Prebiotic Reaction Networks in Water


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. What Is a Chemical Reaction Network?
3. The Miller-Urey Experiment
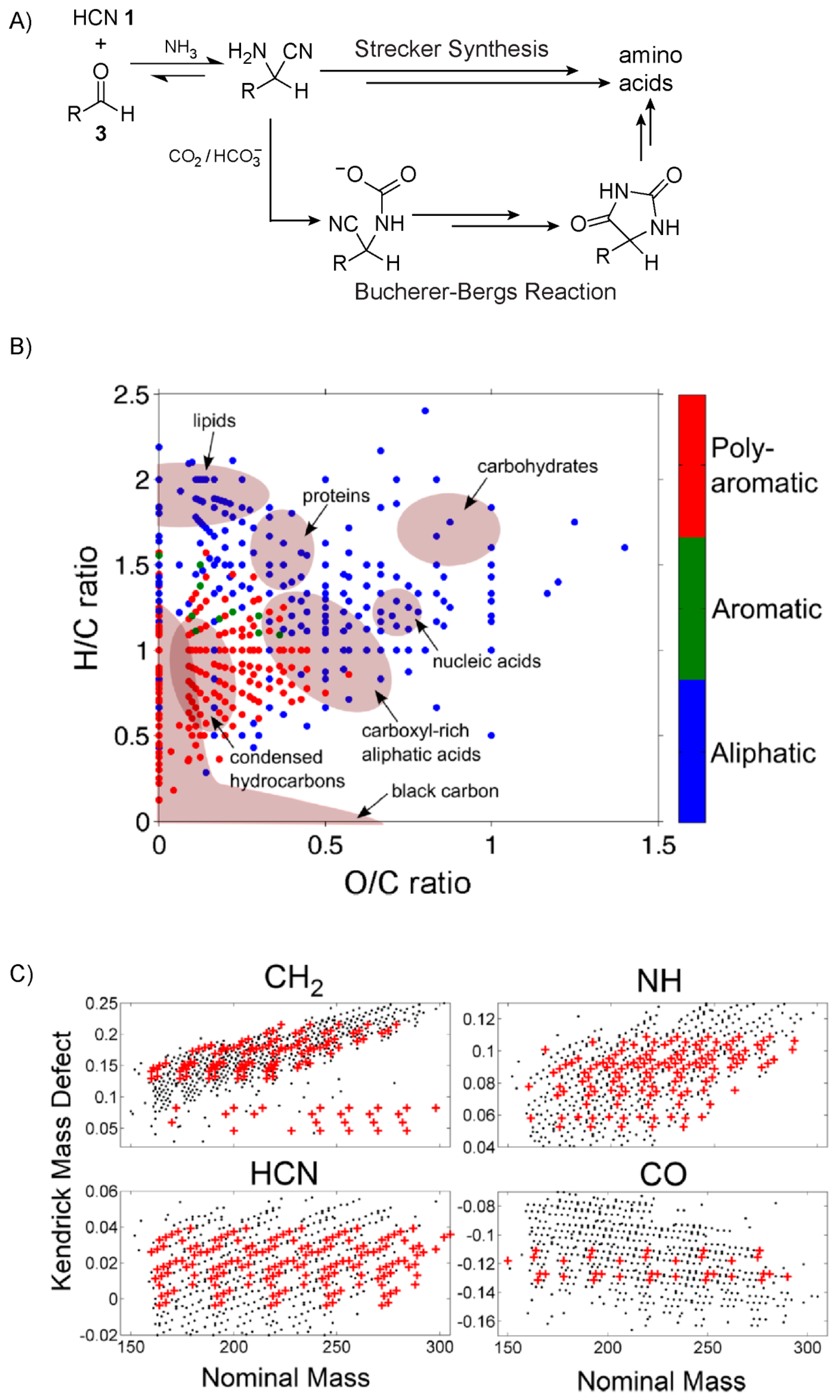
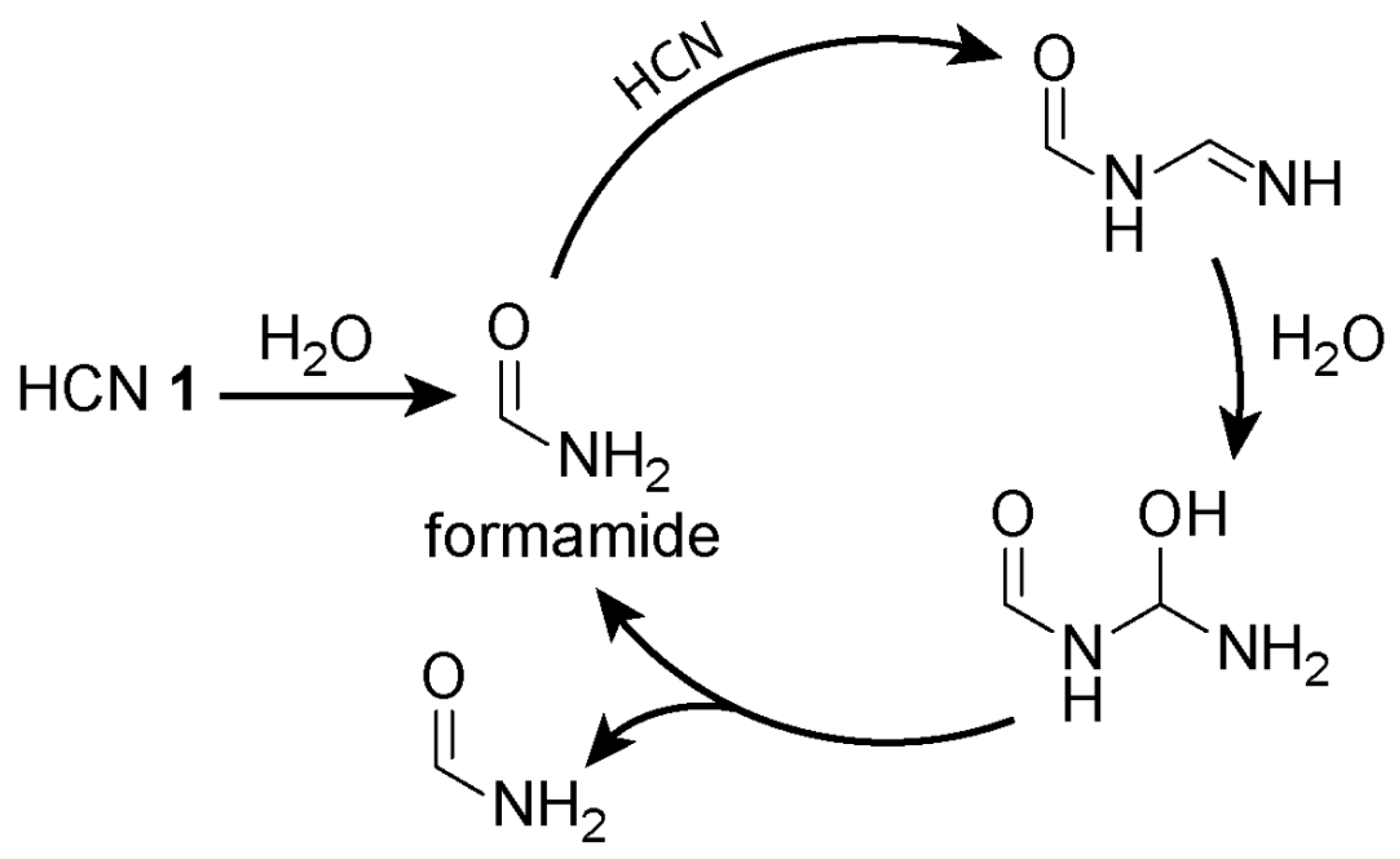
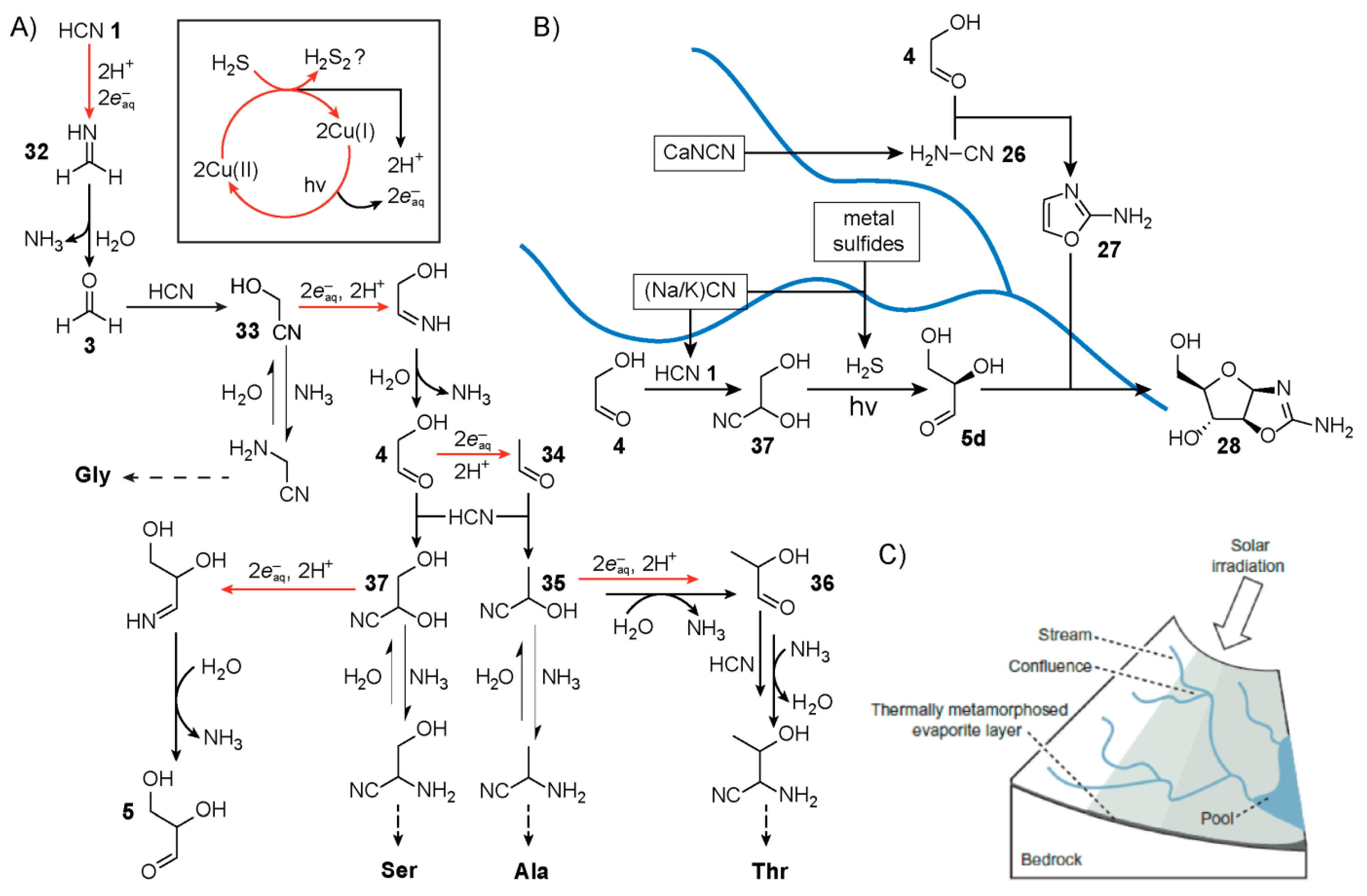
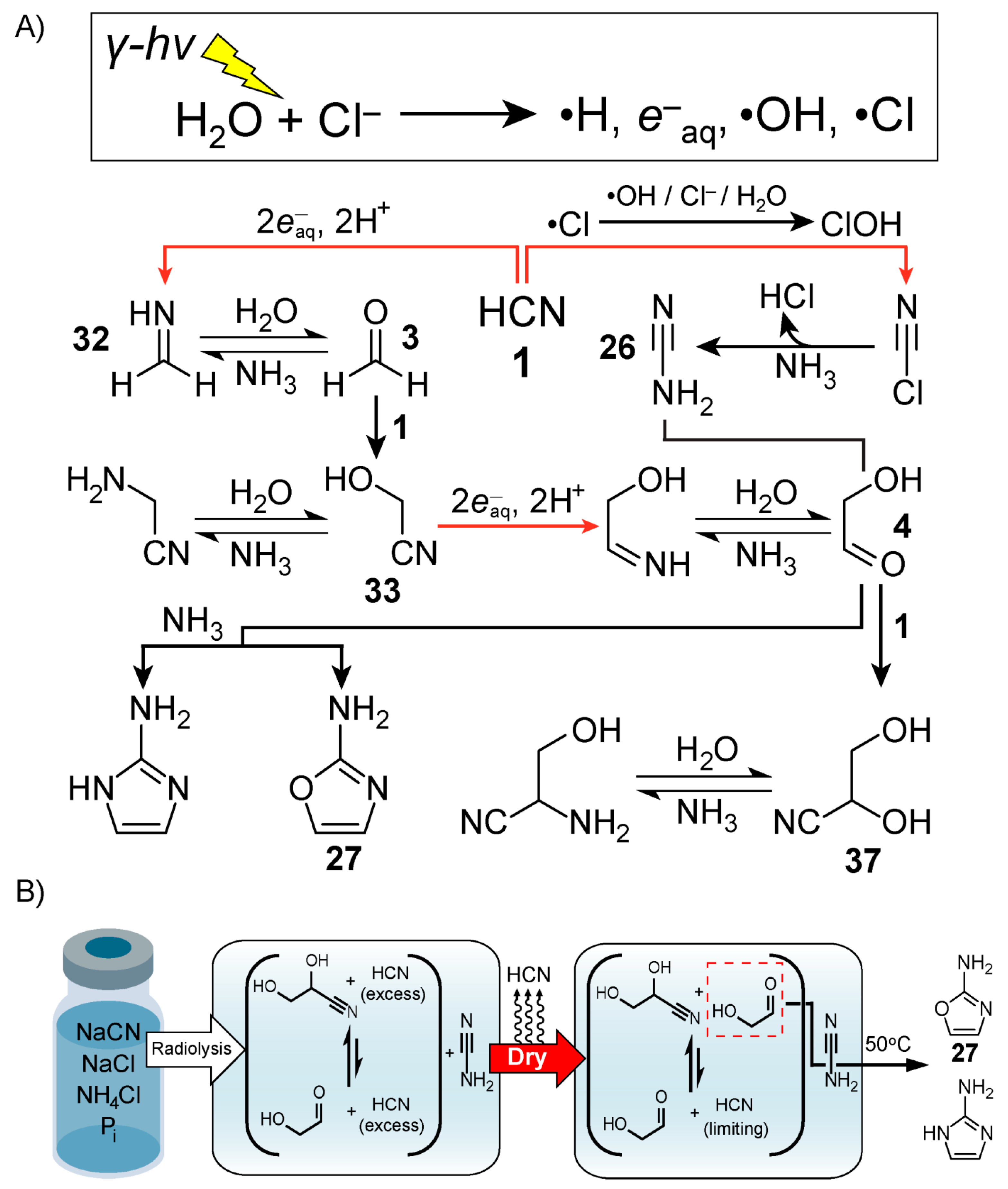
4. Hydrogen Cyanide Polymerisation
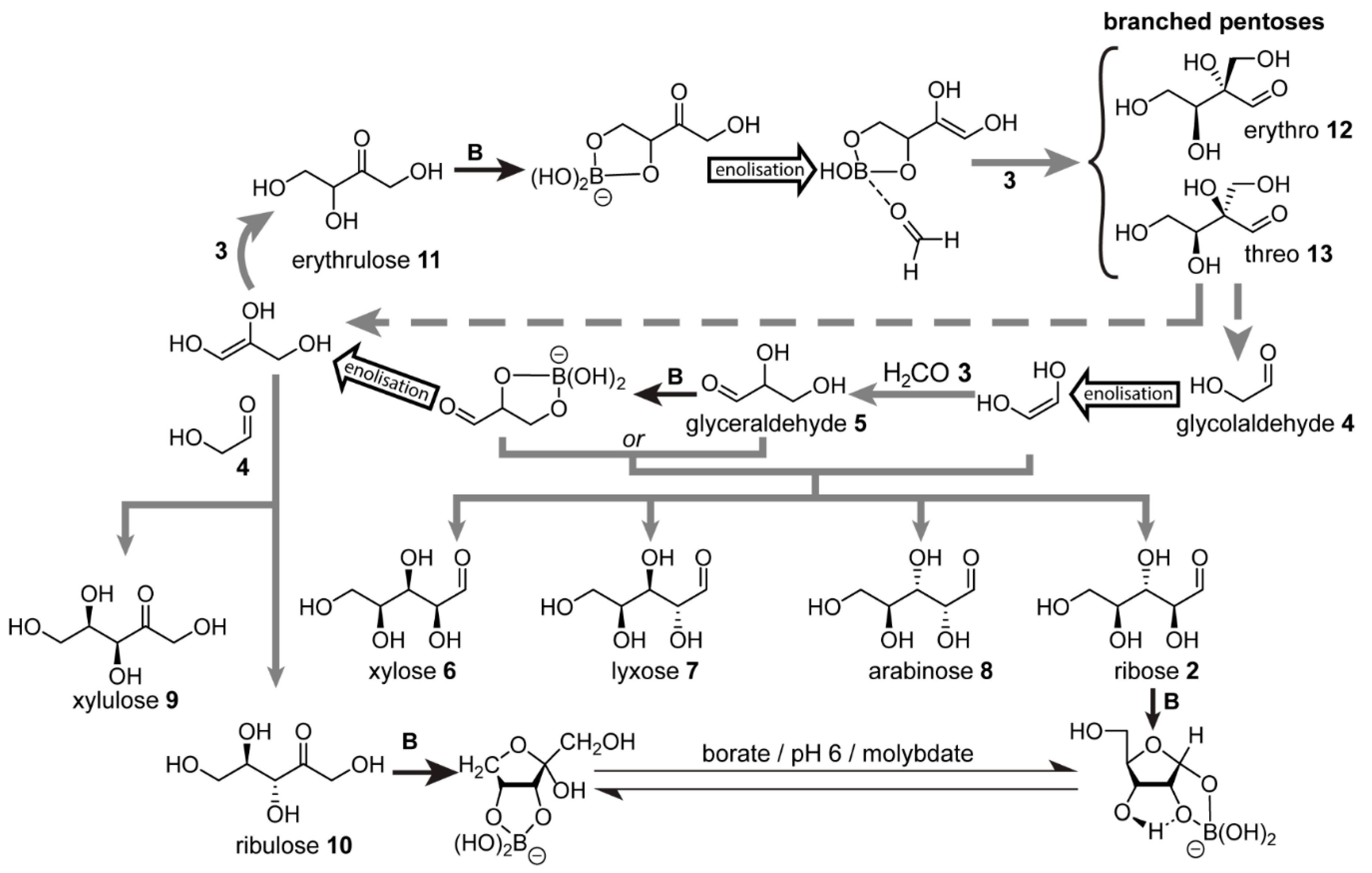
5. The Formose Reaction
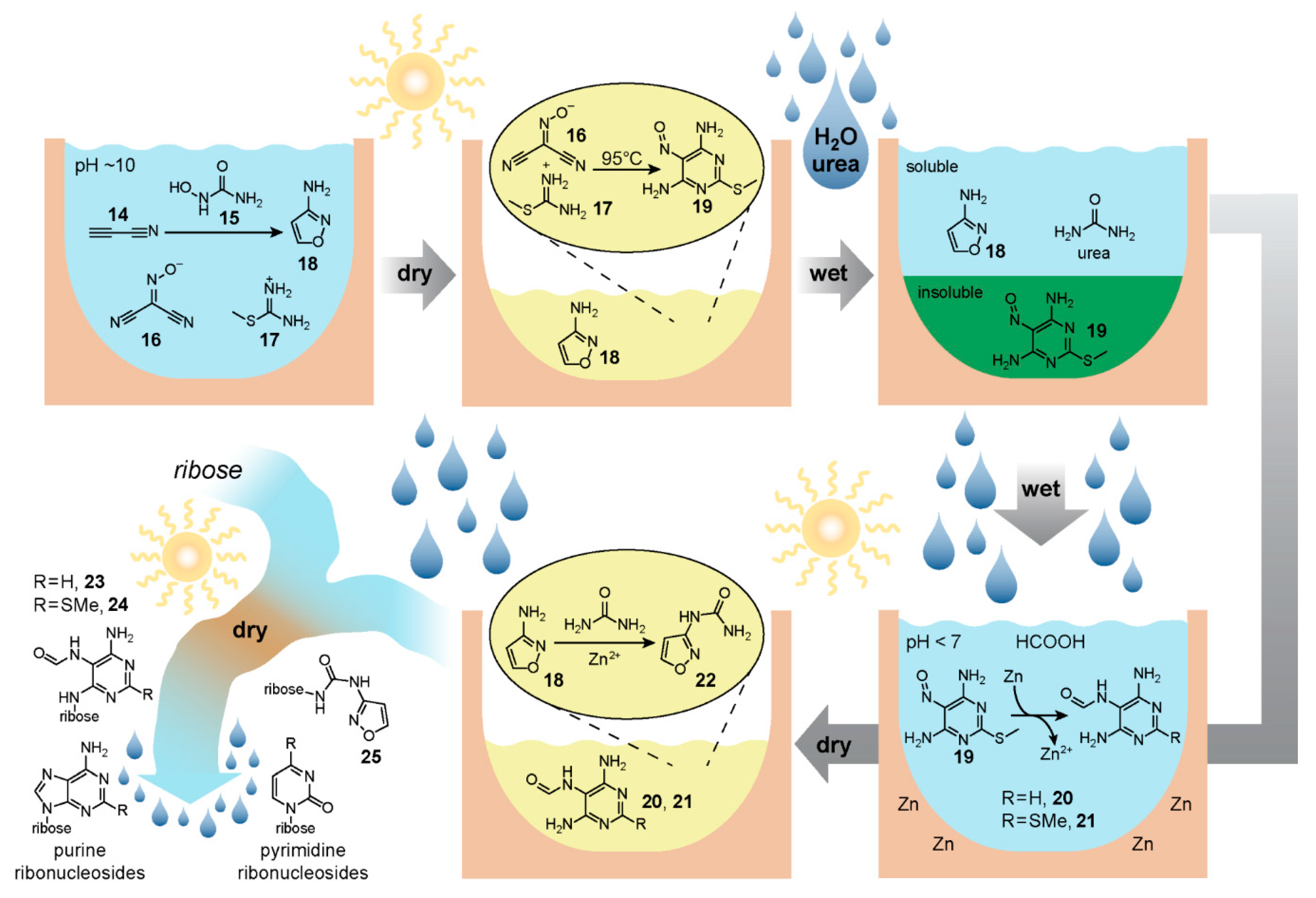
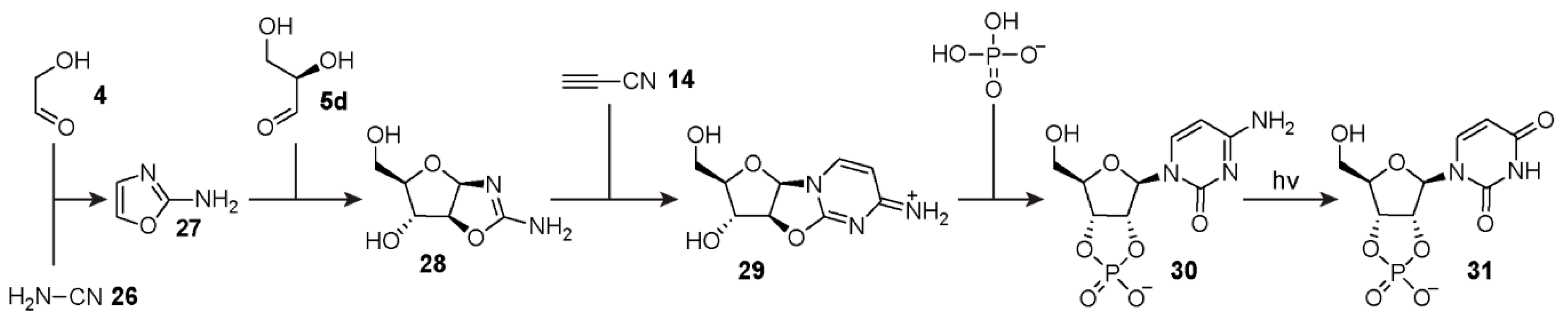
6. Reaction Networks for Ribonucleotide Synthesis
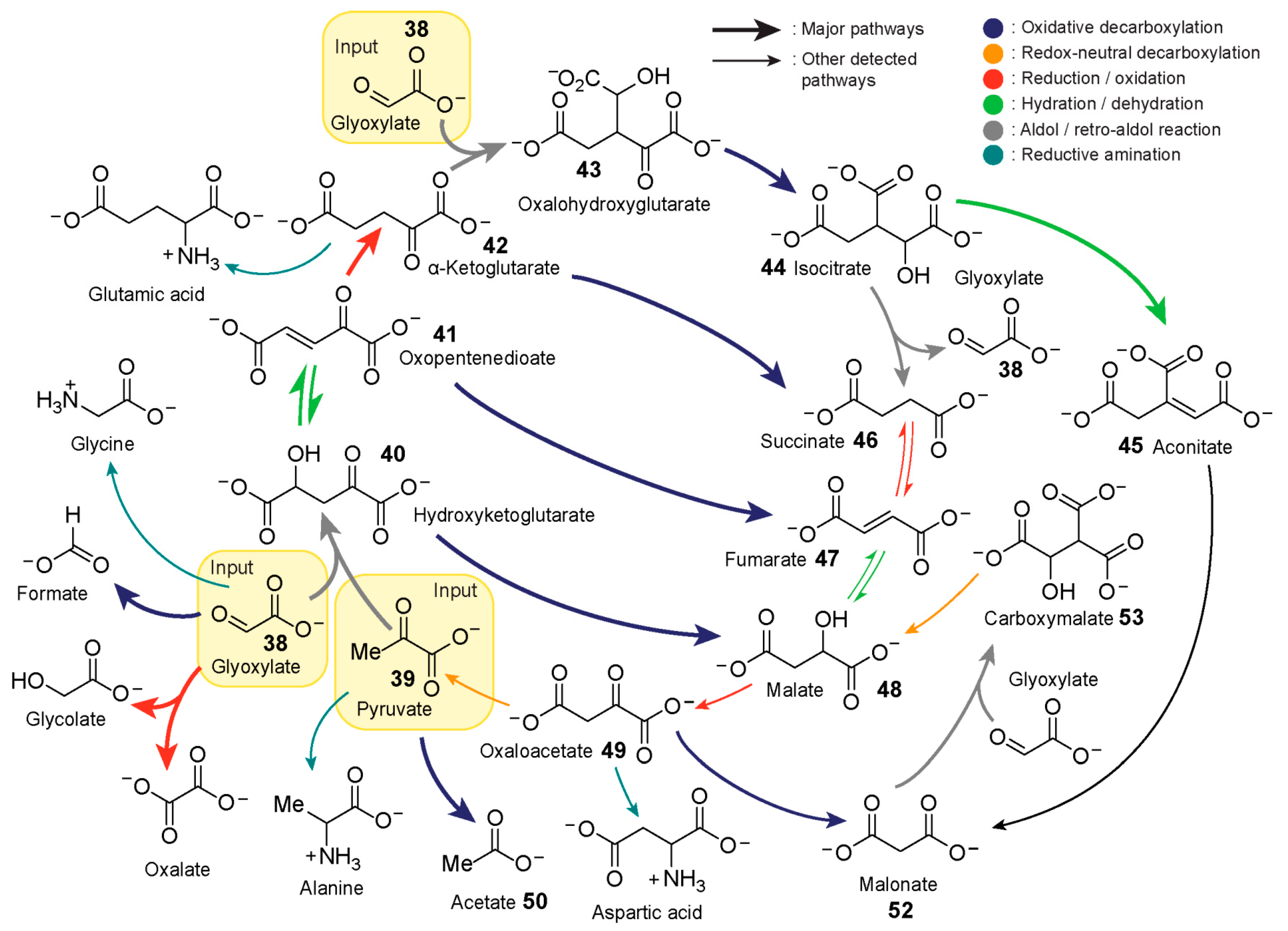
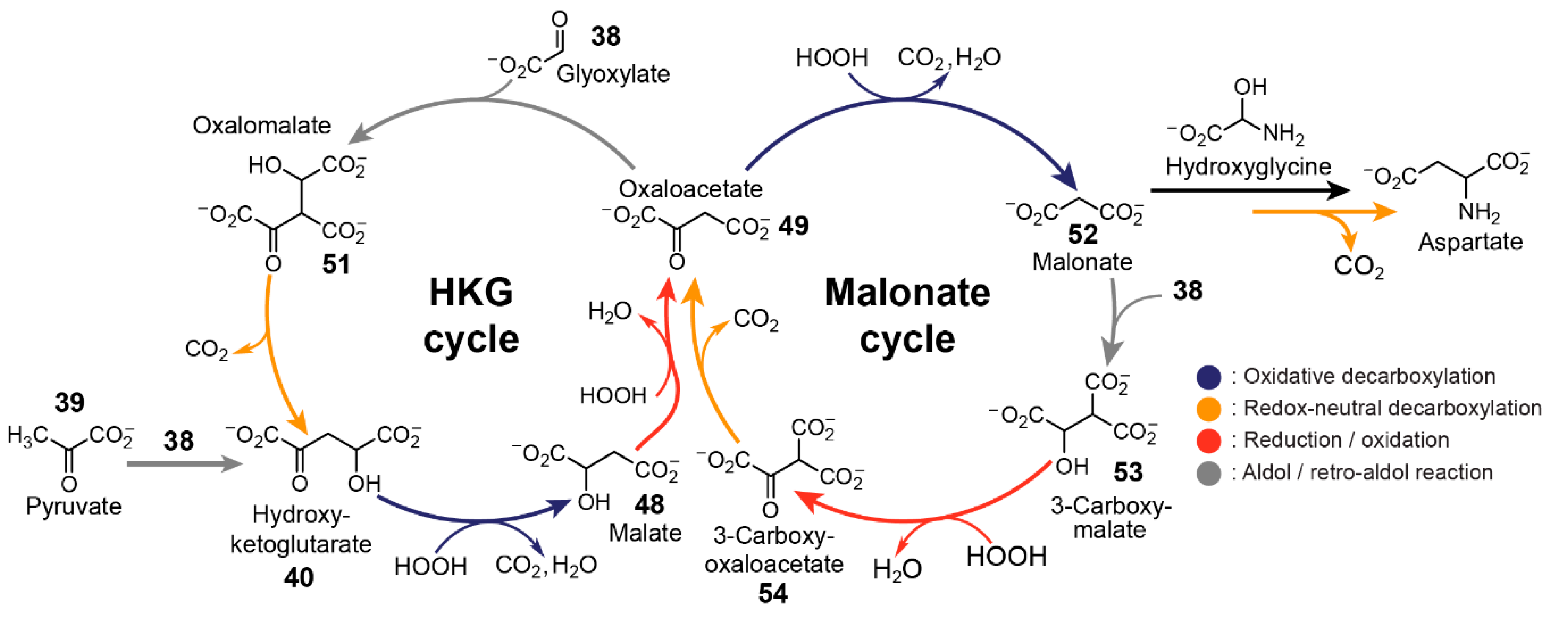
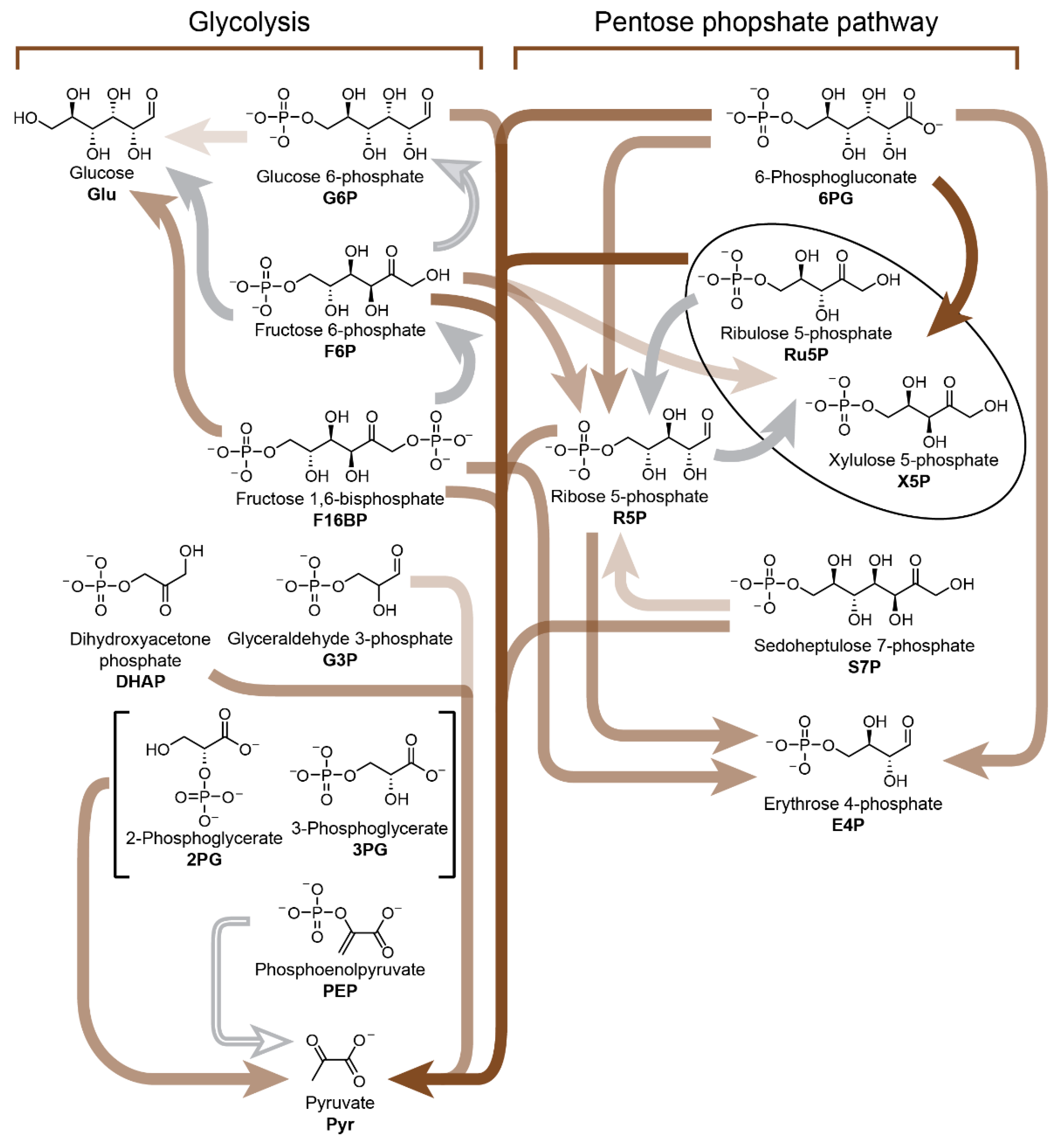
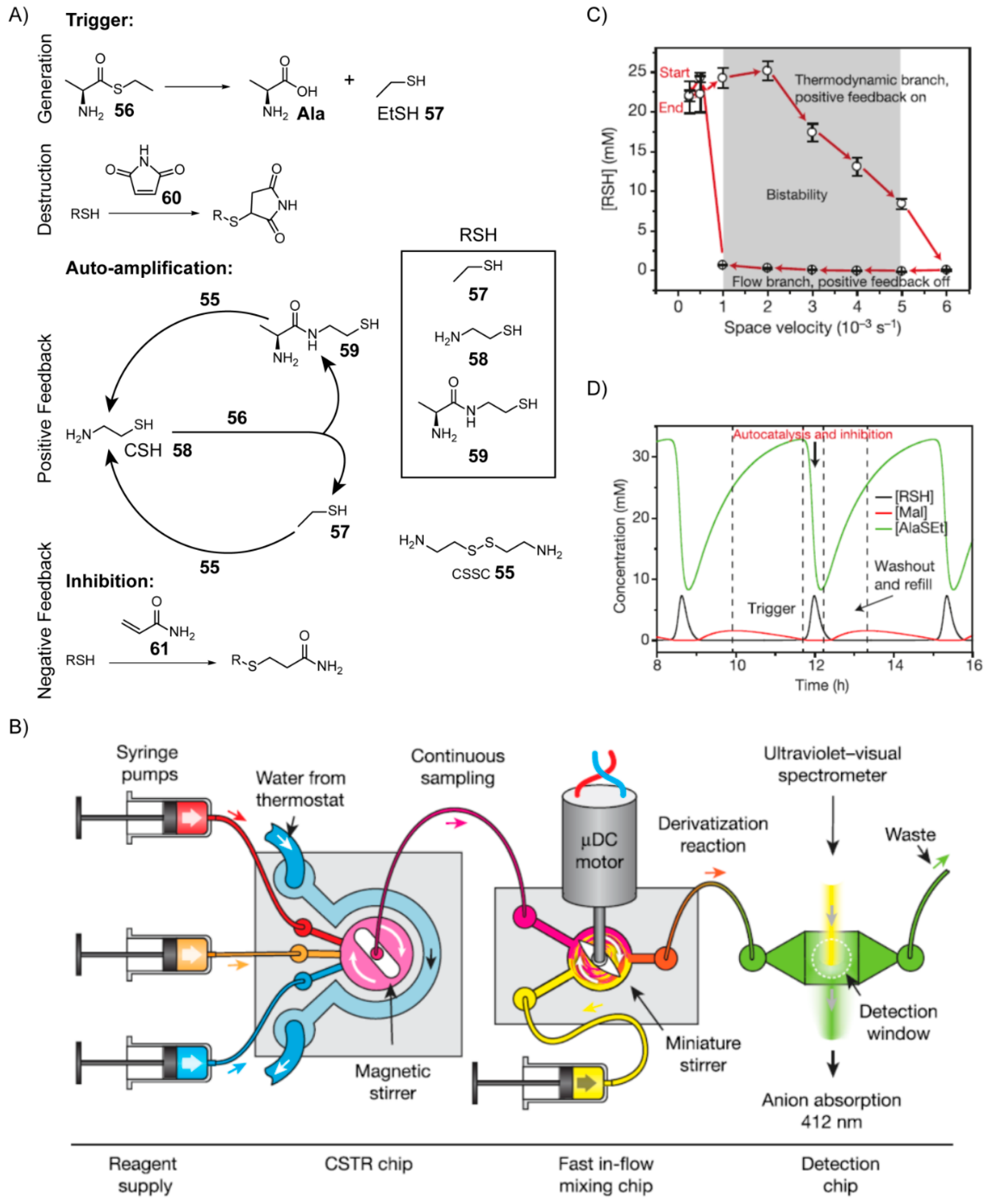
7. Nonenzymatic Analogues and Models of Metabolic Cycles
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smith, E.; Morowitz, H.J. Universality in Intermediary Metabolism. Proc. Natl. Acad. Sci. USA 2004, 101, 13168–13173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, G.; Priami, C. Transactions on Computational Systems Biology VI; Springer: Berlin/Heidelberg, Germany, 2005; Volume 3380, ISBN 978-3-540-25422-5. [Google Scholar]
- Ferrell, J.E., Jr.; Tsai, T.Y.-C.; Yang, Q. Modeling the Cell Cycle: Why Do Certain Circuits Oscillate? Cell 2011, 144, 874–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolley, C.; Douglas, T. Topological Biosignatures: Large-Scale Structure of Chemical Networks from Biology and Astrochemistry. Astrobiology 2012, 12, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Semenov, S.N.; Wong, A.S.Y.; van der Made, R.M.; Postma, S.G.J.; Groen, J.; van Roekel, H.W.H.; de Greef, T.F.A.; Huck, W.T.S. Rational Design of Functional and Tunable Oscillating Enzymatic Networks. Nat. Chem. 2015, 7, 160–165. [Google Scholar] [CrossRef] [Green Version]
