.O.; de Brito, G.A.M.; Moreira, R.G.; Furtado, C.; de Menezes, G.C.A.; Rosa, C.A.; Rosa, L.H.; Franco, G.R. Whole-genome sequencing of the endemic Antarctic fungus Antarctomyces pellizariae reveals an ice-binding protein, a scarce set of secondary metabolites gene clusters and provides insights on Thelebolales phylogeny. Genomics 2020, 112, 2915–2921. [Google Scholar] [CrossRef]
- Sterflinger, K.; Lopandic, K.; Pandey, R.V.; Blasi, B.; Kriegner, A. Nothing special in the specialist? Draft genome sequence of Cryomyces antarcticus, the most extremophilic fungus from Antarctica. PLoS ONE 2014, 9, e109908. [Google Scholar] [CrossRef] [Green Version]
- Lenassi, M.; Gostinčar, C.; Jackman, S.; Turk, M.; Sadowski, I.; Nislow, C.; Jones, S.; Birol, I.; Gunde-Cimerman, N.; Plemenitaš, A. Whole genome duplication and enrichment of metal cation transporters revealed by de novo genome sequencing of extremely halotolerant black yeast Hortaea werneckii. PLoS ONE 2013, 8, e71328. [Google Scholar] [CrossRef] [Green Version]
- Gostinčar, C.; Ohm, R.A.; Kogej, T.; Sonjak, S.; Turk, M.; Zajc, J.; Zalar, P.; Grube, M.; Sun, H.; Han, J.; et al. Genome sequencing of four Aureobasidium pullulans varieties: Biotechnological potential, stress tolerance, and description of new species. BMC Genom. 2014, 15, 549. [Google Scholar] [CrossRef] [Green Version]
- Coleine, C.; Masonjones, S.; Selbmann, L.; Zucconi, L.; Onofri, S.; Pacelli, C.; Stajich, J.E. Draft genome sequences of the antarctic endolithic fungi Rachicladosporium antarcticum CCFEE 5527 and Rachicladosporium sp. CCFEE 5018. Genome Announc. 2017, 5, e00397-17. [Google Scholar] [CrossRef] [Green Version]
- Ametrano, C.G.; Grewe, F.; Crous, P.W.; Goodwin, S.B.; Liang, C.; Selbmann, L.; Lumbsch, H.T.; Leavitt, S.D.; Muggia, L. Genome-scale data resolve ancestral rock-inhabiting lifestyle in Dothideomycetes (Ascomycota). IMA Fungus 2019, 10, 19. [Google Scholar] [CrossRef]
- Blasi, B.; Tafer, H.; Kustor, C.; Poyntner, C.; Lopandic, K.; Sterflinger, K. Genomic and transcriptomic analysis of the toluene degrading black yeast Cladophialophora immunda. Sci. Rep. 2017, 7, 11436. [Google Scholar] [CrossRef] [Green Version]
- Coleine, C.; Selbmann, L.; Masonjones, S.; Onofri, S.; Zucconi, L.; Stajich, J.E. Draft genome sequence of an Antarctic isolate of the black yeast fungus Exophiala mesophila. Microbiol. Res. Announc. 2019, 8, e00142-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesei, D.; Tafer, H.; Poyntner, C.; Piñar, G.; Lopandic, K.; Sterflinger, K. Draft genome sequences of the black rock fungus Knufia petricola and its spontaneous nonmelanized mutant. Genome Announc. 2017, 5, e01242-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, S.; Flibotte, S.; Neira, M.; Formby, S.; Plemenitaš, A.; Cimerman, N.G.; Lenassi, M.; Gostinčar, C.; Stajich, J.E.; Nislow, C. Insight into the recent genome duplication of the halophilic yeast Hortaea werneckii: Combining an improved genome with gene expression and chromatin structure. G3 Genes Genomes Genet. 2017, 7, 2015–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gostinčar, C.; Stajich, J.E.; Zupančič, J.; Zalar, P.; Gunde-Cimerman, N. Genomic evidence for intraspecific hybridization in a clonal and extremely halotolerant yeast. BMC Genom. 2018, 19, 1471–2164. [Google Scholar] [CrossRef] [PubMed]
- Onofri, S.; Pagano, S.; Zucconi, L.; Tosi, S. Friedmanniomyces endolithicus (Fungi, Hyphomycetes), a new gen. and sp. nov. from continental Antarctica. Nova Hedwigia 1999, 68, 175–182. [Google Scholar] [CrossRef]
- Coleine, C.; Masonjones, S.; Sterflinger, K.; Onofri, S.; Selbmann, L.; Stajich, E.J. Peculiar genomic traits in the stress-adapted cryptoendolithic Antarctic fungus Friedmanniomyces endolithicus. Fungal Biol. 2020, 124, 458–467. [Google Scholar] [CrossRef]
- Johnston, C.; Rayner, J.; Patterson, B.; Davis, G. Volatilisation and biodegradation during air sparging of dissolved btex-contaminated groundwater. J. Contam. Hydrol. 1998, 33, 377–404. [Google Scholar] [CrossRef]
- Badali, H.; Gueidan, C.; Najafzadeh, M.J.; Bonifaz, A.; van den Ende, A.H.; de Hoog, G.S. Biodiversity of the genus Cladophialophora. Stud. Mycol. 2008, 61, 175–191. [Google Scholar] [CrossRef]
- Prenafeta-Boldu, F.X.; Summerbell, R.; De Hoog, G.S. Fungi growing on aromatic hydrocarbons: Biotechnology’s unexpected encounter with biohazard? FEMS Microbiol. Rev. 2006, 30, 109–130. [Google Scholar] [CrossRef] [Green Version]
- Vasse, M.; Voglmayr, H.; Mayer, V.; Gueidan, C.; Nepel, M.; Moreno, L.; de Hoog, S.; Selosse, M.A.; McKey, D.; Blatrix, R. A phylogenetic perspective on the association between ants (Hymenoptera: Formicidae) and black yeasts (Ascomycota: Chaetothyriales). Proc. R. Soc. B Biol. Sci. 2017, 284, 20162519. [Google Scholar] [CrossRef]
- Moreno, L.F.; Stielow, J.B.; de Vries, M.; Weiss, V.A.; Vicente, V.A.; de Hoog, S. Draft genome sequence of the ant-associated fungus Phialophora attae (CBS 131958). Genome Announc. 2015, 3, e01099-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, L.F.; Mayer, V.; Voglmayr, H.; Blatrix, R.; Stielow, B.J.; Teixeira, M.M.; Vicente, V.A.; de Hoog, S. Genomic analysis of ant domatia-associated melanized fungi (Chaetothyriales, Ascomycota). Mycol. Progress 2019, 18, 541–552. [Google Scholar] [CrossRef] [Green Version]
- De Fine Licht, H.H.; Schiott, M.; Mueller, U.G.; Boomsma, J. Evolutionary transitions in enzyme activity of ant fungus gardens. Evolution 2010, 64, 2055–2069. [Google Scholar] [CrossRef] [PubMed]
- Honegger, R. The symbiotic phenotype of lichen-forming Ascomycetes. In Fungal Associations. The Mycota (A Comprehensive Treatise on Fungi as Experimental Systems for Basic and Applied Research); Hock, B., Ed.; Springer: Berlin/Heidelberg, Germany, 2001; Volume 9. [Google Scholar]
- Hawksworth, D.L.; Grube, M. Lichens redefined as complex ecosystem. New Phytol. 2020, 227, 1281–1283. [Google Scholar] [CrossRef] [PubMed]
- Lücking, R.; Hodkinson, B.P.; Leavitt, S.D. The 2016 classification of lichenized fungi in the Ascomycota and Basidiomycota—Approaching one thousand genera. Bryologist 2016, 119, 361–416. [Google Scholar] [CrossRef]
- Spribille, T. Relative symbiont input and the lichen symbiotic outcome. Curr. Opin. Plant Biol. 2018, 44, 57–63. [Google Scholar] [CrossRef]
- Muggia, L.; Grube, M. Fungal diversity in lichens: From extremotolerance to interaction with algae. Life 2018, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Moya, P.; Molins, A.A.; Ânez-Alberola, F.M.; Muggia, L.; Barreno, E. Unexpected associated microalgal diversity in the lichen Ramalina farinacea is uncovered by pyrosequencing analyses. PLoS ONE 2017, 12, e0175091. [Google Scholar] [CrossRef] [Green Version]
- Meiser, A.; Otte, J.; Schmit, I.; Dal Grande, F. Sequencing genomes from mixed DNA samples—Evaluating the metagenome skimming approach in lichenized fungi. Scie. Rep. 2017, 7, 14881. [Google Scholar] [CrossRef]
- Allen, J.L.; McKenzie, S.K.; Sleith, R.S.; Alter, S.E. First genome-wide analysis of the endangered, endemic lichen Cetradonia linearis reveals isolation by distance and strong population structure. Am. J. Bot. 2018, 105, 1556–1567. [Google Scholar] [CrossRef]
- Kirk, P.; Cannon, P.; Stalpers, J.; Minter, D.W. Dictionary of the Fungi, 10th ed.; CABI Publishing: Great Britain, UK, 2008. [Google Scholar]
