Atomistic View of Mercury Cycling in Polar Snowpacks: Probing the Role of Hg2+ Adsorption Using Ab Initio Calculations


Abstract
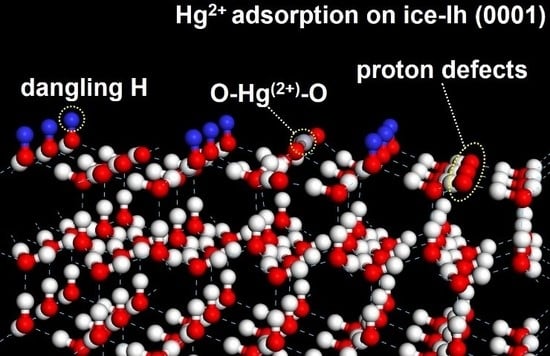
:
1. Introduction
2. Methods
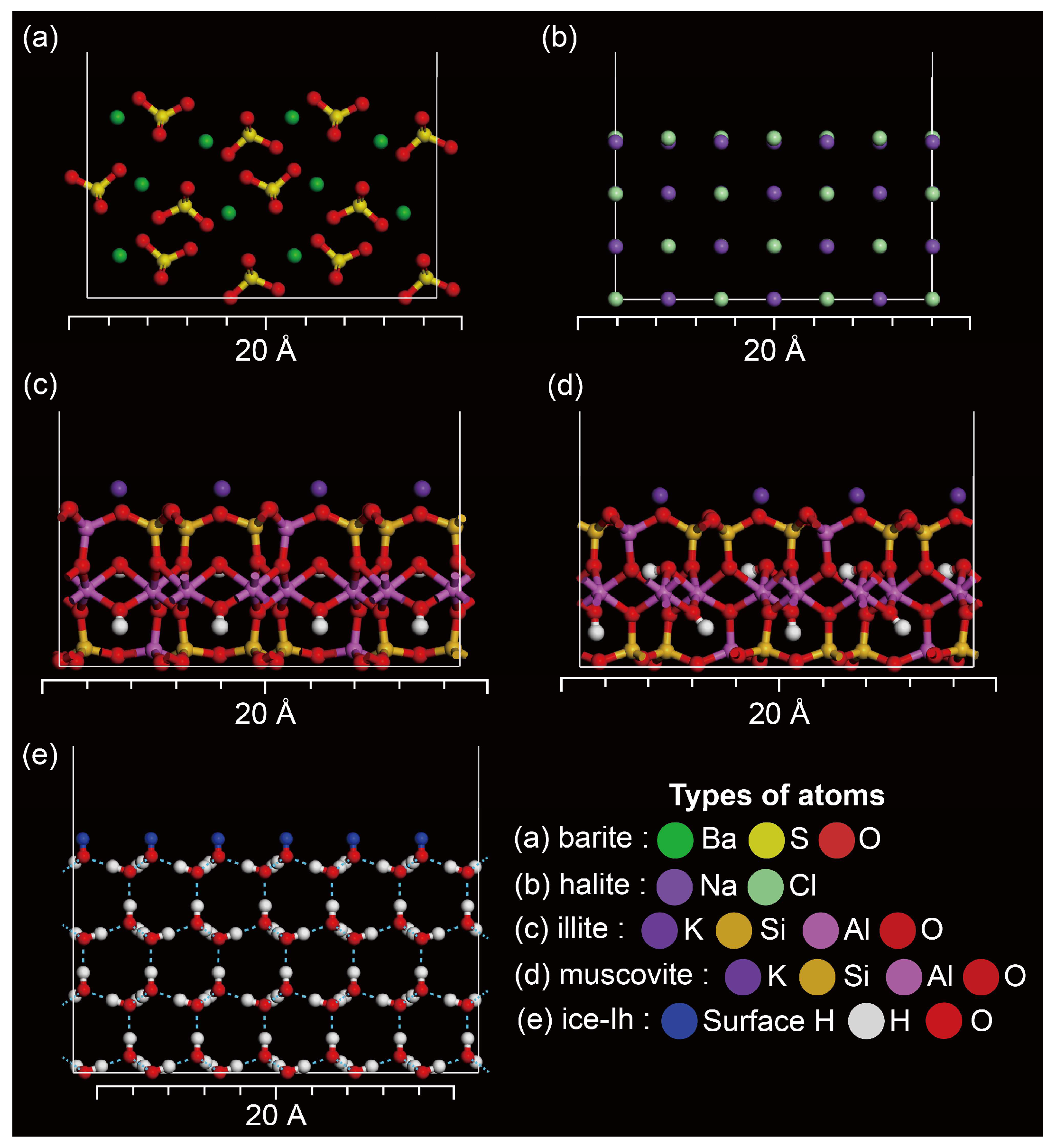
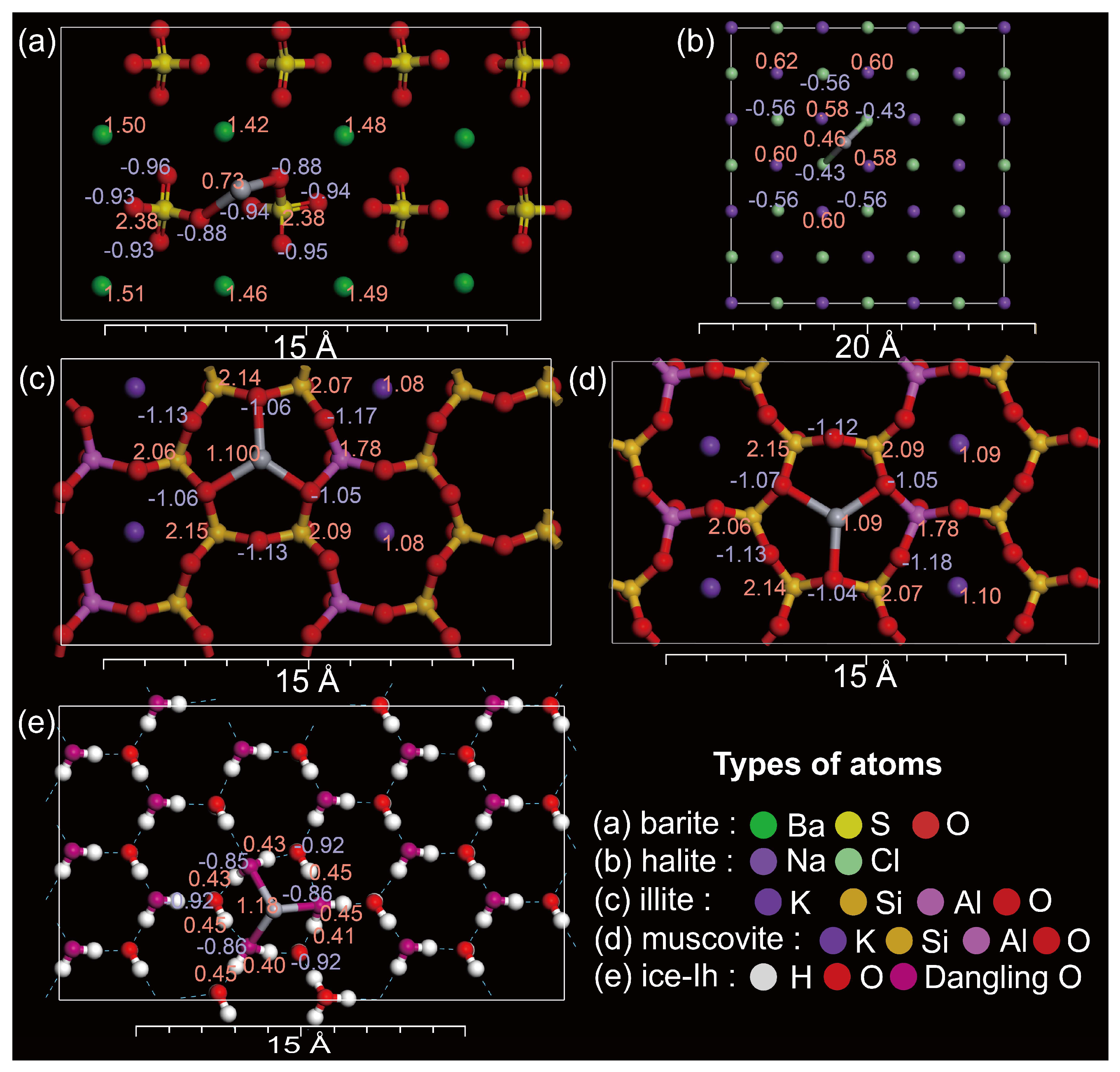
2.1. Selected Adsorbents in the Polar Snowpack
2.1.1. Cleaved Surface Structure of Barite
2.1.2. Cleaved Surface Structure of Halite
2.1.3. Cleaved Surface Structures of Illite and Muscovite
2.1.4. Cleaved Surface Structure of Ice-Ih
2.1.5. Constructing the Cleaved Surface Structures
2.2. Binding Energy Calculation
2.3. Structure Optimization and Electronic Structure Calculations
3. Results and Discussion
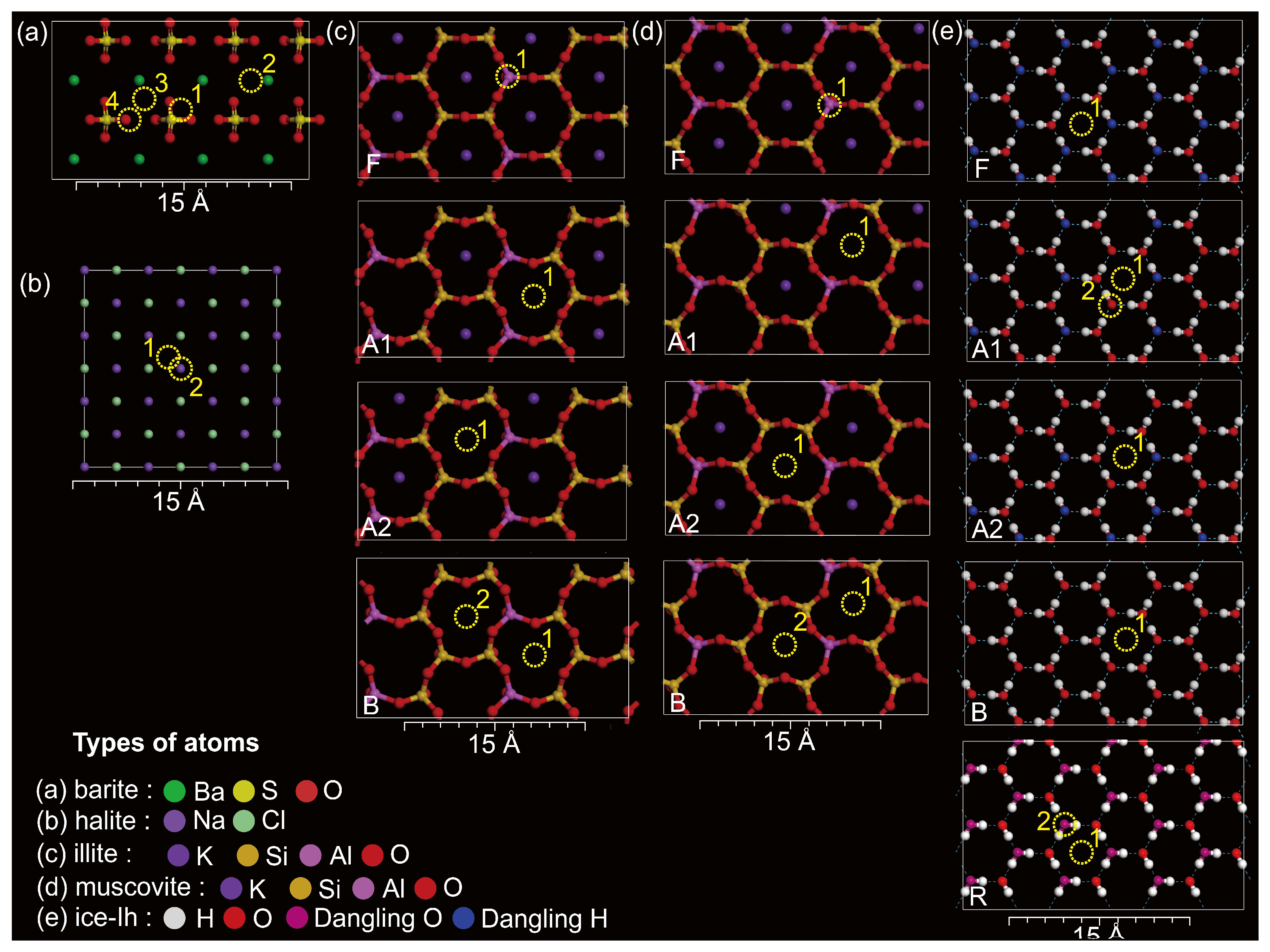
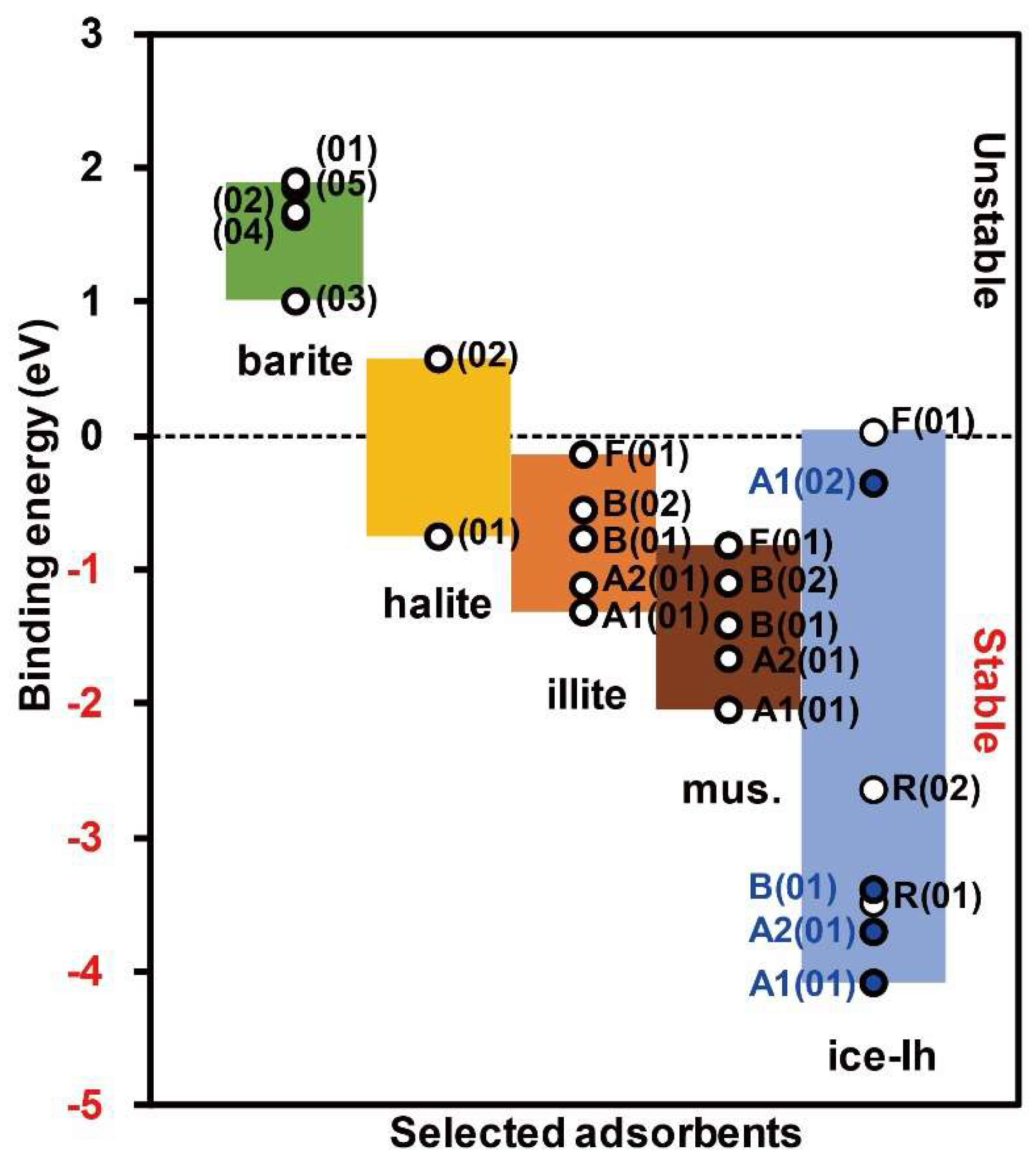
3.1. Hg2+ on Barite
3.2. Hg2+ on Halite
3.3. Hg2+ on Illite and Muscovite
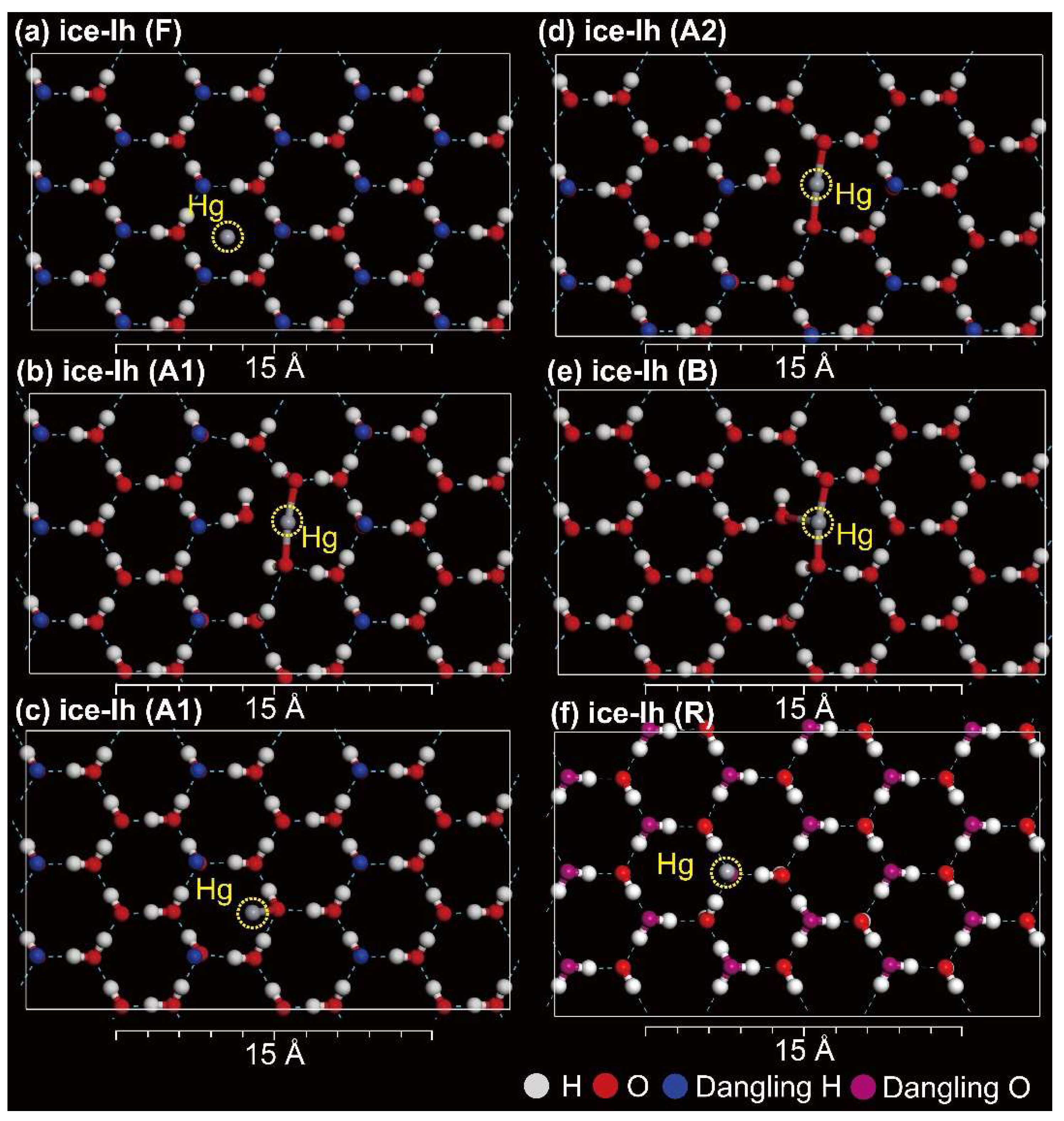
3.4. Hg2+ on Ice-Ih
3.5. Relative Adsorption Affinities to the Selected Adsorbents
3.6. Implication for Hg Cycling between the Polar Atmosphere and Snowpack
3.7. Limitation and Future Study
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

References
- Han, Y.; Huh, Y.; Hur, S.D.; Hong, S.; Chung, J.W.; Motoyama, H. Net deposition of mercury to the Antarctic Plateau enhanced by sea salt. Sci. Total Environ. 2017, 583, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Jitaru, P.; Gabrielli, P.; Marteel, A.; Plane, J.M.C.; Planchon, F.A.M.; Gauchard, P.A.; Ferrari, C.P.; Boutron, C.F.; Adams, F.C.; Hong, S.; et al. Atmospheric depletion of mercury over Antarctica during glacial periods. Nat. Geosci. 2009, 2, 505. [Google Scholar] [CrossRef]
- Dommergue, A.; Bahlmann, E.; Ebinghaus, R.; Ferrari, C.; Boutron, C. Laboratory simulation of Hg0 emissions from a snowpack. Anal. Bioanal. Chem. 2007, 388, 319–327. [Google Scholar] [CrossRef] [PubMed]
- AMAP. AMAP Assessment 2011: Mercury in the Arctic Arctic Monitoring and Assessment Programme (AMAP); AMAP: Oslo, Norway, 2011; p. 193. [Google Scholar]
- Lindberg, S.E.; Brooks, S.; Lin, C.J.; Scott, K.J.; Landis, M.S.; Stevens, R.K.; Goodsite, M.; Richter, A. Dynamic oxidation of gaseous mercury in the arctic troposphere at polar sunrise. Environ. Sci. Technol. 2002, 36, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Steffen, A.; Douglas, T.; Amyot, M.; Ariya, P.; Aspmo, K.; Berg, T.; Bottenheim, J.; Brooks, S.; Cobbett, F.; Dastoor, A.; et al. A synthesis of atmospheric mercury depletion event chemistry linking atmosphere, snow and water. Atmos. Chem. Phys. Discuss. 2007, 7, 10837–10931. [Google Scholar] [CrossRef]