- Zotenko, E.; Mestre, J.; O’Leary, D.P.; Przytycka, T.M. Why Do Hubs in the Yeast Protein Interaction Network Tend To Be Essential: Reexamining the Connection between the Network Topology and Essentiality. PLoS Comput. Biol. 2008, 4, e1000140:1–e1000140:16. [Google Scholar] [CrossRef]
- Vasas, V.; Fernando, C.; Santos, M.; Kauffman, S.; Szathmáry, E. Evolution Before Genes. Biol. Direct 2012, 7, 1:1–1:14. [Google Scholar] [CrossRef] [Green Version]
- Hordijk, W.; Steel, M.; Kauffman, S. The Structure of Autocatalytic Sets: Evolvability, Enablement, and Emergence. Acta Biotheor. 2012, 60, 379–392. [Google Scholar] [CrossRef]
- Nghe, P.; Hordijk, W.; Kauffman, S.A.; Walker, S.I.; Schmidt, F.J.; Kemble, H.; Yeates, J.A.M.; Lehman, N. Prebiotic Network Evolution: Six Key Parameters. Mol. Biosyst. 2015, 11, 3206–3217. [Google Scholar] [CrossRef]
- Sutherland, J.D. The Origin of Life–Out of the Blue. Angew. Chem. Int. Ed. 2016, 55, 104–121. [Google Scholar] [CrossRef]
- Kitadai, N.; Maruyama, S. Origins of Building Blocks of Life: A Review. Geosci. Front. 2018, 9, 1117–1153. [Google Scholar] [CrossRef]
- Pascal, R.; Boiteau, L.; Commeyras, A. From the Prebiotic Synthesis of α-Amino Acids Towards a Primitive Translation Apparatus for the Synthesis of Peptides. In Prebiotic Chemistry. Topics in Current Chemistry; Walde, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 259, pp. 69–122. ISBN 978-3-540-27759-0. [Google Scholar]
- Plankensteiner, K.; Reiner, H.; Rode, B.M. Prebiotic Chemistry: The Amino Acid and Peptide World. Curr. Org. Chem. 2005, 9, 1107–1114. [Google Scholar] [CrossRef]
- Canavelli, P.; Islam, S.; Powner, M.W. Peptide Ligation by Chemoselective Aminonitrile Coupling in Water. Nature 2019, 571, 546–549. [Google Scholar] [CrossRef]