- McDonald, T.R.; Mueller, O.; Dietrich, F.S.; Lutzoni, F. High-throughput genome sequencing of lichenizing fungi to assess gene loss in the ammonium transporter/ammonia permease gene family. BMC Genom. 2013, 14, 225. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-Y.; Choi, J.; Kim, J.A.; Yu, N.-H.; Kim, S.; Kondratyuk, S.Y.; Lee, Y.-H.; Hur, J.-S. (a) Draft genome sequence of lichen-forming fungus Caloplaca flavorubescens strain KoLRI002931. Genome Announc. 2013, 1, e00678-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.-Y.; Choi, J.; Kim, J.A.; Jeong, M.-H.; Kim, S.; Lee, Y.-H.; Hur, J.-S. (b) Draft genome sequence of Cladonia macilenta KoLRI003786, a lichen-forming fungus producing biruloquinone. Genome Announc. 2013, 1, e00695-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.-Y.; Choi, J.; Lee, G.-W.; Jeong, M.-H.; Kim, J.A.; Oh, S.-O.; Lee, Y.-H.; Hur, J.-S. (a) Draft genome sequence of Umbilicaria muehlenbergii KoLRILF000956, a lichen-forming fungus amenable to genetic manipulation. Genome Announc. 2014, 2, e00357-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.-Y.; Choi, J.; Lee, G.-W.; Kim, J.A.; Oh, S.-O.; Jeong, M.-H.; Yu, N.-H.; Kim, S.; Lee, Y.-H.; Hur, J.-S. (b) Draft genome sequence of lichen-forming fungus Cladonia metacorallifera strain KoLRI002260. Genome Announc. 2014, 2, e01065-13. [Google Scholar] [CrossRef] [Green Version]
- Armaleo, D.; Müller, O.; Lutzoni, F.; Andrésson, O.S.; Blanc, G.; Bode, H.B.; Collart, F.R.; Dal Grande, F.; Dietrich, F.; Grigoriev, I.V.; et al. The lichen symbiosis re-viewed through the genomes of Cladonia grayi and its algal partner Asterochloris glomerata. BMC Genom. 2019, 20, 605. [Google Scholar] [CrossRef] [Green Version]
- Junttila, S.; Rudd, S. Characterization of a transcriptome from a non-model organism, Cladonia rangiferina, the grey reindeer lichen, using high-throughput next generation sequencing and EST sequence data. BMC Genom. 2012, 13, 575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brigham, L.M.; Allende, L.M.; Shipley, B.R.; Boyd, K.C.; Higgins, T.J.; Kelly, N.; Anderson Stewart, C.R.; Keepers, K.G.; Pogoda, C.S.; Lendemer, J.C.; et al. Genomic insights into the mitochondria of 11 eastern North American species of Cladonia. Mitochondrial DNA Part B 2018, 3, 508–512. [Google Scholar] [CrossRef]
- Xavier, B.B.; Miao, V.P.; Jónsson, Z.O.; Andrésson, Ó.S. Mitochondrial genomes from the lichenized fungi Peltigera membranacea and Peltigera malacea: Features and phylogeny. Fungal Biol. 2012, 116, 802–814. [Google Scholar] [CrossRef]
- Funk, E.R.; Adams, A.N.; Spotten, S.M.; Van Hove, R.A.; Whittington, K.T.; Keepers, K.G.; Pogoda, C.S.; Lendemer, J.C.; Tripp, E.A.; Kane, N.C. The complete mitochondrial genomes of five lichenized fungi in the genus Usnea (Ascomycota: Parmeliaceae). Mitochondrial DNA Part B 2018, 3, 305–308. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yuan, X.; Chen, L.; Wang, X.; Li, C. Draft genome sequence of the lichen forming fungus Ramalina intermedia strain YAF0013. Genome Announc. 2018, 6, e00478-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilken, P.M.; Aylward, J.; Chand, R.; Lane, F.A.; Sinha, S.; Ametrano, C.G.; Distefano, I.; Divakar, P.K.; Duong, T.A.; Huhndorf, S.; et al. IMA Genome—F13: Draft genome sequences of Ambrosiella cleistominuta, Cercospora brassicicola, C. citrullina, Physcia stellaris, and Teratosphaeria pseudoeucalypti. IMA Fungus 2020, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Liu, B.; Zhang, X.Y.; Zhou, Q.-M.; Zhang, T.; Li, H.; Yu, Y.-F.; Zhang, X.-L.; Hao, X.-Y.; Wang, M.; et al. Genome characteristics reveal the impact of lichenization on lichen-forming fungus Endocarpon pusillum Hedwig (Verrucariales, Ascomycota). BMC Genom. 2014, 15, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal Grande, F.; Meiser, A.; Greshake-Tzovaras, B.; Otte, J.; Ebersberger, I.; Schmitt, I. The draft genome of the lichen-forming fungus Lasallia hispanica (Frey) Sancho & A. Crespo. Lichenologist 2018, 50, 329–340. [Google Scholar]
- Pizarro, D.; Dal Grande, F.; Leavitt, S.D.; Dyer, P.S.; Schmitt, I.; Crespo, A.; Lumbsch, H.T.; Divakar, P.K. Whole-genome sequence data uncover widespread heterothallism in the largest group of lichen-forming fungi. Genome Biol. Evol. 2019, 11, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Keuler, R.; Garretson, A.; Saunders, T.; Erickson, R.J.; St. Andre, N.; Grew, F.; Smith, H.; Lumbsch, T.H.; Huang, J.P.; St. Clair, L.L.; et al. Genome-scale data reveal the role of hybridization in lichen-forming fungi. Sci. Rep. 2020, 10, 1497. [Google Scholar] [CrossRef] [Green Version]
- Leavitt, S.D.; Grewe, F.; Widhelm, T.; Muggia, L.; Wray, B.; Lumbsch, T.H. Resolving evolutionary relationships in lichen-forming fungi using diverse phylogenomic datasets and analytical approaches. Sci. Rep. 2016, 6, 22262. [Google Scholar] [CrossRef]
- Grewe, F.; Huang, J.-P.; Leavitt, S.D.; Lumbsch, H.T. Reference-based RADseq resolves robust relationships among closely related species of lichen-forming fungi using metagenomic DNA. Sci. Rep. 2017, 7, 9884. [Google Scholar] [CrossRef] [Green Version]
- Grewe, F.; Lagostina, E.; Wu, H.; Printzen, C.; Lumbsch, H.T. Population genomic analyses of RAD sequences resolves the phylogenetic relationship of the lichen-forming fungal species Usnea antarctica and Usnea aurantiacoatra. MycoKeys 2018, 43, 61–113. [Google Scholar] [CrossRef]
- Lutzoni, F.; Pagel, M.; Reeb, V. Major fungal lineages are derived from lichen symbiotic ancestors. Nature 2001, 411, 937–940. [Google Scholar] [CrossRef]
- Greshake, B.; Zehr, S.; Dal Grande, F.; Meiser, A.; Schmitt, I.; Ebersberger, I. Potential and pitfalls of eukaryotic metagenome skimming: A test case for lichens. Mol. Ecol. Resour. 2016, 16, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keepers, K.G.; Pogoda, C.S.; White, K.H.; Anderson Stewart, C.R.; Hoffman, J.R.; Ruiz, A.M.; McCain, C.M.; Lendemer, J.C.; Kane, N.C.; Tripp, E.A. Whole genome shotgun sequencing detects greater lichen fungal diversity than amplicon-based methods in environmental samples. Front. Ecol. Evol. 2019, 7, 484. [Google Scholar] [CrossRef] [Green Version]
- Greshake-Tzovaras, B.; Segers, F.H.I.D.; Bicker, A.; Dal Grande, F.; Ott, J.; Anvar, S.Y.; Hankeln, T.; Schmitt, I.; Ebersberger, I. What is in Umbilicaria pustulata? A metagenomic approach to reconstruct the holo-genome of a lichen. Genome Biol. Evol. 2020, 12, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Chen, S.F.; Ferreira, M.; Chang, R.; Sayari, M.; Kanzi, A.; Wingfield, B.; Wingfield, M.; Pizarro, D.; Crespo, A.; et al. Draft genome sequences of five Calonectria species from Eucalyptus plantations in China, Celoporthe dispersa, Sporothrix phasma and Alectoria sarmentosa. IMA Fungus 2019, 10, 22. [Google Scholar] [CrossRef]
- Pogoda, C.S.; Keepers, K.G.; Nadiadi, A.Y.; Bailey, D.W.; Lendemer, J.C.; Tripp, E.A.; Kane, N.C. Genome streamlining via complete loss of introns has occurred multiple times in lichenized fungal mitochondria. Ecol. Evol. 2019, 9, 4245–4263. [Google Scholar] [CrossRef] [Green Version]
- Kuramae, E.E.; Robert, V.; Snel, B.; Weiss, M.; Boekhout, T. Phylogenomics reveal a robust fungal tree of life. FEMS Yeast Res. 2006, 6, 1213–1220. [Google Scholar] [CrossRef]
- Liu, Y.; Leigh, J.; Brinkmann, H.; Cushion, M.; Naiara, E.Z.; Hervé, P.; Lang, B. Phylogenomic analyses support the monophyly of Taphrinomycotina, including Schizosaccharomyces fission yeasts. Mol. Biol. Evol. 2008, 26, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, Q.; Xia, X.; Liu, X.; Ge, L.; Yang, L. Phylogenetic relationships of ascomycetes and basidiomycetes based on comparative genomics analysis. Genes Genom. 2017, 39, 1307–1316. [Google Scholar] [CrossRef]
- Ohm, R.A.; Feau, N.; Henrissat, B.; Schoch, C.L.; Horwitz, B.A.; Barry, K.W.; Condon, B.J.; Copeland, A.C.; Dhillon, B.; Glaser, F.; et al. Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes Fungi. PLoS Pathol. 2012, 8, e1003037. [Google Scholar] [CrossRef] [Green Version]
- Haridas, S.; Albert, R.; Binder, M.; Bloem, J.; LaButti, K.; Salamov, A.; Andreopoulos, B.; Baker, S.E.; Barry, K.; Bills, G.; et al. 101 Dothideomycetes genomes: A test case for predicting lifestyles and emergence of pathogens. Stud. Mycol. 2020, 96, 141–153. [Google Scholar] [CrossRef]
- ncbi-genome-download script, Apache License, Version 2.0, January 2004. Available online: http://www.apache.org/licenses/; https://github.com/kblin/ncbi-genome-download (accessed on 12 November 2020).