- Bartels-Rausch, T.; Krysztofiak, G.; Bernhard, A.; Schläppi, M.; Schwikowski, M.; Ammann, M. Photoinduced reduction of divalent mercury in ice by organic matter. Chemosphere 2011, 82, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Douglas, T.A.; Sturm, M.; Simpson, W.R.; Blum, J.D.; Alvarez-Aviles, L.; Keeler, G.J.; Perovich, D.K.; Biswas, A.; Johnson, K. Influence of snow and ice crystal formation and accumulation on mercury deposition to the arctic. Environ. Sci. Technol. 2008, 42, 1542–1551. [Google Scholar] [CrossRef]
- Bartels-Rausch, T.; Huthwelker, T.; Jöri, M.; Gäggeler, H.W.; Ammann, M. Interaction of gaseous elemental mercury with snow surfaces: Laboratory investigation. Environ. Res. Lett. 2008, 3, 045009. [Google Scholar] [CrossRef]
- Mann, E.A.; Mallory, M.L.; Ziegler, S.E.; Avery, T.S.; Tordon, R.; O’Driscoll, N.J. Photoreducible mercury loss from arctic snow is influenced by temperature and snow age. Environ. Sci. Technol. 2015, 49, 12120–12126. [Google Scholar] [CrossRef]
- Angot, H.; Magand, O.; Helmig, D.; Ricaud, P.; Quennehen, B.; Gallée, H.; Del Guasta, M.; Sprovieri, F.; Pirrone, N.; Savarino, J.; et al. New insights into the atmospheric mercury cycling in central Antarctica and implications on a continental scale. Atmos. Chem. Phys. 2016, 16, 8249–8264. [Google Scholar] [CrossRef] [Green Version]
- Lalonde, J.D.; Poulain, A.J.; Amyot, M. The role of mercury redox reactions in snow on snow-to-air mercury transfer. Environ. Sci. Technol. 2002, 36, 174–178. [Google Scholar] [CrossRef] [PubMed]
- King, M.D.; Simpson, W.R. Extinction of UV radiation in Arctic snow at Alert, Canada (82 °N). J. Geophys. Res. Atmos. 2001, 106, 12499–12507. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Tse, J.S. Ab Initio molecular dynamics with density functional theory. Annu. Rev. Phys. Chem. 2002, 53, 249–290. [Google Scholar] [CrossRef] [PubMed]
- Marx, D.; Hutter, J. Ab initio molecular dynamics: Theory and implementation. Mod. Methods Algorithms Quantum Chem. 2000, 1, 301–449. [Google Scholar]
- Payne, M.C.; Teter, M.P.; Allan, D.C.; Arias, T.A.; Joannopoulos, J.D. Iterative minimization techniques for ab initio total-energy calculations: Molecular dynamics and conjugate gradients. Rev. Mod. Phys. 1992, 64, 1045–1096. [Google Scholar] [CrossRef]