- Frenkel-Pinter, M.; Samanta, M.; Ashkenasy, G.; Leman, L.J. Prebiotic Peptides: Molecular Hubs in the Origin of Life. Chem. Rev. 2020, 120, 4707–4765. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.G.; Yu, S.-S.; Mamajanov, I.; Grover, M.A.; Krishnamurthy, R.; Fernández, F.M.; Hud, N.V. Ester-Mediated Amide Bond Formation Driven by Wet-Dry Cycles: A Possible Path to Polypeptides on the Prebiotic Earth. Angew. Chem. Int. Ed. 2015, 54, 9871–9875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, E.T.; Cleaves II, H.J.; Bada, J.L.; Fernández, F.M. Quantitation of α-Hydroxy Acids in Complex Prebiotic Mixtures via Liquid Chromatography/Tandem Mass Spectrometry. Rapid Commun. Mass Spectrom. 2016, 30, 2043–2051. [Google Scholar] [CrossRef]
- Chandru, K.; Guttenberg, N.; Giri, C.; Hongo, Y.; Butch, C.; Mamajanov, I.; Cleaves II, H.J. Simple Prebiotic Synthesis of High Diversity Dynamic Combinatorial Polyester Libraries. Commun. Chem. 2018, 1, 30:1–30:8. [Google Scholar] [CrossRef]
- Eschenmoser, A. On a Hypothetical Generational Relationship Between HCN and Constituents of the Reductive Citric Acid Cycle. Chem. Biodivers. 2007, 4, 554–573. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Rappoport, D.; Aspuru-Guzik, A. Uncertainty of Prebiotic Scenarios: The Case of the Non-Enzymatic Reverse Tricarboxylic Acid Cycle. Sci. Rep. 2015, 5, 8009:1–8009:7. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, F.S.; Chen, K.; Mojica, M.; Conley, M.; Napoline, J.W.; Butch, C.; Pollet, P.; Krishnamurthy, R.; Liotta, C.L. A Plausible Prebiotic Origin of Glyoxylate: Nonenzymatic Transamination Reactions of Glycine with Formaldehyde. Synlett 2017, 28, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Springsteen, G.; Yerabolu, J.R.; Nelson, J.; Rhea, C.J.; Krishnamurthy, R. Linked Cycles of Oxidative Decarboxylation of Glyoxylate as Protometabolic Analogs of the Citric Acid Cycle. Nat. Commun. 2018, 9, 91:1–91:8. [Google Scholar] [CrossRef] [Green Version]