- Ruibal, C.; Gueidan, C.; Selbmann, L.; Gorbushina, A.A.; Crous, P.W.; Groenewald, J.Z.; Muggia, L.; Grube, M.; Isola, D.; Schoch, C.L.; et al. Phylogeny of rock-inhabiting fungi related to Dothideomycetes. Stud. Mycol. 2009, 64, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Steenwyk, J.L.; Shen, X.-X.; Lind, A.L.; Goldman, G.H.; Rokas, A. A robust phylogenomic time tree for biotechnologically and medically important fungi in the genera Aspergillus and Penicillium. mBio 2019, 10, e00925-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, P.R.; Quijada, L.; Smith, C.A.; Baral, H.O.; Hosoya, T.; Baschien, C.; Paertel, K.; Zhuang, W.Y.; Haelewaters, D.; Park, D.; et al. A multigene phylogeny toward a new phylogenetic classification of Leotiomycetes. IMA Fungus 2019, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Luikart, G.; England, P.R.; Tallmon, D.; Jordan, S.; Taberlet, P. The power and promise of population genomics: From genotyping to genome typing. Nat. Rev. Genet. 2003, 4, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Grünwald, N.J.; McDonald, B.A.; Milgroom, M.G. Population genomics of fungal and oomycete pathogens. Ann. Rev. Phytopat. 2016, 54, 323–346. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Chen, M.; Shang, Y.; Tang, G.; Tao, Y.; Zeng, L.; Huang, B.; Li, Z.; Zhan, S.; Wang, C. Population genomics and evolution of a fungal pathogen after releasing exotic strains to control insect pests for 20 years. ISME J. 2020, 14, 1422–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, A.C.; Ward, T.J. Population genomics of Fusarium graminearum reveals signatures of divergent evolution within a major cereal pathogen. PLoS ONE 2018, 13, e0194616. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Zhang, H.; van der Lee, T.A.J.; Waalwijk, C.; van Diepeningen, A.D.; Feng, J.; Brankovics, B.; Chen, W. Population genomic analysis reveals a highly conserved mitochondrial genome in Fusarium asiaticum. Front. Microbiol. 2020, 11, 839. [Google Scholar] [CrossRef]
- Zhang, D.D.; Wang, J.; Wang, D.; Kong, Z.Q.; Zhou, L.; Zhang, G.Y.; Gui, Y.J.; Li, J.J.; Huang, J.Q.; Wang, B.L.; et al. Population genomics demystifies the defoliation phenotype in the plant pathogen Verticillium dahliae. New Phytol. 2019, 222, 1012–1029. [Google Scholar] [CrossRef] [Green Version]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 genes. Science 1996, 274, 546–567. [Google Scholar] [CrossRef] [Green Version]
- Green, E.D. Strategies for the systematic sequencing of complex genomes. Nat. Rev. 2001, 2, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Galagan, J.E.; Henn, M.R.; Ma, L.J.; Cuomo, C.A.; Birren, B. Genomics of the fungal kingdom: Insights into eukaryotic biology. Genome Res. 2005, 15, 1620–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulski, J.K. Next-Generation Sequencing—An. Overview of the History, Tools, and “Omic” Applications. Next Generation Sequencing–Advances, Applications and Challenges; Kulski, J.K., Ed.; InTech: London, UK, 2016; pp. 3–60. [Google Scholar]
- Kozarewa, I.; Ning, Z.; Quail, M.A.; Sanders, M.J.; Berriman, M.; Turner, D.J. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G + C)-biased genomes. Nat. Methods 2009, 6, 291–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haridas, S.; Breuill, C.; Bohlmann, J.; Hsiang, T. A biologist’s guide to de novo genome assembly using next-generation sequence data: A test with fungal genomes. J. Microbiol. Met. 2011, 86, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Di Ventra, M.; Garaj, S.; Hibbs, A.; Huang, X.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef]
- Pushkarev, D.; Neff, N.F.; Quake, S.R. Single-molecule sequencing of an individual human genome. Nat. Biotechnol. 2009, 27, 847–850. [Google Scholar] [CrossRef]
- Deamer, D.; Akeson, M.; Branton, D. Three decades of nanopore sequencing. Nat. Biotechnol. 2016, 34, 518–524. [Google Scholar] [CrossRef]
- Branton, D.; Deamer, D.W.; Marziali, A.; Bayley, H.; Benner, S.A.; Butler, T.; Di Ventra, M.; Garaj, S.; Hibbs, A.; Huang, X.; et al. The potential and challenges of nanopore sequencing. Nat. Biotechnol. 2008, 27, 1146–1153. [Google Scholar] [CrossRef]
- Derrington, I.M.; Butler, T.Z.; Collins, M.D.; Manrao, E.; Pavlenok, M.; Niederweis, M.; Gundlach, J.H. Nanopore DNA sequencing with MspA. Proc. Nat. Acad. Sci. USA 2010, 107, 16060–16065. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef] [Green Version]
- Manrao, E.A.; Derrington, I.M.; Laszlo, A.H.; Langford, K.W.; Hopper, M.K.; Gillgren, N.; Pavlenok, M.; Niederweis, M.; Gundlach, J.H. Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase. Nat. Biotech. 2012, 30, 349–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The third revolution in sequencing technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, M.; Motooka, D.; Gotoh, K.; Imai, T.; Yoshitake, K.; Goto, N.; Iida, T.; Yasunaga, T.; Horii, T.; Arakawa, K.; et al. Performance comparison of second-and third-generation sequencers using a bacterial genome with two chromosomes. BMC Genom. 2014, 15, 699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenjaroenpun, P.; Wongsurawat, T.; Pereira, R.; Patumcharoenpol, P.; Ussery, D.W.; Nielsen, J.; Nookaew, I. Complete genomic and transcriptional landscape analysis using third-generation sequencing: A case study of Saccharomyces cerevisiae CEN. PK113-7D. Nucleic Acids 2018, 46, e38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutreux, F.; Da Silva, C.; d’Agata, L.; Couloux, A.; Gay, E.J.; Istace, B.; Lapalu, N.; Lemainque, A.; Linglin, J.; Noel, B.; et al. De novo assembly and annotation of three Leptosphaeria genomes using Oxford Nanopore MinION sequencing. Sci. Data 2018, 5, 180235. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 12 November 2020).
- Jaklitsch, W.; Baral, H.-O.; Lücking, R.; Lumbsch, T.H. Ascomycota. In Syllabus of Plant Families, 23rd ed.; Frey, W., Ed.; Borntraeger Science Publishers: Stuttgart, Germany, 2016. [Google Scholar]
- Sohn, J.I.; Nam, J.W. The present and future of de novo whole-genome assembly. Briefings Bioinform. 2018, 19, 23–40. [Google Scholar]
- Miller, J.R.; Koren, S.; Sutton, G. Assembly algorithms for next-generation sequencing data. Genomics 2010, 95, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Chen, J.; Yang, Y.; Tang, Y.; Shang, J.; Shen, B. A practical comparison of de novo genome assembly software tools for next-generation sequencing technologies. PLoS ONE 2011, 6, e17915. [Google Scholar] [CrossRef]
- Wajid, B.; Serpedin, E. Review of general algorithmic features for genome assemblers for next generation sequencers. Genom. Proteom. Bioinform. 2012, 10, 58–73. [Google Scholar] [CrossRef] [Green Version]
- Del Angel, V.D.; Hjerde, E.; Sterck, L.; Capella-Gutierrez, S.; Notredame, C.; Pettersson, O.V.; Amselem, J.; Bouri, L.; Bocs, S.; Klopp, C.; et al. Ten steps to get started in genome assembly and annotation. F1000Research 2018, 7, ELIXIR-148. [Google Scholar] [CrossRef] [Green Version]
- Mohanta, T.K.; Bae, H. The diversity of fungal genome. Biol. Proced. Online 2015, 17, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 2047–2217. [Google Scholar] [CrossRef]
- Koren, S.; Phillippy, A.M. One chromosome, one contig: Complete microbial genomes from long-read sequencing and assembly. Curr. Op. Microbiol. 2015, 23, 110–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef] [Green Version]
- Boetzer, M.; Pirovano, W. SSPACE-LongRead: Scaffolding bacterial draft genomes using long read sequence information. BMC Bioinform. 2014, 15, 211. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Antipov, D.; Korobeynikov, A.; McLean, J.S.; Pevzner, P.A. hybridSPAdes: An algorithm for hybrid assembly of short and long reads. Bioinformatics 2016, 32, 1009–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef] [Green Version]
- Salzberg, S.L. Next-generation genome annotation: We still struggle to get it right. Genome Biol. 2019, 20, 92. [Google Scholar] [CrossRef] [Green Version]
- Sczyrba, A.; Hofmann, P.; Belmann, P.; Koslicki, D.; Janssen, S.; Dröge, J.; Gregor, I.; Majda, S.; Fiedler, J.; Dahms, E.; et al. Critical assessment of metagenome interpretation—A benchmark of metagenomics software. Nat. Methods 2017, 14, 1063–1071. [Google Scholar] [CrossRef] [Green Version]
- Strous, M.; Kraft, B.; Bisdorf, R.; Tegetmeyer, H. The binning of metagenomic contigs for microbial physiology of mixed cultures. Front. Microbiol. 2012, 3, 410. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.W.; Tang, Y.H.; Tringe, S.G.; Simmons, B.A.; Singer, S.W. MaxBin: An automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2014, 2, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donovan, P.D.; Gonzalez, G.; Higgins, D.G.; Butler, G.; Ito, K. Identification of fungi in shotgun metagenomics datasets. PLoS ONE 2018, 13, e0192898. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castanera, R.; Lopez-Varas, L.; Borgognone, A.; LaButti, K.; Lapidus, A.; Schmutz, J.; Grimwood, J.; Perez, G.; Pisabarro, A.G.; Grigoriev, I.V.; et al. Transposable elements versus the fungal genome: Impact on whole-genome architecture and transcriptional profiles. PLoS Genetics 2016, 12, e1006108. [Google Scholar] [CrossRef] [Green Version]