- Pastore, G.; Smargiassi, E.; Buda, F. Theory of ab initio molecular-dynamics calculations. Phys. Rev. A 1991, 44, 6334–6347. [Google Scholar] [CrossRef]
- Asaduzzaman, A.M.; Schreckenbach, G. Adsorption of Na and Hg on the ice (Ih) surface: A density-functional study. J. Phys. Chem. C 2010, 114, 2941–2946. [Google Scholar] [CrossRef]
- Li, Y.H.; Schoonmaker, J.E. Chemical composition and mineralogy of marine sediments. In Treatise on Geochemistry; Holland, H.D., Turekian, K.K., Eds.; Pergamon: Oxford, UK, 2003; pp. 1–35. [Google Scholar]
- Obenholzner, J.; Schroettner, H.; Delgado, H. Barite aerosol particles from volcanic plumes and fumaroles-FESEM/EDS analysis. In Proceedings of the EGS-AGU-EUG Joint Assembly, Nice, France, 6–11 April 2003; p. 8119. [Google Scholar]
- Gaudichet, A.; De Angelis, M.; Lefevre, R.; Petit, J.; Korotkevitch, Y.; Petrov, V. Mineralogy of insoluble particles in the Vostok Antarctic ice core over the last climatic cycle (150 kyr). Geophys. Res. Lett. 1988, 15, 1471–1474. [Google Scholar] [CrossRef]
- Sala, M.; Dapiaggi, M.; Delmonte, B.; Marino, F.; Artioli, G.; Maggi, V.; Revel-Rolland, M.; Petit, J. Mineralogical composition of EPICA Dome C aeolian ice core dust. In Proceedings of the EGU General Assembly, Vienna, Austria, 19–24 April 2009; p. 6230. [Google Scholar]
- Kuwahara, Y. Muscovite surface structure imaged by fluid contact mode AFM. Phys. Chem. Miner. 1999, 26, 198–205. [Google Scholar] [CrossRef]
- Ostendorf, F.; Schmitz, C.; Hirth, S.; Kühnle, A.; Kolodziej, J.J.; Reichling, M. How flat is an air-cleaved mica surface? Nanotechnology 2008, 19, 305705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, K.; Chang, C.C. Electric dipoles on clean mica surfaces. Surf. Sci. 1969, 14, 39–51. [Google Scholar] [CrossRef]
- Pan, D.; Liu, L.M.; Tribello, G.A.; Slater, B.; Michaelides, A.; Wang, E. Surface energy and surface proton order of ice-Ih. Phys. Rev. Lett. 2008, 101, 155703. [Google Scholar] [CrossRef] [PubMed]
- Ding, P.; Li-Min, L.; Gareth, A.T.; Ben, S.; Angelos, M.; Enge, W. Surface energy and surface proton order of the ice Ih basal and prism surfaces. J. Phys. Condens. Matter 2010, 22, 074209. [Google Scholar] [CrossRef] [Green Version]