- Muchowska, K.B.; Varma, S.J.; Moran, J. Synthesis and Breakdown of Universal Metabolic Precursors Promoted by Iron. Nature 2019, 569, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, R.T.; Yadav, M.; Krishnamurthy, R.; Springsteen, G. A Plausible Metal-Free Ancestral Analogue of the Krebs Cycle Composed Entirely of α-Ketoacids. Nat. Chem. 2020, 12, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Cleaves II, H.J. The Prebiotic Geochemistry of Formaldehyde. Precambrian Res. 2008, 164, 111–118. [Google Scholar] [CrossRef]
- Ritson, D.; Sutherland, J.D. Prebiotic Synthesis of Simple Sugars by Photoredox Systems Chemistry. Nat. Chem. 2012, 4, 895–899. [Google Scholar] [CrossRef] [Green Version]
- Benner, S.A.; Kim, H.-J.; Biondi, E. Prebiotic Chemistry that Could Not Not Have Happened. Life 2019, 9, 84. [Google Scholar] [CrossRef] [Green Version]
- Lamour, S.; Pallmann, S.; Haas, M.; Trapp, O. Prebiotic Sugar Formation Under Nonaqueous Conditions and Mechanochemical Acceleration. Life 2019, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Cleaves, H.J. Nucleobases on the Primitive Earth: Their Sources and Stabilities. In Prebiotic Chemistry and Chemical Evolution of Nucleic Acids. Nucleic Acids and Molecular Biology; Menor-Salván, C., Ed.; Springer: Cham, Switzerland, 2018; Volume 35, pp. 1–19. ISBN 978-3-319-93583-6. [Google Scholar]
- Benner, S.A.; Bell, E.A.; Biondi, E.; Brasser, R.; Carell, T.; Kim, H.-J.; Mojzsis, S.J.; Omran, A.; Pasek, M.A.; Trail, D. When Did Life Likely Emerge on Earth in an RNA-First Process? ChemSystemsChem 2020, 2, e1900035:1–e1900035:20. [Google Scholar] [CrossRef]
- Yadav, M.; Kumar, R.; Krishnamurthy, R. Chemistry of Abiotic Nucleotide Synthesis. Chem. Rev. 2020, 120, 4766–4805. [Google Scholar] [CrossRef]
- Fialho, D.M.; Roche, T.P.; Hud, N.V. Prebiotic Syntheses of Noncanonical Nucleosides and Nucleotides. Chem. Rev. 2020, 120, 4806–4830. [Google Scholar] [CrossRef]