- Quesneville, H.; Bergman, C.M.; Andrieu, O.; Autard, D.; Nouaud, D.; Ashburner, M.; Anxolabehere, D. Combined evidence annotation of transposable elements in genome sequences. PLoS Comput. Biol. 2005, 1, e22. [Google Scholar] [CrossRef]
- Yandell, M.; Ence, D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genetics 2012, 13, 329–342. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Korf, I.; Robb, S.M.; Parra, G.; Ross, E.; Moore, B. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008, 18, 188–196. [Google Scholar] [CrossRef] [Green Version]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER1: Unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 2016, 32, 767–769. [Google Scholar] [CrossRef]
- Lomsadze, A.; Burns, P.D.; Borodovsky, M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 2014, 42, e119. [Google Scholar] [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [Green Version]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, A.C.; Hane, J.K.; Ellwood, S.R.; Oliver, R.P. CodingQuarry: Highly accurate hidden Markov model gene prediction in fungal genomes using RNA-seq transcripts. BMC Genom. 2015, 16, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekpinar, A.D.; Kalmer, A. Utility of various molecular markers in fungal identification and phylogeny. Nova Hedwigia 2019, 109, 187–224. [Google Scholar] [CrossRef]
- McCarthy, C.G.; Fitzpatrick, D.A. Multiple approaches to phylogenomic reconstruction of the fungal kingdom. Adv. Genet. 2017, 100, 211–266. [Google Scholar]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Zdobnov, E.M.; Tegenfeldt, F.; Kuznetsov, D.; Waterhouse, R.M.; Simao, F.A.; Ioannidis, P.; Seppey, M.; Loetscher, A.; Kriventseva, E.V. OrthoDB v9. 1: Cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 2017, 45, D744–D749. [Google Scholar] [CrossRef]
- Jeffroy, O.; Brinkmann, H.; Delsuc, F.; Philippe, H. Phylogenomics: The beginning of incongruence? Trends Genet. 2006, 22, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Gribaldo, S.; Philippe, H. Ancient phylogenetic relationships. Theoret. Pop. Biol. 2002, 61, 391–408. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Lemey, P. Assessing substitution saturation with DAMBE. In The Phylogenetic Handbook: A Practical Approach to Phylogenetic Analysis and Hypothesis Testing; Lemey, P., Salemi, M., Vandamme, A., Eds.; Cambridge University Press: Cambridge, UK, 2009; pp. 615–630. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirarab, S.; Reaz, R.; Bayzid, M.S.; Zimmermann, T.; Swenson, M.S.; Warnow, T. ASTRAL: Genome-scale coalescent-based species tree estimation. Bioinformatics 2014, 30, i541–i548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delsuc, F.; Brinkmann, H.; Philippe, H. Phylogenomics and the reconstruction of the tree of life. Nat. Rev. Genet. 2005, 6, 361–375. [Google Scholar] [CrossRef]
- Bernard, G.; Chan, C.X.; Chan, Y.B.; Chua, X.Y.; Cong, Y.; Hogan, J.M.; Maetschke, S.R.; Ragan, M.A. Alignment-free inference of hierarchical and reticulate phylogenomic relationships. Briefings Bioinform. 2019, 20, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Marcais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Qi, J.; Luo, H.; Hao, B. CVTree: A phylogenetic tree reconstruction tool based on whole genomes. Nucleic Acids Res. 2004, 32, W45–W47. [Google Scholar] [CrossRef] [Green Version]
- Yi, H.; Jin, L. Co-phylog: An assembly-free phylogenomic approach for closely related organisms. Nucleic Acids Res. 2013, 41, e75. [Google Scholar] [CrossRef]
- Wan, L.; Reinert, G.; Sun, F.; Waterman, M.S. Alignment-free sequence comparison (II): Theoretical power of comparison statistics. J. Comp. Biol. 2010, 17, 1467–1490. [Google Scholar] [CrossRef]
- Bejerano, G.; Pheasant, M.; Makunin, I.; Stephen, S.; Kent, W.J.; Mattick, J.S.; Haussler, D. Ultraconserved elements in the human genome. Science 2004, 304, 1321–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.; Brasileiro, A.C.M.; Souza, D.S.L.; Romano, E.; Campos, M.A.; Grossi-De-Sá, M.F.; Silva, M.S.; Franco, O.L.; Fragoso, R.R.; Bevitori, R.; et al. Plant-pathogen interactions: What is proteomics telling us? FEBS J. 2008, 275, 3731–3746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, S.V.; Fonseca, F.L.; Rodrigues, M.L.; Mundodi, V.; Abi-Chacra, E.; Winters, M.S.; Alderete, J.F.; de Almeida Soares, C.M. Paracoccidioides brasiliensis enolase is a surface protein that binds plasminogen and mediates interaction of yeast forms with host cells. Infect. Immun. 2010, 78, 4040–4050. [Google Scholar] [CrossRef] [Green Version]
- Kniemeyer, O.; Schmidt, A.D.; Vödisch, M.; Wartenberg, D.; Brakhage, A. a Identification of virulence determinants of the human pathogenic fungi Aspergillus fumigatus and Candida albicans by proteomics. Int. J. Med. Microbiol. 2011, 301, 368–377. [Google Scholar] [CrossRef]
- Ashwin, N.M.R.; Barnabas, L.; Ramesh Sundar, A.; Malathi, P.; Viswanathan, R.; Masi, A.; Agrawal, G.K.; Rakwal, R. Advances in proteomic technologies and their scope of application in understanding plant–pathogen interactions. J. Plant. Biochem. Biotechnol. 2017, 26, 371–386. [Google Scholar] [CrossRef]
- Gonzalez-Fernandez, R.; Jorrin-Novo, J.V. Contribution of proteomics to the study of plant pathogenic fungi. J. Proteome Res. 2012, 11, 3–16. [Google Scholar] [CrossRef]
- Lau, A.F.; Drake, S.K.; Calhoun, L.B.; Henderson, C.M.; Zelazny, A.M. Development of a clinically comprehensive database and a simple procedure for identification of molds from solid media by matrix-assisted laser desorption ionization-Time of flight mass spectrometry. J. Clin. Microbiol. 2013, 51, 828–834. [Google Scholar] [CrossRef] [Green Version]
- Bhadauria, V.; Zhao, W.; Wang, L.; Zhang, Y.; Liu, J.; Yang, J.; Kong, L.; Peng, Y.-L. Advances in fungal proteomics. Microbiol. Res. 2007, 162, 193–200. [Google Scholar] [CrossRef]
- De Oliveira, J.M.P.F.; De Graaff, L.H.; Miguel, J.; De Oliveira, P.F.; De Graaff, L.H. Proteomics of industrial fungi: Trends and insights for biotechnology. Appl. Microbiol. Biotechnol. 2011, 89, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Banks, C.; Lakshminarasimhan, M.; Washburn, M.P. Shotgun Proteomics. eLS 2014, 1156, 1–7. [Google Scholar] [CrossRef]
- Marcotte, E.M. How do shotgun proteomics algorithms identify proteins? Nat. Biotechnol. 2007, 25, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Sinitcyn, P.; Daniel Rudolph, J.; Cox, J. Computational methods for understanding Mass Spectrometry–Based Shotgun proteomics data. Annu. Rev. Biomed. Data Sci. 2018, 1, 207–234. [Google Scholar] [CrossRef]
- Kim, Y.; Nandakumar, M.P.; Marten, M.R. Proteomics of filamentous fungi. Trends Biotechnol. 2007, 25, 395–400. [Google Scholar] [CrossRef]
- Krishnaswamy, A.; Barnes, N.; Lotlikar, N.P.; Damare, S.R. An improved method for protein extraction from minuscule quantities of fungal biomass. Indian J. Microbiol. 2019, 59, 100–104. [Google Scholar] [CrossRef]
- Champer, J.; Ito, J.; Clemons, K.; Stevens, D.; Kalkum, M. Proteomic analysis of pathogenic fungi reveals highly expressed conserved cell wall proteins. J. Fungi 2016, 2, 6. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, M.; Wariishi, H. Development of a sample preparation method for fungal proteomics. FEMS Microbiol. Lett. 2005, 247, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Jami, M.-S.; Barreiro, C.; García-Estrada, C.; Martín, J.-F. Proteome analysis of the penicillin producer Penicillium chrysogenum: Characterization of protein changes during the industrial strain improvement. Mol. Cell. Proteomics 2010, 9, 1182–1198. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Fernandez, R.; Prats, E.; Jorrin-Novo, J.V. Proteomics of plant pathogenic fungi. J. Biomed. Biotechnol. 2010, 2010, 1–36. [Google Scholar] [CrossRef] [Green Version]
- Bianco, L.; Perrotta, G. Methodologies and perspectives of proteomics applied to filamentous fungi: From sample preparation to secretome analysis. Int. J. Mol. Sci. 2015, 16, 5803–5829. [Google Scholar] [CrossRef] [Green Version]
- Crichton, P.G.; Harding, M.; Ruprecht, J.J.; Lee, Y.; Kunji, E.R.S. Lipid, detergent, and coomassie blue G-250 affect the migration of small membrane proteins in blue native gels: Mitochondrial carriers migrate as monomers not dimers. J. Biol. Chem. 2013, 288, 22163–22173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenman, H.C.; Casadevall, A. Synthesis and assembly of fungal melanin. Appl. Microbiol. Biotechnol. 2012, 93, 931–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaves, E.G.A.; Parente-Rocha, J.A.; Baeza, L.C.; Araújo, D.S.; Borges, C.L.; de Oliveira, M.A.P.; de Soares, C.M.A. Proteomic analysis of Paracoccidioides brasiliensis during Infection of alveolar macrophages primed or not by interferon-gamma. Front. Microbiol. 2019, 10, 96. [Google Scholar] [CrossRef] [PubMed]
- Do Amaral, C.C.; Fernandes, G.F.; Rodrigues, A.M.; Burger, E.; De Camargo, Z.P. Proteomic analysis of Paracoccidioides brasiliensis complex isolates: Correlation of the levels of differentially expressed proteins with in vivo virulence. PLoS ONE 2019, 14, e0218013. [Google Scholar] [CrossRef]
- Muthamil, S.; Prasath, K.G.; Priya, A.; Precilla, P.; Pandian, S.K. Global proteomic analysis deciphers the mechanism of action of plant derived oleic acid against Candida albicans virulence and biofilm formation. Sci. Rep. 2020, 10, 5113. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Yin, Q. Comprehensive proteomic analysis of the wheat pathogenic fungus Zymoseptoria tritici. Proteomics 2015, 16, 98–101. [Google Scholar] [CrossRef]