- Buch, V.; Groenzin, H.; Li, I.; Shultz, M.J.; Tosatti, E. Proton order in the ice crystal surface. Proc. Natl. Acad. Sci. USA 2008, 105, 5969–5974. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.J. A further refinement of the barite structure. Can. Mineral. 1977, 15, 522–526. [Google Scholar]
- Walker, D.; Verma, P.K.; Cranswick, L.M.D.; Jones, R.L.; Clark, S.M.; Buhre, S. Halite-sylvite thermoelasticity. Am. Mineral. 2004, 89, 204. [Google Scholar] [CrossRef]
- Drits, V.A.; Zviagina, B.B.; McCarty, D.K.; Salyn, A.L. Factors responsible for crystal-chemical variations in the solid solutions from illite to aluminoceladonite and from glauconite to celadonite. Am. Mineral. 2010, 95, 348–361. [Google Scholar] [CrossRef]
- Catti, M.; Ferraris, G.; Hull, S.; Pavese, A. Powder neutron diffraction study of 2M1 muscovite at room pressure and at 2 GPa. Eur. J. Mineral. 1994, 6, 171–178. [Google Scholar] [CrossRef]
- Goto, A.; Hondoh, T.; Mae, S. The electron density distribution in ice-Ih determined by single-crystal X-ray diffractometry. J. Chem. Phys. 1990, 93, 1412–1417. [Google Scholar] [CrossRef]
- Bernal, J.D.; Fowler, R.H. A theory of water and ionic solution, with particular reference to hydrogen and hydroxyl ions. J. Chem. Phys. 1933, 1, 515–548. [Google Scholar] [CrossRef]
- Fenter, P.; McBride, M.T.; Srajer, G.; Sturchio, N.C.; Bosbach, D. Structure of barite (001)- and (210)- water interfaces. J. Phys. Chem. B 2001, 105, 8112–8119. [Google Scholar] [CrossRef]
- Davey, R.J.; Black, S.N.; Bromley, L.A.; Cottier, D.; Dobbs, B.; Rout, J.E. Molecular design based on recognition at inorganic surfaces. Nature 1991, 353, 549–550. [Google Scholar] [CrossRef]
- Black, S.N.; Bromley, L.A.; Cottier, D.; Davey, R.J.; Dobbs, B.; Rout, J.E. Interactions at the organic/inorganic interface - Binding motifs for phosphonates at the surface of barite crystals. J. Chem. Soc. Faraday Trans. 1991, 87, 3409–3414. [Google Scholar] [CrossRef]
- Hartman, P.; Strom, C.S. Structural morphology of crystals with the barite (BaSO4) structure: A revision and extension. J. Cryst. Growth 1989, 97, 502–512. [Google Scholar] [CrossRef]
- Murray, H.H. Structure and composition of the clay minerals and their physical and chemical properties. In Developments in Clay Science; Murray, H.H., Ed.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 2, pp. 7–31. [Google Scholar]
- Stixrude, L.; Peacor, D.R. First-principles study of illite–smectite and implications for clay mineral systems. Nature 2002, 420, 165. [Google Scholar] [CrossRef] [PubMed]