- Deamer, D. The Role of Lipid Membranes in Life’s Origin. Life 2017, 7, 5. [Google Scholar] [CrossRef]
- Szostak, J.W. The Narrow Road to the Deep Past: In Search of the Chemistry of the Origin of Life. Angew. Chem. Int. Ed. 2017, 56, 11037–11043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toparlak, O.D.; Mansy, S.S. Progress in Synthesizing Protocells. Exp. Biol. Med. 2019, 244, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.; Fiore, M. Investigating Prebiotic Protocells for a Comprehensive Understanding of the Origins of Life: A Prebiotic Systems Chemistry Perspective. Life 2019, 9, 49. [Google Scholar] [CrossRef] [Green Version]
- Gözen, İ. A Hypothesis for Protocell Division on the Early Earth. ACS Nano 2019, 13, 10869–10871. [Google Scholar] [CrossRef] [PubMed]
- Wołos, A.; Roszak, R.; Żądło-Dobrowolska, A.; Beker, W.; Mikulak-Klucznik, B.; Spólnik, G.; Dygas, M.; Szymkuć, S.; Grzybowski, B.A. Synthetic Connectivity, Emergence, and Self-Regeneration in the Network of Prebiotic Chemistry. Science 2020, 369, eaaw1955:1–eaaw1955:12. [Google Scholar] [CrossRef]
- Stoddart, J.F. From Supramolecular to Systems Chemistry: Complexity Emerging out of Simplicity. Angew. Chem. Int. Ed. 2012, 51, 12902–12903. [Google Scholar] [CrossRef] [Green Version]
- Ashkenasy, G.; Hermans, T.M.; Otto, S.; Taylor, A.F. Systems Chemistry. Chem. Soc. Rev. 2017, 46, 2543–2554. [Google Scholar] [CrossRef]
- Plasson, R. Chemical Reaction Network. In Encyclopedia of Astrobiology, 2011 Edition; Gargaud, M., Amils, R., Quintanilla, J.C., Cleaves, H.J., II, Irvine, W.M., Pinti, D.L., Viso, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 287–288. ISBN 978-3-642-11271-3. [Google Scholar]
- Legal Information Institute. JACOBELLIS v. OHIO. 1964, Volume 378, p. 184. Available online: https://www.law.cornell.edu/supremecourt/text/378/184 (accessed on 16 December 2020).