- Chen, X.L.; Liu, C.; Tang, B.; Ren, Z.; Wang, G.L.; Liu, W. Quantitative proteomics analysis reveals important roles of N-glycosylation on ER quality control system for development and pathogenesis in Magnaporthe oryzae. PLoS Pathog. 2020, 16, e1008355. [Google Scholar] [CrossRef]
- Wang, J.; Li, L.; Chai, R.; Zhang, Z.; Qiu, H.; Mao, X.; Hao, Z.; Wang, Y.; Sun, G. Succinyl-proteome profiling of Pyricularia oryzae, a devastating phytopathogenic fungus that causes rice blast disease. Sci. Rep. 2019, 9, 3490. [Google Scholar] [CrossRef] [Green Version]
- Cagas, S.E.; Jain, M.R.; Li, H.; Perlin, D.S. Profiling the Aspergillus fumigatus proteome in response to caspofungin. Antimicrob. Agents Chemother. 2011, 55, 146–154. [Google Scholar] [CrossRef] [Green Version]
- Benndorf, D.; Müller, A.; Bock, K.; Manuwald, O.; Herbarth, O.; Von Bergen, M. Identification of spore allergens from the indoor mould Aspergillus versicolor. Allergy Eur. J. Allergy Clin. Immunol. 2008, 63, 454–460. [Google Scholar] [CrossRef]
- Shi, L.; Ge, B.; Wang, J.; Liu, B.; Ma, J.; Wei, Q.; Zhang, K. ITRAQ-based proteomic analysis reveals the mechanisms of Botrytis cinerea controlled with Wuyiencin. BMC Microbiol. 2019, 19, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aumer, T.; Voisin, S.N.; Knobloch, T.; Landon, C.; Bulet, P. Impact of an antifungal insect defensin on the proteome of the phytopathogenic fungus Botrytis cinerea. J. Proteome Res. 2020, 19, 1131–1146. [Google Scholar] [CrossRef] [PubMed]
- Anjo, S.I.; Figueiredo, F.; Fernandes, R.; Manadas, B.; Oliveira, M. A proteomic and ultrastructural characterization of Aspergillus fumigatus’ conidia adaptation at different culture ages. J. Proteomics 2017, 161, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, Q.; Li, Y.; Qiao, L.; Pang, Q.; Huang, B. iTRAQ-based quantitative proteomic analysis of conidia and mycelium in the filamentous fungus Metarhizium robertsii. Fungal Biol. 2018, 122, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Xi, L.; Xu, X.; Liu, W.; Li, X.; Liu, Y.; Li, M.; Zhang, J.; Li, M. Differentially expressed proteins of pathogenic Penicillium marneffei in yeast and mycelial phases. J. Med. Microbiol. 2007, 56, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Tse, H.; Chan, J.S.Y.; Zhou, A.C.; Curreem, S.O.T.; Lau, C.C.Y.; Yuen, K.Y.; Woo, P.C.Y. Proteome profiling of the dimorphic fungus Penicillium marneffei extracellular proteins and identification of glyceraldehyde-3-phosphate dehydrogenase as an important adhesion factor for conidial attachment. FEBS J. 2013, 280, 6613–6626. [Google Scholar] [CrossRef]
- Otun, S.; Ntushelo, K. Proteomic analysis of the phytogenic fungus Sclerotinia sclerotiorum. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2020, 1144, 122053. [Google Scholar] [CrossRef]
- Parente, A.F.A.; de Rezende, T.C.V.; de Castro, K.P.; Bailão, A.M.; Parente, J.A.; Borges, C.L.; Silva, L.P.; de Soares, C.M.A. A proteomic view of the response of Paracoccidioides yeast cells to zinc deprivation. Fungal Biol. 2013, 117, 399–410. [Google Scholar] [CrossRef]
- De Arruda Grossklaus, D.; Bailão, A.M.; Vieira Rezende, T.C.; Borges, C.L.; de Oliveira, M.A.P.; Parente, J.A.; de Almeida Soares, C.M.; Cristina, T.; Rezende, V.; De Arruda, D.; et al. Response to oxidative stress in Paracoccidioides yeast cells as determined by proteomic analysis. Microbes Infect. 2013, 15, 347–364. [Google Scholar] [CrossRef]
- Da Rodrigues, L.N.S.; de Brito, W.A.; Parente, A.F.A.; Weber, S.S.; Bailão, A.M.; Casaletti, L.; Borges, C.L.; de Soares, C.M.A. Osmotic stress adaptation of Paracoccidioides lutzii, Pb01, monitored by proteomics. Fungal Genet. Biol. 2016, 95, 13–23. [Google Scholar] [CrossRef]
- De Lima, P.S.; Casaletti, L.; Bailão, A.M.; de Vasconcelos, A.T.R.; da Fernandes, G.R.; de Soares, C.M.A. Transcriptional and proteomic responses to carbon starvation in Paracoccidioides. PLoS Negl. Trop. Dis. 2014, 8, e2855. [Google Scholar] [CrossRef]
- Parente, A.F.A.; Naves, P.E.C.; Pigosso, L.L.; Casaletti, L.; McEwen, J.G.; Parente-Rocha, J.A.; Soares, C.M.A. The response of Paracoccidioides spp. to nitrosative stress. Microbes Infect. 2015, 17, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Parente-Rocha, J.A.; Parente, A.F.A.; Baeza, L.C.; Bonfim, S.M.R.C.; Hernandez, O.; McEwen, J.G.; Bailão, A.M.; Taborda, C.P.; Borges, C.L.; De Almeida Soares, C.M. Macrophage interaction with Paracoccidioides brasiliensis yeast cells modulates fungal metabolism and generates a response to oxidative stress. PLoS ONE 2015, 10, e0137619. [Google Scholar] [CrossRef] [Green Version]
- Tesei, D.; Marzban, G.; Marchetti-Deschmann, M.; Tafer, H.; Arcalis, E.; Sterflinger, K. Proteome of tolerance fine-tuning in the human pathogen black yeast Exophiala dermatitidis. J. Proteomics 2015, 128, 39–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.; Du, B.; Zhu, F.; Gao, Y.; Li, J. Proteomic analysis of Aspergillus niger 3.316 under heat stress. Microbiol. Open 2020, 9, e1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, Y.; Robertson, S.L.; Parker, J.; Muddiman, D.C.; Dean, R.A. Comparative proteomic analysis between nitrogen supplemented and starved conditions in Magnaporthe oryzae. Proteome Sci. 2017, 15, 20. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, M.D.; Beynon, R.J.; Gethings, L.A.; Claydon, A.J.; Langridge, J.I.; Vissers, J.P.C.; Brown, A.J.P.; Hammond, D.E. Specificity of the osmotic stress response in Candida albicans highlighted by quantitative proteomics. Sci. Rep. 2018, 8, 14492. [Google Scholar] [CrossRef] [Green Version]
- Ingle, S.; Kazi, R.; Patil, R.; Zore, G. Proteome analysis of Candida albicans cells undergoing chlamydosporulation. J. Proteins Proteom. 2019, 10, 269–290. [Google Scholar] [CrossRef]
- Krijger, J.-J.; Thon, M.R.; Deising, H.B.; Wirsel, S.G.R. Compositions of fungal secretomes indicate a greater impact of phylogenetic history than lifestyle adaptation. BMC Genom. 2014, 15, 722. [Google Scholar] [CrossRef] [Green Version]
- Krishnamoorthy, A.S.; Pravin, I.A.; Priyadharshini, B.; Manikandan, R. Perspectives of secretome proteomics in filamentous fungi. Madras Agric. 2017, 104, 107–113. [Google Scholar]
- Vallejo, M.; Nakayasu, E.; Matsuo, A.; Sobreira, T.P.; Longo, L.G.; Ganiko, L.; Almeida, I.; Puccia, R. Vesicle and vesicle-free extracellular proteome of Paracoccidioides brasiliensis: Comparative analysis with other pathogenic fungi. J. Proteome Res. 2012, 11, 1676–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joffe, L.S.; Nimrichter, L.; Rodrigues, M.L.; Del Poeta, M. Potential roles of fungal extracellular vesicles during infection. mSphere 2016, 1, e00099-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellen, A.F.; Albers, S.V.; Huibers, W.; Pitcher, A.; Hobel, C.F.V.; Schwarz, H.; Folea, M.; Schouten, S.; Boekema, E.J.; Poolman, B.; et al. Proteomic analysis of secreted membrane vesicles of archaeal Sulfolobus species reveals the presence of endosome sorting complex components. Extremophiles 2009, 13, 67–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapp, K.; Schrempf, S.; Lemberg, M.K.; Dobberstein, B. Post-targeting functions of signal peptides. In Madame Curie Bioscience Database [Internet]; Zimmermann, R., Ed.; Landes Bioscience: Austin, TX, USA, 2009; pp. 2000–2013. [Google Scholar]
- Pombeiro-Sponchiado, S.R.; Sousa, G.S.; Andrade, J.C.R.; Lisboa, H.F.; Goncalves, R.C.R. Production of melanin pigment by fungi and its biotechnological applications. World’s Larg. Sci. Technol. Med. Open Access Book Publ. 2017. [Google Scholar] [CrossRef] [Green Version]
- Simpson, R.J.; Jensen, S.S.; Lim, J.W.E. Proteomic profiling of exosomes: Current perspectives. Proteomics 2008, 8, 4083–4099. [Google Scholar] [CrossRef]
- Rodrigues, M.L.; Nakayasu, E.S.; Oliveira, D.L.; Nimrichter, L.; Nosanchuk, J.D.; Almeida, I.C.; Casadevall, A. Extracellular vesicles produced by Cryptococcus neoformans contain protein components associated with virulence. Eukaryot. Cell 2008, 7, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Furi, I.; Momen-Heravi, F.; Szabo, G. Extracellular vesicle isolation: Present and future. Ann. Transl. Med. 2017, 5, 263. [Google Scholar] [CrossRef] [Green Version]
- Girard, V.; Dieryckx, C.; Job, C.; Job, D. Secretomes: The fungal strike force. Proteomics 2013, 13, 597–608. [Google Scholar] [CrossRef]
- Espino, J.J.; Gutierrez-Sannchez, G.; Brito, N.; Shah, P.; Ron, O.; Gonzalez, C. The Botrytis cinerea early secretome. Proteomics 2010, 10, 3020–3034. [Google Scholar] [CrossRef] [Green Version]
- De Vallée, A.; Bally, P.; Bruel, C.; Chandat, L.; Choquer, M.; Dieryckx, C.; Dupuy, J.W.; Kaiser, S.; Latorse, M.P.; Loisel, E.; et al. A similar secretome disturbance as a hallmark of non-pathogenic Botrytis cinerea ATMT-mutants? Front. Microbiol. 2019, 10, 2829. [Google Scholar] [CrossRef]
- Monteiro, V.N.; do Nascimento Silva, R.; Steindorff, A.S.; Costa, F.T.; Noronha, E.F.; Ricart, C.A.O.; de Sousa, M.V.; Vainstein, M.H.; Ulhoa, C.J. New insights in Trichoderma harzianum antagonism of fungal plant pathogens by secreted protein analysis. Curr. Microbiol. 2010, 61, 298–305. [Google Scholar] [CrossRef]