- Teich-McGoldrick, S.L.; Greathouse, J.A.; Cygan, R.T. Molecular dynamics simulations of uranyl adsorption and structure on the basal surface of muscovite. Mol. Simul. 2014, 40, 610–617. [Google Scholar] [CrossRef]
- Wang, L.; Liu, R.; Hu, Y.; Sun, W. pH effects on adsorption behavior and self-aggregation of dodecylamine at muscovite/aqueous interfaces. J. Mol. Graph. Model. 2016, 67, 62–68. [Google Scholar] [CrossRef]
- Odelius, M.; Bernasconi, M.; Parrinello, M. Two dimensional ice adsorbed on mica surface. Phys. Rev. Lett. 1997, 78, 2855–2858. [Google Scholar] [CrossRef]
- Purton, J.; Allan, N.; Blundy, J. Impurity cations in the bulk and the {001} surface of muscovite: An atomistic simulation study. J. Mater. Chem. 1997, 7, 1947–1951. [Google Scholar] [CrossRef]
- Kenneth, G.L. The physics of snow crystals. Rep. Prog. Phys. 2005, 68, 855. [Google Scholar] [CrossRef]
- Olijve, L.L.; Meister, K.; DeVries, A.L.; Duman, J.G.; Guo, S.; Bakker, H.J.; Voets, I.K. Blocking rapid ice crystal growth through nonbasal plane adsorption of antifreeze proteins. Proc. Natl. Acad. Sci. USA 2016, 113, 3740–3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, E.A.; Monserrat, B.; Needs, R.J. Vibrational effects on surface energies and band gaps in hexagonal and cubic ice. J. Chem. Phys. 2016, 145, 044703. [Google Scholar] [CrossRef] [PubMed]
- Shoaib, M.A.; Choi, C.H. Adsorptions of HOCl on ice surface: Effects of long-range electrostatics, surface heterogeneity, and hydrogen disorders of ice crystal. J. Phys. Chem. C 2012, 116, 3694–3701. [Google Scholar] [CrossRef]
- Park, S.C.; Moon, E.S.; Kang, H. Some fundamental properties and reactions of ice surfaces at low temperatures. Phys. Chem. Chem. Phys. 2010, 12, 12000–12011. [Google Scholar] [CrossRef] [PubMed]
- Materer, N.; Starke, U.; Barbieri, A.; Van Hove, M.A.; Somorjai, G.A.; Kroes, G.J.; Minot, C. Molecular surface structure of ice (0001): Dynamical low-energy electron diffraction, total-energy calculations and molecular dynamics simulations. Surf. Sci. 1997, 381, 190–210. [Google Scholar] [CrossRef]
- Eisenberg, D.; Coulson, C.A. Energy of formation of D-defects in ice. Nature 1963, 199, 368–369. [Google Scholar] [CrossRef]
- de Koning, M.; Antonelli, A. On the trapping of Bjerrum defects in ice-Ih: The case of the molecular vacancy. J. Phys. Chem. B 2007, 111, 12537–12542. [Google Scholar] [CrossRef]