- Gewirtz, P. On “I Know It When I See It”. Yale Law J. 1996, 105, 1023–1047. [Google Scholar] [CrossRef]
- Jeong, H.; Tombor, B.; Albert, R.; Oltvai, Z.N.; Barabási, A.-L. The Large-Scale Organization of Metabolic Networks. Nature 2000, 407, 651–654. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Smith, H.B.; Mathis, C.; Raymond, J.; Walker, S.I. Universal Scaling Across Biochemical Networks on Earth. Sci. Adv. 2019, 5, eaau0149:1–eaau0149:12. [Google Scholar] [CrossRef] [Green Version]
- Jacob, P.-M.; Lapkin, A. Statistics of the network of organic chemistry. React. Chem. Eng. 2018, 3, 102–118. [Google Scholar] [CrossRef] [Green Version]
- Lima-Mendez, G.; van Helden, J. The Powerful Law of the Power Law and Other Myths in Network Biology. Mol. Biosyst. 2009, 5, 1482–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broido, A.D.; Clauset, A. Scale-Free Networks Are Rare. Nat. Commun. 2019, 10, 1017:1–1017:10. [Google Scholar] [CrossRef] [PubMed]
- Richert, C. Prebiotic Chemistry and Human Intervention. Nat. Commun. 2018, 9, 5177:1–5177:3. [Google Scholar] [CrossRef] [PubMed]
- Benner, S.A.; Kim, H.-J.; Carrigan, M.A. Asphalt, Water, and the Prebiotic Synthesis of Ribose, Ribonucleosides, and RNA. Acc. Chem. Res. 2012, 45, 2025–2034. [Google Scholar] [CrossRef] [PubMed]
- Yi, R.; Tran, Q.P.; Ali, S.; Yoda, I.; Adam, Z.R.; Cleaves II, H.J.; Fahrenbach, A.C. A Continuous Reaction Network that Produces RNA Precursors. Proc. Natl. Acad. Sci. USA 2020, 117, 13267–13274. [Google Scholar] [CrossRef] [PubMed]
- Lazcano, A. What Is Life? A Brief Historical Overview. Chem. Biodivers. 2008, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Muchowska, K.B.; Varma, S.J.; Moran, J. Nonenzymatic Metabolic Reactions and Life’s Origins. Chem. Rev. 2020, 120, 7708–7744. [Google Scholar] [CrossRef]
- Cleaves II, H.J. Prebiotic Chemistry: What We Know, What We Don’t. Evol. Educ. Outreach 2012, 5, 342–360. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.L. A Production of Amino Acids Under Possible Primitive Earth Conditions. Science 1953, 117, 528–529. [Google Scholar] [CrossRef] [Green Version]
- Oparin, A.I. Origin of Life, 2nd ed.; Dover Publications: New York, NY, USA, 1953; ISBN 0486602133. [Google Scholar]
- Urey, H.C. On the Early Chemical History of the Earth and the Origin of Life. Proc. Natl. Acad. Sci. USA 1952, 38, 351–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal, J.D. The Physical Basis of Life. Proc. Phys. Soc. A 1949, 62, 537–558. [Google Scholar] [CrossRef]
- Cleaves, H.J.; Chalmers, J.H.; Lazcano, A.; Miller, S.L.; Bada, J.L. A Reassessment of Prebiotic Organic Synthesis in Neutral Planetary Atmospheres. Orig. Life Evol. Biosph. 2008, 38, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Zahnle, K.; Schaefer, L.; Fegley, B. Earth’s Earliest Atmospheres. Cold Spring Harb. Perspect. Biol. 2010, 2, a004895:1–a004895:17. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.L. Production of Some Organic Compounds under Possible Primitive Earth Conditions. J. Am. Chem. Soc. 1955, 77, 2351–2361. [Google Scholar] [CrossRef]
- Stribling, R.; Miller, S.L. Energy Yields for Hydrogen Cyanide and Formaldehyde Syntheses: The HCN and Amino Acid Concentrations in the Primitive Ocean. Orig. Life Evol. Biosph. 1987, 17, 261–273. [Google Scholar] [CrossRef]
- Taillades, J.; Beuzelin, I.; Garrel, L.; Tabacik, V.; Bied, C.; Commeyras, A. N-Carbamoyl-α-Amino Acids Rather than Free α-Amino Acids Formation in the Primitive Hydrosphere: A Novel Proposal for the Emergence of Prebiotic Peptides. Orig. Life Evol. Biosph. 1998, 28, 61–77. [Google Scholar] [CrossRef]
- Strecker, A. Ueber einen Neuen aus Aldehyd–Ammoniak und Blausäure Entstehenden Körper. Justus Liebigs Ann. Chem. 1854, 91, 349–351. [Google Scholar] [CrossRef] [Green Version]
- Bucherer, H.T.; Lieb, V.A. Über die Bildung Substituierter Hydantoine aus Aldehyden und Ketonen. Synthese von Hydantoinen. J. Prakt. Chem. 1934, 141, 5–43. [Google Scholar] [CrossRef]
- Wollrab, E.; Scherer, S.; Aubriet, F.; Carré, V.; Carlomagno, T.; Codutti, L.; Ott, A. Chemical Analysis of a “Miller-Type” Complex Prebiotic Broth. Part I: Chemical Diversity, Oxygen and Nitrogen Based Polymers. Orig. Life Evol. Biosph. 2016, 46, 149–169. [Google Scholar] [CrossRef]
- Kim, S.; Kramer, R.W.; Hatcher, P.G. Graphical Method for Analysis of Ultrahigh-Resolution Broadband Mass Spectra of Natural Organic Matter, the Van Krevelen Diagram. Anal. Chem. 2003, 75, 5336–5344. [Google Scholar] [CrossRef]
- Hertkorn, N.; Frommberger, M.; Witt, M.; Koch, B.P.; Schmitt-Kopplin, P.; Perdue, E.M. Natural Organic Matter and the Event Horizon of Mass Spectrometry. Anal. Chem. 2008, 80, 8908–8919. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Bermejo, M.; Zorzano, M.-P.; Osuna-Esteban, S. Simple Organics and Biomonomers Identified in HCN Polymers: An Overview. Life 2013, 3, 421–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oró, J. Mechanism of Synthesis of Adenine from Hydrogen Cyanide under Possible Primitive Earth Conditions. Nature 1961, 191, 1193–1194. [Google Scholar] [CrossRef] [PubMed]
- Scherer, S.; Wollrab, E.; Codutti, L.; Carlomagno, T.; da Costa, S.G.; Volkmer, A.; Bronja, A.; Schmitz, O.J.; Ott, A. Chemical Analysis of a “Miller-Type” Complex Prebiotic Broth. Part II: Gas, Oil, Water and the Oil/Water-Interface. Orig. Life Evol. Biosph. 2017, 47, 381–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, E. The Unforeseen Challenge: From Genotype-to-Phenotype in Cell Populations. Reports Prog. Phys. 2015, 78, 036602:1–036602:51. [Google Scholar] [CrossRef] [PubMed]
- Mamajanov, I.; Herzfeld, J. HCN Polymers Characterized by SSNMR: Solid State Reaction of Crystalline Tetramer (Diaminomaleonitrile). J. Chem. Phys. 2009, 130, 134504:1–134504:5. [Google Scholar] [CrossRef] [Green Version]
- Mamajanov, I.; Herzfeld, J. HCN Polymers Characterized by Solid State NMR: Chains and Sheets Formed in the Neat Liquid. J. Chem. Phys. 2009, 130, 134503:1–134503:6. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Lin, G.; Upton, K.T.; Imanaka, H.; Smith, M.A. Structural Investigation of HCN Polymer Isotopomers by Solution-State Multidimensional NMR. J. Phys. Chem. A 2012, 116, 4751–4759. [Google Scholar] [CrossRef]
- Benkö, G.; Flamm, C.; Stadler, P.F. A Graph-Based Toy Model of Chemistry. J. Chem. Inf. Comput. Sci. 2003, 43, 1085–1093. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.L.; Flamm, C.; Merkle, D.; Stadler, P.F. In Silico Support for Eschenmoser’s Glyoxylate Scenario. Isr. J. Chem. 2015, 55, 919–933. [Google Scholar] [CrossRef]
- Meringer, M.; Cleaves, H.J. Exploring Astrobiology Using in Silico Molecular Structure Generation. Philos. Trans. R. Soc. A 2017, 375, 20160344:1–20160344:12. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.L.; Andersen, T.; Flamm, C.; Hanczyc, M.M.; Merkle, D.; Stadler, P.F. Navigating the Chemical Space of HCN Polymerization and Hydrolysis: Guiding Graph Grammars by Mass Spectrometry Data. Entropy 2013, 15, 4066–4083. [Google Scholar] [CrossRef] [Green Version]
- Oró, J.; Kimball, A.P. Synthesis of Purines under Possible Primitive Earth Conditions. I. Adenine from Hydrogen Cyanide. Arch. Biochem. Biophys. 1961, 94, 217–227. [Google Scholar] [CrossRef]
- Oró, J.; Kimball, A.P. Synthesis of Purines under Possible Primitive Earth Conditions: II. Purine Intermediates from Hydrogen Cyanide. Arch. Biochem. Biophys. 1962, 96, 293–313. [Google Scholar] [CrossRef]