- Li, T.; Wu, Q.; Wang, Y.; John, A.; Qu, H.; Gong, L.; Duan, X.; Zhu, H.; Yun, Z.; Jiang, Y. Application of proteomics for the investigation of the effect of initial pH on pathogenic mechanisms of Fusarium proliferatum on banana fruit. Front. Microbiol. 2017, 8, 2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; An, B.; Guo, Y.; Hou, X.; Luo, H.; He, C.; Wang, Q. Label free proteomics and systematic analysis of secretome reveals effector candidates regulated by SGE1 and FTF1 in the plant pathogen Fusarium oxysporum f. sp. cubense tropical race 4. BMC Genom. 2020, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Muria-Gonzalez, M.J.; Zulak, K.G.; Allegaert, E.; Oliver, R.P.; Ellwood, S.R. Profile of the in vitro secretome of the barley net blotch fungus, Pyrenophora teres f. teres. Physiol. Mol. Plant. Pathol. 2020, 109, 101451. [Google Scholar] [CrossRef]
- Wang, J.; Lovett, B.; St Leger, R.J. The secretome and chemistry of Metarhizium; a genus of entomopathogenic fungi. Fungal Ecol. 2019, 38, 7–11. [Google Scholar] [CrossRef]
- Dionisio, G.; Kryger, P.; Steenberg, T. Label-free differential proteomics and quantification of exoenzymes from isolates of the entomopathogenic fungus Beauveria bassiana. Insects 2016, 7, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beys-Da-Silva, W.O.; Santi, L.; Berger, M.; Calzolari, D.; Passos, D.O.; Guimarães, J.A.; Moresco, J.J.; Yates, J.R. Secretome of the biocontrol agent Metarhizium anisopliae induced by the cuticle of the cotton pest Dysdercus peruvianus reveals new insights into infection. J. Proteome Res. 2014, 13, 2282–2296. [Google Scholar] [CrossRef]
- Rasheed, M.; Kumar, N.; Kaur, R. Global secretome characterization of the pathogenic yeast Candida glabrata. J. Proteome Res. 2020, 19, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhang, L.; Zou, H.; Wang, L. Secretome profiling reveals temperature-dependent growth of Aspergillus fumigatus. Sci. China Life Sci. 2018, 61, 578–592. [Google Scholar] [CrossRef]
- Oliveira, D.L.; Nakayasu, E.S.; Joffe, L.S.; Guimarães, A.J.; Sobreira, T.J.P.; Nosanchuk, J.D.; Cordero, R.J.B.; Frases, S.; Casadevall, A.; Almeida, I.C.; et al. Biogenesis of extracellular vesicles in yeast. Commun. Integr. Biol. 2010, 3, 533–535. [Google Scholar] [CrossRef]
- Silva, V.K.A.; Rodrigues, M.L.; May, R.C. Deciphering fungal extracellular vesicles: From cell biology to pathogenesis. Curr. Clin. Microbiol. Rep. 2019, 6, 89–97. [Google Scholar] [CrossRef]
- Bleackley, M.R.; Dawson, C.S.; Anderson, M.A. Fungal extracellular vesicles with a focus on proteomic analysis. Proteomics 2019, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, P.C.; Nakayasu, E.S.; Rodrigues, M.L.; Frases, S.; Casadevall, A.; Zancope-Oliveira, R.M.; Almeida, I.C.; Nosanchuk, J.D. Vesicular transport in Histoplasma capsulatum: An effective mechanism for trans-cell wall transfer of proteins and lipids in ascomycetes. Cell. Microbiol. 2008, 10, 1695–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimrichter, L.; De Souza, M.M.; Del Poeta, M.; Nosanchuk, J.D.; Joffe, L.; Tavares, P.D.M.; Rodrigues, M.L. Extracellular vesicle-associated transitory cell wall components and their impact on the interaction of fungi with host cells. Front. Microbiol. 2016, 7, 1034. [Google Scholar] [CrossRef] [Green Version]
- Karkowska-Kuleta, J.; Kozik, A. Moonlighting proteins as virulence factors of pathogenic fungi, parasitic protozoa and multicellular parasites. Mol. Oral Microbiol. 2014, 29, 270–283. [Google Scholar] [CrossRef]
- Cleare, L.G.; Zamith, D.; Heyman, H.M.; Couvillion, S.P.; Nimrichter, L.; Rodrigues, M.L.; Nakayasu, E.S.; Nosanchuk, J.D. Media Matters! Alterations in the loading and release of Histoplasma capsulatum extracellular vesicles in response to different nutritional milieus. Cell. Microbiol. 2020, 22, e13217. [Google Scholar] [CrossRef]
- Ikeda, M.A.K.; De Almeida, J.R.F.; Jannuzzi, G.P.; Cronemberger-Andrade, A.; Torrecilhas, A.C.T.; Moretti, N.S.; Da Cunha, J.P.C.; De Almeida, S.R.; Ferreira, K.S. Extracellular vesicles from sporothrix brasiliensisare an important virulence factor that induce an increase in fungal burden in experimental sporotrichosis. Front. Microbiol. 2018, 9, 2286. [Google Scholar] [CrossRef]
- De Groot, P.W.J.; de Boer, A.D.; Brandt, B.W.; Valentín, E. 5 The Ascomycetous Cell Wall: From a proteomic perspective. Growth Differ. Sex. 2016, 1, 81–101. [Google Scholar] [CrossRef]
- Karkowska-Kuleta, J.; Kozik, A. Cell wall proteome of pathogenic fungi. Acta Biochim. Pol. 2015, 62, 339–351. [Google Scholar] [CrossRef] [Green Version]
- Pitarch, A.; Nombela, C.; Gil, C. Cell wall fractionation for yeast and fungal proteomics. Methods Mol. Biol. 2008, 425, 217–239. [Google Scholar] [CrossRef]
- Klis, F.M.; de Jong, M.; Brul, S.; de Groot, P.W.J. Extraction of cell surface-associated proteins from living yeast cells. Yeast 2007, 24, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Vialás, V.; Perumal, P.; Gutierrez, D.; Ximénez-Embún, P.; Nombela, C.; Gil, C.; Chaffin, W.L. Cell surface shaving of Candida albicans biofilms, hyphae, and yeast form cells. Proteomics 2012, 12, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Longo, L.V.G.; da Cunha, J.P.C.; Sobreira, T.J.P.; Puccia, R. Proteome of cell wall-extracts from pathogenic Paracoccidioides brasiliensis: Comparison among morphological phases, isolates, and reported fungal extracellular vesicle proteins. EuPA Open Proteom. 2014, 3, 216–228. [Google Scholar] [CrossRef] [Green Version]
- Yin, Q.Y.; de Groot, P.W.J.; de Koster, C.G.; Klis, F.M. Mass spectrometry-based proteomics of fungal wall glycoproteins. Trends Microbiol. 2008, 16, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Hernáez, M.L.; Ximénez-Embún, P.; Martínez-Gomariz, M.; Gutiérrez-Blázquez, M.D.; Nombela, C.; Gil, C. Identification of Candida albicans exposed surface proteins in vivo by a rapid proteomic approach. J. Proteom. 2010, 73, 1404–1409. [Google Scholar] [CrossRef]
- Puccia, R.; Vallejo, M.C.; Matsuo, A.L.; Longo, L.V.G. The Paracoccidioides cell wall: Past and present layers toward understanding interaction with the host. Front. Microbiol. 2011, 2, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araújo, D.S.; de Sousa Lima, P.; Baeza, L.C.; Parente, A.F.A.; Melo Bailão, A.; Borges, C.L.; de Almeida Soares, C.M. Employing proteomic analysis to compare Paracoccidioides lutzii yeast and mycelium cell wall proteins. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1304–1314. [Google Scholar] [CrossRef]
- Félix-Contreras, C.; Alba-Fierro, C.A.; Ríos-Castro, E.; Luna-Martínez, F.; Cuéllar-Cruz, M.; Ruiz-Baca, E. Proteomic analysis of Sporothrix schenckii cell wall reveals proteins involved in oxidative stress response induced by menadione. Microb. Pathog. 2020, 141, 103987. [Google Scholar] [CrossRef]
- Awad, A.; El Khoury, P.; Wex, B.; Khalaf, R.A. Proteomic analysis of a Candida albicans pga1 Null Strain. EuPA Open Proteom. 2018, 18, 1–6. [Google Scholar] [CrossRef]
- Voltersen, V.; Blango, M.G.; Herrmann, S.; Schmidt, F.; Heinekamp, T.; Strassburger, M.; Krüger, T.; Bacher, P.; Lother, J.; Weiss, E.; et al. Proteome analysis reveals the conidial surface protein CcpA essential for virulence of the pathogenic fungus Aspergillus fumigatus. MBio 2018, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- De Carrion, S.J.; Leal, S.M.; Ghannoum, M.A.; Aimanianda, V.; Latgé, J.-P.; Pearlman, E. The RodA Hydrophobin on Aspergillus fumigatus spores masks dectin-1– and dectin-2–dependent responses and enhances fungal survival in vivo. J. Immunol. 2013, 191, 2581–2588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Champer, J.; Diaz-Arevalo, D.; Champer, M.; Hong, T.B.; Wong, M.; Shannahoff, M.; Ito, J.I.; Clemons, K.V.; Stevens, D.A.; Kalkum, M. Protein targets for broad-spectrum mycosis vaccines: Quantitative proteomic analysis of Aspergillus and Coccidioides and comparisons with other fungal pathogens. Ann. N. Y. Acad. Sci. 2012, 1273, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Evilia, C. Understanding protein adaptations can help us solve real problems. Semin. Cell Dev. Biol. 2018, 84, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Marzban, G.; Tesei, D.; Sterflinger, K. A Review beyond the borders: Proteomics of microclonial black fungi and black yeasts. Nat. Sci. 2013, 5, 640–645. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, K.S.; Thomas, T. Protein Adaptation in Extremophiles. In Molecular Anatomy and Physiology of Proteins; Uversky, Ed.; Nova Biomedical Books: New York, NY, USA, 2008. [Google Scholar]
- Sterflinger, K. Black Yeasts and Meristematic Fungi: Ecology, Diversity ad Identification. In Biodiversity and Ecophysiology of Yeasts; Péter, G., Rosa, C., Eds.; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Isola, D.; Marzban, G.; Selbmann, L.; Onofri, S.; Laimer, M.; Sterflinger, K. Sample preparation and 2-DE procedure for protein expression profiling of black microcolonial fungi. Fungal Biol. 2011, 115, 971–977. [Google Scholar] [CrossRef]
- Krishnaswamy, A.; Damare, S.R. Proteomic Approach to Study Fungal Growth under Simulated Deep-Sea Conditions. 2019. Available online: http://hdl.handle.net/10603/286747 (accessed on 12 November 2020).