- Agmon, N. The Grotthuss mechanism. Chem. Phys. Lett. 1995, 244, 456–462. [Google Scholar] [CrossRef]
- Francis, G.P.; Payne, M.C. Finite basis set corrections to total energy pseudopotential calculations. J. Phys. Condens. Matter 1990, 2, 4395. [Google Scholar] [CrossRef]
- Liu, T.; Xue, L.; Guo, X.; Zheng, C.G. DFT study of mercury adsorption on α-Fe2O3 surface: Role of oxygen. Fuel 2014, 115, 179–185. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Tkatchenko, A.; Scheffler, M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [PubMed]
- Gillan, M.J.; Alfè, D.; Michaelides, A. Perspective: How good is DFT for water? J. Chem. Phys. 2016, 144, 130901. [Google Scholar] [CrossRef] [PubMed]
- Parq, J.H.; Lee, S.K.; Lee, S.M.; Yu, J. Ab initio study of elastic properties of high-pressure polymorphs of CO2 phases II and V. J. Phys. Chem. C 2016, 120, 23152–23164. [Google Scholar] [CrossRef]
- Lee, B.H.; Lee, S.K. Effect of lattice topology on the adsorption of benzyl alcohol on kaolinite surfaces: Quantum chemical calculations of geometry optimization, binding energy, and NMR chemical shielding. Am. Mineral. 2009, 94, 1392–1404. [Google Scholar] [CrossRef]
- Barzilai, J.; Borwein, J.M. Two-point step size gradient methods. IMA J. Numer. Anal. 1988, 8, 141–148. [Google Scholar] [CrossRef]
- Schwarz, K.; Blaha, P.; Madsen, G.K.H. Electronic structure calculations of solids using the WIEN2k package for material sciences. Comput. Phys. Commun. 2002, 147, 71–76. [Google Scholar] [CrossRef]
- Hofer, T.S.; Randolf, B.R.; Rode, B.M. The hydration of the mercury(I)-dimer—A quantum mechanical charge field molecular dynamics study. Chem. Phys. 2008, 349, 210–218. [Google Scholar] [CrossRef]
- Rode, B.M.; Hofer, T.S.; Randolf, B.R.; Schwenk, C.F.; Xenides, D.; Vchirawongkwin, V. Ab initio quantum mechanical charge field (QMCF) molecular dynamics: A QM/MM—MD procedure for accurate simulations of ions and complexes. Theor. Chem. Acc. 2006, 115, 77–85. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef] [PubMed]
- Crecelius, E.; Trefry, J.; McKinley, J.; Lasorsa, B.; Trocine, R. Study of Barite Solubility and the Release of Trace Components to the Marine Environment; US Department of the Interior (Minerals Management Service, Gulf of Mexico Region): New Orleans, LA, USA, 2007; Volume OC5 Study MMS 2007-061, pp. 1–176.