- Saladino, R.; Carota, E.; Botta, G.; Kapralov, M.; Timoshenko, G.N.; Rozanov, A.Y.; Krasavin, E.; Di Mauro, E. Meteorite-Catalyzed Syntheses of Nucleosides and of Other Prebiotic Compounds from Formamide Under Proton Irradiation. Proc. Natl. Acad. Sci. USA 2015, 112, E2746–E2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzarello, S.; Shock, E. The Organic Composition of Carbonaceous Meteorites: The Evolutionary Story Ahead of Biochemistry. Cold Spring Harb. Perspect. Biol. 2010, 2, a002105:1–a002105:19. [Google Scholar] [CrossRef]
- Schmitt-Kopplin, P.; Gabelica, Z.; Gougeon, R.D.; Fekete, A.; Kanawati, B.; Harir, M.; Gebefuegi, I.; Eckel, G.; Hertkorn, N. High Molecular Diversity of Extraterrestrial Organic Matter in Murchison Meteorite Revealed 40 Years After Its Fall. Proc. Natl. Acad. Sci. USA 2010, 107, 2763–2768. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, Y.; Chikaraishi, Y.; Ohkouchi, N.; Ogawa, N.O.; Glavin, D.P.; Dworkin, J.P.; Abe, C.; Nakamura, T. Extraterrestrial Ribose and Other Sugars in Primitive Meteorites. Proc. Natl. Acad. Sci. USA 2019, 116, 24440–24445. [Google Scholar] [CrossRef] [Green Version]
- Callahan, M.P.; Smith, K.E.; Cleaves II, H.J.; Ruzicka, J.; Stern, J.C.; Glavin, D.P.; House, C.H.; Dworkin, J.P. Carbonaceous Meteorites Contain a Wide Range of Extraterrestrial Nucleobases. Proc. Natl. Acad. Sci. USA 2011, 108, 13995–13998. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, W. Origin of life: The RNA world. Nature 1986, 319, 618. [Google Scholar] [CrossRef]
- Orgel, L.E. Prebiotic Chemistry and the Origin of the RNA World. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 99–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, J.C.; Hud, N.V.; Williams, L.D. The Ribosome Challenge to the RNA World. J. Mol. Evol. 2015, 80, 143–161. [Google Scholar] [CrossRef] [PubMed]
- Benner, S.A.; Ellington, A.D.; Tauer, A. Modern Metabolism as a Palimpsest of the RNA World. Proc. Natl. Acad. Sci. USA 1989, 86, 7054–7058. [Google Scholar] [CrossRef] [Green Version]
- Joyce, G.F.; Orgel, L.E. Prospects for Understanding the Origin of the RNA World. In The RNA World; Gesteland, R.F., Atkins, J.F., Eds.; Cold Spring Harbor Laboratory Press: Plainview, NY, USA, 1993; pp. 1–25. ISBN 0-87969-380-0. [Google Scholar]
- Butlerow, A. Formation Synthétique d’une Substance Sucrée. CR Acad. Sci. 1861, 53, 145–147. [Google Scholar]
- Breslow, R. On the Mechanism of the Formose Reaction. Tetrahedron Lett. 1959, 1, 22–26. [Google Scholar] [CrossRef]
- Gardner, P.M.; Winzer, K.; Davis, B.G. Sugar Synthesis in a Protocellular Model Leads to a Cell Signalling Response in Bacteria. Nat. Chem. 2009, 1, 377–383. [Google Scholar] [CrossRef]
- Shapiro, R. Prebiotic Ribose Synthesis: A Critical Analysis. Orig. Life Evol. Biosph. 1988, 18, 71–85. [Google Scholar] [CrossRef]
- Ricardo, A.; Carrigan, M.A.; Olcott, A.N.; Benner, S.A. Borate Minerals Stabilize Ribose. Science 2004, 303, 196. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-J.; Ricardo, A.; Illangkoon, H.I.; Kim, M.J.; Carrigan, M.A.; Frye, F.; Benner, S.A. Synthesis of Carbohydrates in Mineral-Guided Prebiotic Cycles. J. Am. Chem. Soc. 2011, 133, 9457–9468. [Google Scholar] [CrossRef]
- Kim, H.-J.; Benner, S.A. Prebiotic Stereoselective Synthesis of Purine and Noncanonical Pyrimidine Nucleotide from Nucleobases and Phosphorylated Carbohydrates. Proc. Natl. Acad. Sci. USA 2017, 114, 11315–11320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, R.; Guntha, S.; Eschenmoser, A. Regioselective α-Phosphorylation of Aldoses in Aqueous Solution. Angew. Chem. Int. Ed. 2000, 39, 2281–2285. [Google Scholar] [CrossRef]
- Orgel, L.E. Prebiotic Adenine Revisited: Eutectics and Photochemistry. Orig. Life Evol. Biosph. 2004, 34, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Saladino, R.; Barontini, M.; Cossetti, C.; Di Mauro, E.; Crestini, C. The Effects of Borate Minerals on the Synthesis of Nucleic Acid Bases, Amino Acids and Biogenic Carboxylic Acids from Formamide. Orig. Life Evol. Biosph. 2011, 41, 317–330. [Google Scholar] [CrossRef]
- Nam, I.; Lee, J.K.; Nam, H.G.; Zare, R.N. Abiotic Production of Sugar Phosphates and Uridine Ribonucleoside in Aqueous Microdroplets. Proc. Natl. Acad. Sci. USA 2017, 114, 12396–12400. [Google Scholar] [CrossRef] [Green Version]
- Becker, S.; Thoma, I.; Deutsch, A.; Gehrke, T.; Mayer, P.; Zipse, H.; Carell, T. A High-Yielding, Strictly Regioselective Prebiotic Purine Nucleoside Formation Pathway. Science 2016, 352, 833–836. [Google Scholar] [CrossRef]
- Becker, S.; Schneider, C.; Okamura, H.; Crisp, A.; Amatov, T.; Dejmek, M.; Carell, T. Wet-Dry Cycles Enable the Parallel Origin of Canonical and Non-Canonical Nucleosides by Continuous Synthesis. Nat. Commun. 2018, 9, 163:1–163:9. [Google Scholar] [CrossRef] [Green Version]
- Becker, S.; Feldmann, J.; Wiedemann, S.; Okamura, H.; Schneider, C.; Iwan, K.; Crisp, A.; Rossa, M.; Amatov, T.; Carell, T. Unified Prebiotically Plausible Synthesis of Pyrimidine and Purine RNA Ribonucleotides. Science 2019, 366, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, R.A.; Ferris, J.P.; Orgel, L.E. Cyanoacetylene in Prebiotic Synthesis. Science 1966, 154, 784–785. [Google Scholar] [CrossRef]
- Anastasi, C.; Crowe, M.A.; Powner, M.W.; Sutherland, J.D. Direct Assembly of Nucleoside Precursors from Two- and Three-Carbon Units. Angew. Chem. Int. Ed. 2006, 45, 6176–6179. [Google Scholar] [CrossRef]
- Powner, M.W.; Gerland, B.; Sutherland, J.D. Synthesis of Activated Pyrimidine Ribonucleotides in Prebiotically Plausible Conditions. Nature 2009, 459, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.A.; Orgel, L.E. Studies in Prebiotic Synthesis. V. Synthesis and Photoanomerization of Pyrimidine Nucleosides. J. Mol. Biol. 1970, 47, 531–543. [Google Scholar] [CrossRef]
- Xu, J.; Chmela, V.; Green, N.J.; Russell, D.A.; Janicki, M.J.; Góra, R.W.; Szabla, R.; Bond, A.D.; Sutherland, J.D. Selective Prebiotic Formation of RNA Pyrimidine and DNA Purine Nucleosides. Nature 2020, 582, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Ritson, D.J.; Sutherland, J.D. Synthesis of Aldehydic Ribonucleotide and Amino Acid Precursors by Photoredox Chemistry. Angew. Chem. Int. Ed. 2013, 52, 5845–5847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, B.H.; Percivalle, C.; Ritson, D.J.; Duffy, C.D.; Sutherland, J.D. Common Origins of RNA, Protein and Lipid Precursors in a Cyanosulfidic Protometabolism. Nat. Chem. 2015, 7, 301–307. [Google Scholar] [CrossRef] [Green Version]
- Kurosawa, K.; Sugita, S.; Ishibashi, K.; Hasegawa, S.; Sekine, Y.; Ogawa, N.O.; Kadono, T.; Ohno, S.; Ohkouchi, N.; Nagaoka, Y.; et al. Hydrogen Cyanide Production Due to Mid-Size Impacts in a Redox-Neutral N2-Rich Atmosphere. Orig. Life Evol. Biosph. 2013, 43, 221–245. [Google Scholar] [CrossRef]
- Pasek, M.A.; Dworkin, J.P.; Lauretta, D.S. A Radical Pathway for Organic Phosphorylation during Schreibersite Corrosion with Implications for the Origin of Life. Geochim. Cosmochim. Acta 2007, 71, 1721–1736. [Google Scholar] [CrossRef]
- Pasek, M.A.; Gull, M.; Herschy, B. Phosphorylation on the Early Earth. Chem. Geol. 2017, 475, 149–170. [Google Scholar] [CrossRef]