- Kniemeyer, O. Proteomics of eukaryotic microorganisms: The medically and biotechnologically important fungal genus Aspergillus. Proteomics 2011, 11, 3232–3243. [Google Scholar] [CrossRef]
- Jacobson, E.S. Pathogenic roles for fungal melanins. Clin. Microbiol. Rev. 2000, 13, 708–717. [Google Scholar] [CrossRef]
- Amin, S.; Rastogi, R.P.; Sonani, R.R.; Ray, A.; Sharma, R.; Madamwar, D. Bioproduction and characterization of extracellular melanin-like pigment from industrially polluted metagenomic library equipped Escherichia coli. Sci. Total Environ. 2018, 635, 323–332. [Google Scholar] [CrossRef]
- Wong, H.J.; Mohamad-Fauzi, N.; Rizman-Idid, M.; Convey, P.; Alias, S.A. Protective mechanisms and responses of micro-fungi towards ultraviolet-induced cellular damage. Polar Sci. 2019, 20, 19–34. [Google Scholar] [CrossRef]
- Breitenbach, R.; Silbernagl, D.; Toepel, J.; Sturm, H.; Broughton, W.J.; Sassaki, G.L.; Gorbushina, A.A. Corrosive extracellular polysaccharides of the rock-inhabiting model fungus Knufia petricola. Extremophiles 2018, 22, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Adav, S.S.; Li, A.A.; Manavalan, A.; Punt, P.; Sze, S.K. Quantitative iTRAQ secretome analysis of Aspergillus niger reveals novel hydrolytic enzymes. J. Proteome Res. 2010, 9, 3932–3940. [Google Scholar] [CrossRef] [PubMed]
- Tesei, D.; Marzban, G.; Zakharova, K.; Isola, D.; Selbmann, L.; Sterflinger, K. Alteration of protein patterns in black rock inhabiting fungi as a response to different temperatures. Fungal Biol. 2012, 116, 932–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakharova, K.; Tesei, D.; Marzban, G.; Dijksterhuis, J.; Wyatt, T.; Sterflinger, K. Microcolonial fungi on rocks: A life in constant drought? Mycopathologia 2013, 175, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakharova, K.; Marzban, G.; de Vera, J.-P.; Lorek, A.; Sterflinger, K. Protein patterns of black fungi under simulated Mars-like conditions. Sci. Rep. 2014, 4, 5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groenewald, M.; Boekhout, T.; Neuvéglise, C.; Gaillardin, C.; Van Dijck, P.W.M.; Wyss, M. Yarrowia lipolytica: Safety assessment of an oleaginous yeast with a great industrial potential. Crit. Rev. Microbiol. 2014, 40, 187–206. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wan, H.; Chen, H.; Fang, F.; Liu, S.; Zhou, J. Proteomic analysis of the response of α-ketoglutarate-producer Yarrowia lipolytica WSH-Z06 to environmental pH stimuli. Appl. Microbiol. Biotechnol. 2016, 100, 8829–8841. [Google Scholar] [CrossRef]
- Martínez, J.L.; Luna, C.; Ramos, J. Proteomic changes in response to potassium starvation in the extremophilic yeast Debaryomyces hansenii. FEMS Yeast Res. 2012, 12, 651–661. [Google Scholar] [CrossRef] [Green Version]
- Gunde-Cimerman, N.; Ramos, J.; Plemenitaš, A. Halotolerant and halophilic fungi. Mycol. Res. 2009, 113, 1231–1241. [Google Scholar] [CrossRef]
- Thomé-Ortiz, P.E.; Peña, A.; Ramírez, J. Monovalent cation fluxes and physiological changes of Debaryomyces hansenii grown at high concentrations of KCl and NaCl. Yeast 1998, 14, 1355–1371. [Google Scholar] [CrossRef]
- Sadaf, A.; Sinha, R.; Khare, S.K. Proteomic profiling of Sporotrichum thermophile under the effect of ionic liquids: Manifestation of an oxidative stress response. 3 Biotech. 2019, 9, 240. [Google Scholar] [CrossRef] [Green Version]
- Romsdahl, J.; Blachowicz, A.; Chiang, A.; Singh, N.K.; Stajich, J.E.; Kalkum, M.; Venkateswaran, K.J.; Wang, C. Characterization of Aspergillus niger isolated from the international space station. mSystems 2018, 3, e00112-18. [Google Scholar] [CrossRef] [Green Version]
- Checinska, A.; Probst, A.J.; Vaishampayan, P.; White, J.R.; Kumar, D.; Stepanov, V.G.; Fox, G.E.; Nilsson, H.R.; Pierson, D.L.; Perry, J.; et al. Microbiomes of the dust particles collected from the international space station and spacecraft assembly facilities. Microbiome 2015, 3, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabib, E.; Arroyo, J. How carbohydrates sculpt cells: Chemical control of morphogenesis in the yeast cell wall. Nat. Rev. Microbiol. 2013, 11, 648–655. [Google Scholar] [CrossRef]
- Kawasaki, L.; Aguirre, J. Multiple catalase genes are differentially regulated in Aspergillus nidulans. J. Bacteriol. 2001, 183, 1434–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singaravelan, N.; Grishkan, I.; Beharav, A.; Wakamatsu, K.; Ito, S.; Nevo, E. Adaptive melanin response of the soil fungus Aspergillus niger to UV radiation stress at “Evolution Canyon”, Mount Carmel, Israel. PLoS ONE 2008, 3, e2993. [Google Scholar] [CrossRef]
- Blachowicz, A.; Chiang, A.J.; Elsaesser, A.; Kalkum, M.; Ehrenfreund, P.; Stajich, J.E.; Torok, T.; Wang, C.C.C.; Venkateswaran, K. Proteomic and metabolomic characteristics of extremophilic fungi under simulated Mars conditions. Front. Microbiol. 2019, 10, 1013. [Google Scholar] [CrossRef] [PubMed]
- Firon, A.; Villalba, F.; Beffa, R.; D’Enfert, C. Identification of essential genes in the human fungal pathogen Aspergillus fumigatus by transposon mutagenesis. Eukaryot. Cell 2003, 2, 247–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, I.; Khaliq, S.; Sajid, S.; Akbar, A. Biotechnological applications of halophilic fungi: Past, present, and future. In Fungi in Extreme Environments: Ecological Role and Biotechnological Significance; Tiquia-Arashiro, S., Grube, M., Eds.; Springer: Cham, Switherlands, 2019; pp. 291–306. [Google Scholar] [CrossRef]
- Park, S.; Lee, B.; Park, K. Extremophilic Carbohydrate Active Enzymes (CAZymes). J. Nutr. Heal. Food Eng. Extrem. 2017, 7, 00230. [Google Scholar] [CrossRef] [Green Version]
- Salvato, F. Proteomics for Bioenergy Production. Plant Based Genet. Tools Biofuels Prod. 2017, 103–121. [Google Scholar] [CrossRef]
- Da Rosa-Garzon, N.G.; Laure, H.J.; Rosa, J.C.; Cabral, H. Fusarium oxysporum cultured with complex nitrogen sources can degrade agricultural residues: Evidence from analysis of secreted enzymes and intracellular proteome. Renew. Energy 2019, 133, 941–950. [Google Scholar] [CrossRef]
- Jin, M.; Gai, Y.; Guo, X.; Hou, Y.; Zeng, R. Properties and applications of extremozymes from deep-sea extremophilic microorganisms. Mar. Drugs 2019, 17, 656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sani, R.K.; Krishnaraj, R.N. Extremophilic Enzymatic Processing of Lignocellulosic Feedstocks to Bioenergy; Springer: Cham, Swizerland, 2017; ISBN 9783319546841. [Google Scholar]
- Arfi, Y.; Chevret, D.; Henrissat, B.; Berrin, J.G.; Levasseur, A.; Record, E. Characterization of salt-adapted secreted lignocellulolytic enzymes from the mangrove fungus Pestalotiopsis sp. Nat. Commun. 2013, 4, 1810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, C.; Basotra, N.; Singh, S.; Di Falco, M.; Tsang, A.; Chadha, B.S. Malbranchea cinnamomea: A thermophilic fungal source of catalytically efficient lignocellulolytic glycosyl hydrolases and metal dependent enzymes. Bioresour. Technol. 2016, 200, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Basotra, N.; Kaur, B.; Di Falco, M.; Tsang, A.; Chadha, B.S. Mycothermus thermophilus (Syn. Scytalidium thermophilum): Repertoire of a diverse array of efficient cellulases and hemicellulases in the secretome revealed. Bioresour. Technol. 2016, 222, 413–421. [Google Scholar] [CrossRef]
- Winger, A.M.; Heazlewood, J.L.; Chan, L.J.G.; Petzold, C.J.; Permaul, K.; Singh, S. Secretome analysis of the thermophilic xylanase hyper-producer Thermomyces lanuginosus SSBP cultivated on corn cobs. J. Ind. Microbiol. Biotechnol. 2014, 41, 1687–1696. [Google Scholar] [CrossRef]
- Peterson, R.; Grinyer, J.; Nevalainen, H. Secretome of the coprophilous fungus Doratomyces stemonitis C8, isolated from koala feces. Appl. Environ. Microbiol. 2011, 77, 3793–3801. [Google Scholar] [CrossRef] [Green Version]
- Tsang, A.; Butler, G.; Powlowski, J.; Panisko, E.A.; Baker, S.E. Analytical and computational approaches to define the Aspergilus niger secretome. Fungal Genet. Biol. 2009, 46, S153–S160. [Google Scholar] [CrossRef]
- Tesei, D.; Quartinello, F.; Guebitz, G.M.; Ribitsch, D.; Nöbauer, K.; Razzazi-fazeli, E.; Sterflinger, K. Shotgun proteomics reveals putative polyesterases in the secretome of the rock-inhabiting fungus Knufia chersonesos. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Nai, C.; Wong, H.Y.; Pannenbecker, A.; Broughton, W.J.; Benoit, I.; de Vries, R.P.; Gueidan, C.; Gorbushina, A. Nutritional physiology of a rock-inhabiting, model microcolonial fungus from an ancestral lineage of the Chaetothyriales (Ascomycetes). Fungal Genet. Biol. 2013, 56, 54–66. [Google Scholar] [CrossRef]
- Noack-Schönmann, S.; Bus, T.; Banasiak, R.; Knabe, N.; Broughton, W.J.; Den Dulk-Ras, H.; Hooykaas, P.J.J.; Gorbushina, A.A. Genetic transformation of Knufia petricola A95—A model organism for biofilm-material interactions. AMB Express 2014, 4, 80. [Google Scholar] [CrossRef] [Green Version]
- Lucero Camacho-Morales, R.; García-Fontana, C.; Fernández-Irigoyen, J.; Santamaría, E.; González-López, J.; Manzanera, M.; Aranda, E. Anthracene drives sub-cellular proteome-wide alterations in the degradative system of Penicillium oxalicum. Ecotoxicol. Environ. Saf. 2018, 159, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Kondratiuk, A.S.; Savchuk, O.M.; Hur, J.S. Optimization of protein extraction for lichen thalli. Mycobiology 2015, 43, 157–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Vignani, R.; Scali, M.; Cresti, M. A universal and rapid protocol for protein extraction from recalcitrant plant tissues for proteomic analysis. Electrophoresis 2006, 27, 2782–2786. [Google Scholar] [CrossRef] [PubMed]
- Rustichelli, C.; Visioli, G.; Kostecka, D.; Vurro, E.; Sanità di Toppi, L.; Marmiroli, N. Proteomic analysis in the lichen Physcia adscendens exposed to cadmium stress. Environ. Pollut. 2008, 156, 1121–1127. [Google Scholar] [CrossRef]