- Denney, D. Fate of mercury in drilling-fluid barite in the marine environment. J. Pet. Tech. 2003, 55, 66–67. [Google Scholar] [CrossRef]
- Uddin, M.K. A review on the adsorption of heavy metals by clay minerals, with special focus on the past decade. Chem. Eng. J. 2017, 308, 438–462. [Google Scholar] [CrossRef]
- Churchman, G.J.; Gates, W.P.; Theng, B.K.G.; Yuan, G. Clays and clay minerals for pollution control. In Developments in Clay Science; Bergaya, F., Theng, B.K.G., Lagaly, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 1, pp. 625–675. [Google Scholar]
- Shetaya, W.H.; Huang, J.H.; Osterwalder, S.; Mestrot, A.; Bigalke, M.; Alewell, C. Sorption kinetics of isotopically labelled divalent mercury (196Hg2+) in soil. Chemosphere 2019, 221, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Nagy, K.L.; Park, C.Y.; Fenter, P. Enhanced uptake and modified distribution of mercury(II) by fulvic acid on the muscovite (001) surface. Environ. Sci. Technol. 2009, 43, 5295–5300. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Nagy, K.L.; Park, C.; Fenter, P. Heavy metal sorption at the muscovite (001)–fulvic acid interface. Environ. Sci. Technol. 2011, 45, 9574–9581. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.; Wu, P.; Zhu, Y.; Liu, S.; Zhu, N. Comparative study of Hg(II) adsorption by thiol- and hydroxyl-containing bifunctional montmorillonite and vermiculite. Appl. Surf. Sci. 2015, 356, 91–101. [Google Scholar] [CrossRef]
- Lumsdon, D.G.; Evans, L.J.; Bolton, K.A. The influence of pH and chloride on the retention of cadmium, lead, mercury, and zinc by soils. J. Soil Contam. 1995, 4, 137–150. [Google Scholar] [CrossRef]
- Biester, H.; Zimmer, H. Solubility and changes of mercury binding forms in contaminated soils after immobilization treatment. Environ. Sci. Technol. 1998, 32, 2755–2762. [Google Scholar] [CrossRef]
- Moon, E.S.; Kim, Y.S.; Shin, S.H.; Kang, H. Asymmetric transport efficiencies of positive and negative ion defects in amorphous ice. Phys. Rev. Lett. 2012, 108, 226103. [Google Scholar] [CrossRef]
- Lee, C.W.; Lee, P.R.; Kang, H. Protons at ice surfaces. Angew. Chem. Int. Ed. 2006, 45, 5529–5533. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Kang, H.; Kang, H. Tunneling diffusion of excess protons in amorphous solid water at 10 and 80 K. J. Phys. Chem. C 2019, 123, 3657–3663. [Google Scholar] [CrossRef]
- Moon, E.S.; Kang, H.; Oba, Y.; Watanabe, N.; Kouchi, A. Direct evidence for ammonium ion formation in ice through ultraviolet-induced acid-base reaction of NH3 with H3O+. Astrophys. J. 2010, 713, 906–911. [Google Scholar] [CrossRef]
- Lee, C.W.; Kang, H. UV Photolysis of glycine on ice films: Implication for photosynthesis and photodestruction of amino acids in interstellar medium. Bull. Korean Chem. Soc. 2015, 36, 784–788. [Google Scholar] [CrossRef]
- Kang, H. Chemistry of ice surfaces: Elementary reaction steps on ice studied by reactive ion scattering. Acc. Chem. Res. 2005, 38, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Shoaib, M.A.; Choi, C.H. Adsorptions of formic and acetic acids on ice surface: Surface binding configurations and a possibility of interfacial proton transfer. J. Phys. Chem. C 2013, 117, 4181–4188. [Google Scholar] [CrossRef]
- Shoaib, M.A.; Choi, C.H. Na+, F–, Br–, and Cl– adsorptions and penetrations on an ice surface. ACS Earth Space Chem. 2018, 2, 56–63. [Google Scholar] [CrossRef]
- Moon, E.S.; Yoon, J.; Kang, H. Energy barrier of proton transfer at ice surfaces. J. Chem. Phys. 2010, 133, 044709. [Google Scholar] [CrossRef]
- Tonigold, K.; Groß, A. Adsorption of small aromatic molecules on the (111) surfaces of noble metals: A density functional theory study with semiempirical corrections for dispersion effects. J. Chem. Phys. 2010, 132, 224701. [Google Scholar] [CrossRef] [Green Version]
- Fukazawa, H.; Ikeda, S.; Mae, S. Incoherent inelastic neutron scattering measurements on ice-XI; the proton-ordered phase of ice-Ih doped with KOH. Chem. Phys. Lett. 1998, 282, 215–218. [Google Scholar] [CrossRef]
- Watkins, M.; VandeVondele, J.; Slater, B. Point defects at the ice (0001) surface. Proc. Natl. Acad. Sci. USA 2010, 107, 12429. [Google Scholar] [CrossRef] [PubMed]