- Ritson, D.J.; Battilocchio, C.; Ley, S.V.; Sutherland, J.D. Mimicking the Surface and Prebiotic Chemistry of Early Earth Using Flow Chemistry. Nat. Commun. 2018, 9, 1821:1–1821:10. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Ritson, D.J.; Ranjan, S.; Todd, Z.R.; Sasselov, D.D.; Sutherland, J.D. Photochemical Reductive Homologation of Hydrogen Cyanide Using Sulfite and Ferrocyanide. Chem. Commun. 2018, 54, 5566–5569. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, L.-F.; Bond, A.D.; Sutherland, J.D. Photoredox Chemistry in the Synthesis of 2-Aminoazoles Implicated in Prebiotic Nucleic Acid Synthesis. Chem. Commun. 2020, 56, 13563–13566. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.W.J.; Fahrenbach, A.C. Radicals in Prebiotic Chemistry. Pure Appl. Chem. 2020, 92, 1971–1986. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals (OH/O−) in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.K.; Walker, K.L.; Han, H.S.; Kang, J.; Prinz, F.B.; Waymouth, R.M.; Nam, H.G.; Zare, R.N. Spontaneous Generation of Hydrogen Peroxide from Aqueous Microdroplets. Proc. Natl. Acad. Sci. USA 2019, 116, 19294–19298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, W.M.; Morrison, D.C.; Hamilton, J.G.; Benson, A.A.; Calvin, M. Reduction of Carbon Dioxide in Aqueous Solutions by Ionizing Radiation. Science 1951, 114, 416–418. [Google Scholar] [CrossRef]
- Ebisuzaki, T.; Maruyama, S. Nuclear Geyser Model of the Origin of Life: Driving Force to Promote the Synthesis of Building Blocks of Life. Geosci. Front. 2017, 8, 275–298. [Google Scholar] [CrossRef] [Green Version]
- Adam, Z.R.; Hongo, Y.; Cleaves II, H.J.; Yi, R.; Fahrenbach, A.C.; Yoda, I.; Aono, M. Estimating the Capacity for Production of Formamide by Radioactive Minerals on the Prebiotic Earth. Sci. Rep. 2018, 8, 265:1–265:8. [Google Scholar] [CrossRef]
- Draganić, I.; Draganić, Z.; Petković, L.; Nikolić, A. The Radiation Chemistry of Aqueous Solutions of Simple RCN Compounds. J. Am. Chem. Soc. 1973, 95, 7193–7199. [Google Scholar] [CrossRef]
- Draganić, Z.D.; Draganić, I.G.; Borovičanin, M. The Radiation Chemistry of Aqueous Solutions of Hydrogen Cyanide in the Megarad Dose Range. Radiat. Res. 1976, 66, 42–53. [Google Scholar] [CrossRef]
- Draganić, Z.D.; Niketić, V.; Jovanović, S.; Draganić, I.G. The Radiolysis of Aqueous Ammonium Cyanide: Compounds of Interest to Chemical Evolution Studies. J. Mol. Evol. 1980, 15, 239–260. [Google Scholar] [CrossRef]
- Niketić, V.; Draganić, Z.D.; Nešković, S.; Jovanović, S.; Draganić, I.G. Radiolysis of Aqueous Solutions of Hydrogen Cyanide (pH∼6): Compounds of Interest in Chemical Evolution Studies. J. Mol. Evol. 1983, 19, 184–191. [Google Scholar] [CrossRef]
- Draganić, Z.D.; Draganić, I.G.; Azamar, J.A.; Vujošević, S.I.; Berber, M.D.; Negrón-Mendoza, A. Radiation Chemistry of Overirradiated Aqueous Solutions of Hydrogen Cyanide and Ammonium Cyanide. J. Mol. Evol. 1985, 21, 356–363. [Google Scholar] [CrossRef]
- Draganić, I.G. Radiolysis of Water: A Look at Its Origin and Occurrence in the Nature. Radiat. Phys. Chem. 2005, 72, 181–186. [Google Scholar] [CrossRef]
- Fahrenbach, A.C.; Giurgiu, C.; Tam, C.P.; Li, L.; Hongo, Y.; Aono, M.; Szostak, J.W. Common and Potentially Prebiotic Origin for Precursors of Nucleotide Synthesis and Activation. J. Am. Chem. Soc. 2017, 139, 8780–8783. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Prywes, N.; Tam, C.P.; O’Flaherty, D.K.; Lelyveld, V.S.; Izgu, E.C.; Pal, A.; Szostak, J.W. Enhanced Nonenzymatic RNA Copying with 2-Aminoimidazole Activated Nucleotides. J. Am. Chem. Soc. 2017, 139, 1810–1813. [Google Scholar] [CrossRef]
- Yi, R.; Hongo, Y.; Yoda, I.; Adam, Z.R.; Fahrenbach, A.C. Radiolytic Synthesis of Cyanogen Chloride, Cyanamide and Simple Sugar Precursors. ChemistrySelect 2018, 3, 10169–10174. [Google Scholar] [CrossRef]
- Shapiro, R. Small Molecule Interactions Were Central to the Origin of Life. Q. Rev. Biol. 2006, 81, 105–126. [Google Scholar] [CrossRef] [Green Version]
- Dyson, F. Origins of Life; Cambridge University Press: Cambridge, UK, 1999; ISBN 0-521-62668-4. [Google Scholar]
- Braakman, R.; Smith, E. The Compositional and Evolutionary Logic of Metabolism. Phys. Biol. 2012, 10, 011001:1–011001:62. [Google Scholar] [CrossRef]
- Buchanan, B.B.; Arnon, D.I. A Reverse KREBS Cycle in Photosynthesis: Consensus at Last. Photosynth. Res. 1990, 24, 47–53. [Google Scholar] [CrossRef]
- Huynen, M.A.; Dandekar, T.; Bork, P. Variation and Evolution of the Citric-Acid Cycle: A Genomic Perspective. Trends Microbiol. 1999, 7, 281–291. [Google Scholar] [CrossRef]
- Orgel, L.E. The Implausibility of Metabolic Cycles on the Prebiotic Earth. PLoS Biol. 2008, 6, e18:005–e18:0013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggins, B.R.; Brown, E.M.; McNeill, E.A.; Grimshaw, J. Carbon Dioxide Fixation by Electrochemical Reduction in Water to Oxalate and Glyoxylate. Tetrahedron Lett. 1988, 29, 945–948. [Google Scholar] [CrossRef]
- Varma, S.J.; Muchowska, K.B.; Chatelain, P.; Moran, J. Native Iron Reduces CO2 to Intermediates and End-Products of the Acetyl-CoA Pathway. Nat. Ecol. Evol. 2018, 2, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Goldford, J.E.; Hartman, H.; Smith, T.F.; Segrè, D. Remnants of an Ancient Metabolism without Phosphate. Cell 2017, 168, 1126–1134. [Google Scholar] [CrossRef] [Green Version]
- Canfield, D.E.; Habicht, K.S.; Thamdrup, B. The Archean Sulfur Cycle and the Early History of Atmospheric Oxygen. Science 2000, 288, 658–661. [Google Scholar] [CrossRef]
- Rouxel, O.J.; Bekker, A.; Edwards, K.J. Iron Isotope Constraints on the Archean and Paleoproterozoic Ocean Redox State. Science 2005, 307, 1088–1091. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.A.; Turchyn, A.V.; Ralser, M. Non-Enzymatic Glycolysis and Pentose Phosphate Pathway-Like Reactions in a Plausible Archean Ocean. Mol. Syst. Biol. 2014, 10, 725:1–725:12. [Google Scholar] [CrossRef]
- Keller, M.A.; Zylstra, A.; Castro, C.; Turchyn, A.V.; Griffin, J.L.; Ralser, M. Conditional Iron and pH-Dependent Activity of a Non-Enzymatic Glycolysis and Pentose Phosphate Pathway. Sci. Adv. 2016, 2, e1501235:1–e1501235:11. [Google Scholar] [CrossRef] [Green Version]
- Semenov, S.N.; Kraft, L.J.; Ainla, A.; Zhao, M.; Baghbanzadeh, M.; Campbell, V.E.; Kang, K.; Fox, J.M.; Whitesides, G.M. Autocatalytic, Bistable, Oscillatory Networks of Biologically Relevant Organic Reactions. Nature 2016, 537, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Scharf, C.; Virgo, N.; Cleaves II, H.J.; Aono, M.; Aubert-Kato, N.; Aydinoglu, A.; Barahona, A.; Barge, L.M.; Benner, S.A.; Biehl, M.; et al. A Strategy for Origins of Life Research. Astrobiology 2015, 15, 1031–1042. [Google Scholar] [CrossRef] [Green Version]
- Adam, Z.; Fahrenbach, A.C.; Jacobson, S.M.; Kacar, B.; Zubarev, D.Y. Radiolysis Generates a Complex Organosynthetic Chemical Network. ChemRxiv Prepr. 2019. [Google Scholar] [CrossRef]
- Schwieterman, E.W.; Kiang, N.Y.; Parenteau, M.N.; Harman, C.E.; DasSarma, S.; Fisher, T.M.; Arney, G.N.; Hartnett, H.E.; Reinhard, C.T.; Olson, S.L.; et al. Exoplanet Biosignatures: A Review of Remotely Detectable Signs of Life. Astrobiology 2018, 18, 663–708. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, Q.P.; Adam, Z.R.; Fahrenbach, A.C. Prebiotic Reaction Networks in Water. Life 2020, 10, 352. https://doi.org/10.3390/life10120352
Tran QP, Adam ZR, Fahrenbach AC. Prebiotic Reaction Networks in Water. Life. 2020; 10(12):352. https://doi.org/10.3390/life10120352
Chicago/Turabian StyleTran, Quoc Phuong, Zachary R. Adam, and Albert C. Fahrenbach. 2020. "Prebiotic Reaction Networks in Water" Life 10, no. 12: 352. https://doi.org/10.3390/life10120352