- Bubrick, P.; Frensdorff, A.; Galun, M. Proteins from the lichen Xanthoria parietina (L.)Th.Fr. which bind to phycobiont cell walls. Isolation and partial purification of an algal-binding protein. Symbiosis 1985, 1, 85–95. [Google Scholar]
- Hageman, C.; Fahselt, D.A. Intraspecific variability of isozymes of the lichen Umbilicaria mammulata. Can. J. Bot. 1984, 62, 617–623. [Google Scholar] [CrossRef]
- Kershaw, K.A.; MacFarlane, J.D.; Fovargue, M.R.; Webber, A. Phenotypic differences in the seasonal pattern of net photosynthesis in Cladonia stellaris. Can. J. Bot. 1983, 61, 2169–2180. [Google Scholar] [CrossRef]
- Skult, H.; Hagg, M.; Djupsund, B. Seasonal variation of isozyme and total protein phenotypes in populations of the lichen Parmelia omphalodes (Ascomycetes). Ann. Bot. Fenn. 1990, 27, 47–52. [Google Scholar]
- Sanità di Toppi, L.; Gabbrielli, R. Response to cadmium in higher plants. Environ. Exp. Bot. 1999, 41, 105–130. [Google Scholar] [CrossRef]
- Nicolardi, V.; Cai, G.; Parrotta, L.; Puglia, M.; Bianchi, L.; Bini, L.; Gaggi, C. The adaptive response of lichens to mercury exposure involves changes in the photosynthetic machinery. Environ. Pollut. 2012, 160, 1–10. [Google Scholar] [CrossRef]
- Schneider, T.; Schmid, E.; de Castro, J.V.; Cardinale, M.; Eberl, L.; Grube, M.; Berg, G.; Riedel, K. Structure and function of the symbiosis partners of the lung lichen (Lobaria pulmonaria L. Hoffm.) analyzed by metaproteomics. Proteomics 2011, 11, 2752–2756. [Google Scholar] [CrossRef] [PubMed]
- Grube, M.; Cernava, T.; Soh, J.; Fuchs, S.; Aschenbrenner, I.; Lassek, C.; Wegner, U.; Becher, D.; Riedel, K.; Sensen, C.W.; et al. Exploring functional contexts of symbiotic sustain within lichen-associated bacteria by comparative omics. ISME J. 2015, 9, 412–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eymann, C.; Lassek, C.; Wegner, U.; Bernhardt, J.; Fritsch, O.A.; Fuchs, S.; Otto, A.; Albrecht, D.; Schiefelbein, U.; Cernava, T.; et al. Symbiotic interplay of fungi, algae, and bacteria within the lung lichen Lobaria pulmonaria L. Hoffm. as assessed by state-of-the-art metaproteomics. J. Proteome Res. 2017, 16, 2160–2173. [Google Scholar] [CrossRef] [PubMed]
- Munzi, S.; Sheppard, L.J.; Leith, I.D.; Cruz, C.; Branquinho, C.; Bini, L.; Gagliardi, A.; Cai, G.; Parrotta, L. The cost of surviving nitrogen excess: Energy and protein demand in the lichen Cladonia portentosa as revealed by proteomic analysis. Planta 2017, 245, 819–833. [Google Scholar] [CrossRef]
- Paul, S.; Singh, P.; Rudramurthy, S.M.; Chakrabarti, A.; Ghosh, A.K. Matrix-assisted laser desorption/ionization–time of flight mass spectrometry: Protocol standardization and database expansion for rapid identification of clinically important molds. Future Microbiol. 2017, 12, 1457–1466. [Google Scholar] [CrossRef]
- Zvezdanova, M.E.; Escribano, P.; Ruiz, A.; Martínez-Jiménez, M.C.; Peláez, T.; Collazos, A.; Guinea, J.; Bouza, E.; Rodríguez-Sánchez, B. Increased species-assignement of filamentous fungi using MALDI-TOF MS coupled with a simplifies sample processing and an in-house library. Med. Mycol. 2019, 57, 63–70. [Google Scholar] [CrossRef]
- Patel, R. A moldy application of MALDI: MALDI-ToF mass spectrometry for fungal identification. J. Fungi 2019, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Putignani, L.; Del Chierico, F.; Onori, M.; Mancinelli, L.; Argentieri, M.; Bernaschi, P.; Coltella, L.; Lucignano, B.; Pansani, L.; Ranno, S.; et al. MALDI-TOF mass spectrometry proteomic phenotyping of clinically relevant fungi. Mol. Biosyst. 2011, 7, 620–629. [Google Scholar] [CrossRef]
- Chalupová, J.; Raus, M.; Sedlářová, M.; Šebela, M.; Sebela, M. Identification of fungal microorganisms by MALDI-TOF mass spectrometry. Biotechnol. Adv. 2014, 32, 230–241. [Google Scholar] [CrossRef]
- Ranque, S.; Normand, A.C.; Cassagne, C.; Murat, J.B.; Bourgeois, N.; Dalle, F.; Gari-Toussaint, M.; Fourquet, P.; Hendrickx, M.; Piarroux, R. MALDI-TOF mass spectrometry identification of filamentous fungi in the clinical laboratory. Mycoses 2014, 57, 135–140. [Google Scholar] [CrossRef]
- Del Chierico, F.; Masotti, A.; Onori, M.; Fiscarelli, E.; Mancinelli, L.; Ricciotti, G.; Alghisi, F.; Dimiziani, L.; Manetti, C.; Urbani, A.; et al. MALDI-TOF MS proteomic phenotyping of filamentous and other fungi from clinical origin. J. Proteomics 2012, 75, 3314–3330. [Google Scholar] [CrossRef] [PubMed]
- Borman, A.M.; Fraser, M.; Szekely, A.; Larcombe, D.E.; Johnson, E.M. Rapid identification of clinically relevant members of the genus exophiala by matrix-assisted laser desorption ionization-time of flight mass spectrometry and description of two novel species, Exophiala campbellii and Exophiala lavatrina. J. Clin. Microbiol. 2017, 55, 1162–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondori, N.; Erhard, M.; Welinder-Olsson, C.; Groenewald, M.; Verkley, G.; Moore, E.R.B. Analyses of black fungi by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS): Species-level identification of clinical isolates of Exophiala dermatitidis. FEMS Microbiol. Lett. 2015, 362, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Buskirk, A.D.; Hettick, J.M.; Chipinda, I.; Law, B.F.; Siegel, P.D.; Slaven, J.E.; Green, B.J.; Beezhold, D.H. Fungal pigments inhibit the matrix-assisted laser desorption/ionization time-of-flight mass spectrometry analysis of darkly pigmented fungi. Anal. Biochem. 2011, 411, 122–128. [Google Scholar] [CrossRef]
- Kumar, C.; Mann, M. Bioinformatics analysis of mass spectrometry-based proteomics data sets. FEBS Lett. 2009, 583, 1703–1712. [Google Scholar] [CrossRef] [Green Version]
- Sheynkman, G.M.; Shortreed, M.R.; Cesnik, A.J.; Smith, L.M. Proteogenomics: Integrating next-generation sequencing and Mass Spectrometry to characterize human proteomic variation. Annu. Rev. Anal. Chem. 2016, 9, 521–545. [Google Scholar] [CrossRef] [Green Version]
- Hoff, K.J.; Stanke, M. WebAUGUSTUS—A web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Res. 2013, 41, 123–128. [Google Scholar] [CrossRef]
- Basenko, E.Y.; Pulman, J.A.; Shanmugasundram, A.; Harb, O.S.; Crouch, K.; Starns, D.; Warrenfeltz, S.; Aurrecoechea, C.; Stoeckert, C.J.; Kissinger, J.C.; et al. FungiDB: An integrated bioinformatic resource for fungi and oomycetes. J. Fungi 2018, 4, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Blum, T.; Briesemeister, S.; Kohlbacher, O. MultiLoc2: Integrating phylogeny and Gene Ontology terms improves subcellular protein localization prediction. BMC Bioinform. 2009, 10, 274. [Google Scholar] [CrossRef] [Green Version]
- Briesemeister, S.; Rahnenführer, J.; Kohlbacher, O. Going from where to why-interpretable prediction of protein subcellular localization. Bioinformatics 2010, 26, 1232–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, e111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooney, C.; Wang, Y.H.; Pollastri, G. SCLpred: Protein subcellular localization prediction by N-to-1 neural networks. Bioinformatics 2011, 27, 2812–2819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.W.; Liu, Y.F.; Yu, Y.; Zhang, T.H.; Fan, X.N. MSLoc-DT: A new method for predicting the protein subcellular location of multispecies based on decision templates. Anal. Biochem. 2014, 449, 164–171. [Google Scholar] [CrossRef]
- Savojardo, C.; Martelli, P.L.; Fariselli, P.; Profiti, G.; Casadio, R. BUSCA: An integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 2018, 46, W459–W466. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Sønderby, C.K.; Petersenl, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielson, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Käll, L.; Krogh, A.; Sonnhammer, E.L. Advantages of combined transmembrane topology and signal peptide prediction—The Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef] [Green Version]
- Lum, G.; Min, X.J. FunSecKB: The Fungal Secretome KnowledgeBase. Database J. Biol. Databases Curation 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Ngounou, A.G.; Izabela, W.; Woods, A.G.; Roy, U.; Deinhardt, K.; Darie, C.C.; Desorption-esi, D. Protein—Protein interactions: Switch from classical methods to proteomics and bioinformatics-based approaches Electron transfer dissociation. Cell. Mol. Life Sci. 2014, 41, 205–228. [Google Scholar] [CrossRef]
- Pascovici, D.; Wu, J.X.; Mckay, M.J.; Joseph, C.; Noor, Z.; Kamath, K.; Wu, Y.; Ranganathan, S.; Gupta, V. Clinically relevant post-translational modification analyses—Maturing workflows and bioinformatics tools. Int. J. Mol. Sci. 2019, 20, 16. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, E.E.; Prost, S.; Ertz, D.; Westberg, M.; Frisch, A.; Bendiksby, M. Draft genome sequence and annotation of the lichen-forming fungus Arthonia radiata. Genome Announc. 2018, 6, e00281-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parente-Rocha, J.A.; Tomazett, M.V.; Pigosso, L.L.; Bailão, A.M.; Ferreira de Souza, A.; Paccez, J.D.; Baeza, L.C.; Pereira, M.; Silva Bailão, M.G.; Borges, C.L.; et al. In vitro, ex vivo and in vivo models: A comparative analysis of Paracoccidioides spp. proteomic studies. Fungal Biol. 2018, 122, 505–513. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muggia, L.; Ametrano, C.G.; Sterflinger, K.; Tesei, D. An Overview of Genomics, Phylogenomics and Proteomics Approaches in Ascomycota. Life 2020, 10, 356. https://doi.org/10.3390/life10120356
Muggia L, Ametrano CG, Sterflinger K, Tesei D. An Overview of Genomics, Phylogenomics and Proteomics Approaches in Ascomycota. Life. 2020; 10(12):356. https://doi.org/10.3390/life10120356
Chicago/Turabian StyleMuggia, Lucia, Claudio G. Ametrano, Katja Sterflinger, and Donatella Tesei. 2020. "An Overview of Genomics, Phylogenomics and Proteomics Approaches in Ascomycota" Life 10, no. 12: 356. https://doi.org/10.3390/life10120356