- Watkins, M.; Pan, D.; Wang, E.G.; Michaelides, A.; VandeVondele, J.; Slater, B. Large variation of vacancy formation energies in the surface of crystalline ice. Nat. Mater. 2011, 10, 794–798. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zhang, W.; Li, Y. The glass transition behaviors of low-density amorphous ice films with different thicknesses. J. Chem. Phys. 2010, 133, 204504. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Yoo, C.S. High density amorphous ice at room temperature. Proc. Natl. Acad. Sci. USA 2011, 108, 7685–7688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojamae, L. Crystalline ice: Amorphous on the surface. Nat. Mater. 2011, 10, 725–726. [Google Scholar] [CrossRef] [PubMed]
- Refson, K.; Tulip, P.R.; Clark, S.J. Variational density-functional perturbation theory for dielectrics and lattice dynamics. Phys. Rev. B 2006, 73, 155114. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal | Barite | Halite | Illite | Muscovite | Ice-Ih |
| System | Orthorhombic | Cubic | Monoclinic | Monoclinic | Monoclinic |
| Space group | (62) PNMA | (225) FMM | (12) C2/M | (15) C2/C | (9) CC |
| Lattice parameters of the unit cell | |||||
| a (Å) | 8.8842 | 5.6400 | 5.2021 | 5.2108 | 7.8219 |
| b (Å) | 5.4559 | 5.6400 | 8.9797 | 9.0399 | 8.6299 |
| c (Å) | 7.1569 | 5.6400 | 10.2260 | 20.0210 | 8.6299 |
| α (°) | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 |
| β (°) | 90.00 | 90.00 | 95.76 | 101.57 | 121.55 |
| γ (°) | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 |
| Supercell sizes used to construct surface structure models | |||||
| N × N × N | 2 × 2 × 2 | 3 × 3 × 3 | 2 × 2 × 1 | 2 × 2 × 1 | 3 × 3 × 3 |
| Direction of cleavage plane | |||||
| h k l | (001) | (001) | (001) | (001) | (0001) |
| References | |||||
| [30] | [31] | [32] | [33] | [34,35] | |
| Cleaved surface structures | |||||
| a (Å) | 17.9322 | 16.1735 | 10.4042 | 10.4233 | 13.1921 |
| b (Å) | 10.8498 | 16.1735 | 17.9594 | 18.1650 | 22.8061 |
| c (Å) | 50.1536 | 48.0867 | 48.4581 | 43.5444 | 52.6997 |
| Area of surface (Å2) | 194.5606 | 261.5820 | 186.8532 | 189.3395 | 300.8610 |
| Thickness of layer (Å) | 10.1882 | 8.2059 | 7.9319 | 7.8718 | 13.0837 |
| Vacuum thickness (Å) | 39.9655 | 39.8808 | 40.5262 | 35.6725 | 39.6160 |
| Number of atoms (depending on atomic configurations) | |||||
| 140 | 196 | 160–168 | 160–168 | 414–432 | |
| Energy (eV) | ECD − EAB 1 | EC − EA 2 | ED − EB 3 | EBind4 |
|---|---|---|---|---|
| Barite | ||||
| Site 01 | 1.9653 | 0.0699 | 0.0002 | 1.8952 |
| Site 02 | 1.7905 | 0.1160 | 0.0001 | 1.6744 |
| Site 03 | 1.5100 | 0.4945 | 0.0002 | 1.0154 |
| Site 04 | 1.7683 | 0.1315 | 0.0002 | 1.6367 |
| Site 05 | 1.8801 | 0.0286 | 0.0003 | 1.8513 |
| Halite | ||||
| Site 01 | 0.3853 | 1.1386 | 0.0001 | −0.7535 |
| Site 02 | 0.6281 | 0.0511 | −0.0001 | 0.5771 |
| Illite | ||||
| Type F site 01 | 0.8477 | 0.9866 | 0.0001 | −0.1390 |
| Type A1 site 01 | 0.0675 | 1.3775 | −0.0001 | −1.3098 |
| Type A2 site 01 | 0.2617 | 1.3778 | −0.0001 | −1.1160 |
| Type B site 01 | 0.0844 | 0.8579 | −0.0002 | −0.7732 |
| Type B site 02 | 0.2820 | 0.8387 | 0.0001 | −0.5568 |
| Muscovite | ||||
| Type F site 01 | 0.1645 | 0.9753 | 0.0002 | −0.8109 |
| Type A1 site 01 | −0.5770 | 1.4648 | −0.0004 | −2.0415 |
| Type A2 site 01 | −0.2466 | 1.4187 | −0.0001 | −1.6653 |
| Type B site 01 | −0.5237 | 0.8904 | −0.0003 | −1.4138 |
| Type B site 02 | −0.2129 | 0.8790 | −0.0001 | −1.0918 |
| Ice-Ih5 | ||||
| Type F site 01 | 0.0440 | −0.0001 | −0.0001 | 0.0442 |
| Type R site 01 | −2.2870 | 1.1908 | 0.0000 | −3.4778 |
| Type R site 02 | −1.8707 | 0.7601 | 0.0001 | −2.6309 |
| Type A1 site 01 | −3.0650 | 1.0133 | 0.0002 | −4.0785 |
| Type A1 site 02 | −0.2325 | 0.1247 | −0.0001 | −0.3571 |
| Type A2 site 01 | −2.7403 | 0.9509 | 0.0003 | −3.6916 |
| Type B site 01 | −2.7651 | 0.6175 | −0.0002 | −3.3824 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, Y.S.; Han, Y.; Lee, S.K.; Hur, S.D. Atomistic View of Mercury Cycling in Polar Snowpacks: Probing the Role of Hg2+ Adsorption Using Ab Initio Calculations. Minerals 2019, 9, 459. https://doi.org/10.3390/min9080459
Yi YS, Han Y, Lee SK, Hur SD. Atomistic View of Mercury Cycling in Polar Snowpacks: Probing the Role of Hg2+ Adsorption Using Ab Initio Calculations. Minerals. 2019; 9(8):459. https://doi.org/10.3390/min9080459
Chicago/Turabian StyleYi, Yoo Soo, Yeongcheol Han, Sung Keun Lee, and Soon Do Hur. 2019. "Atomistic View of Mercury Cycling in Polar Snowpacks: Probing the Role of Hg2+ Adsorption Using Ab Initio Calculations" Minerals 9, no. 8: 459. https://doi.org/10.3390/min9080459