Sequential Bioleaching of Phosphorus and Uranium


Abstract
:1. Introduction
2. Materials and Methods
2.1. Studied Ores
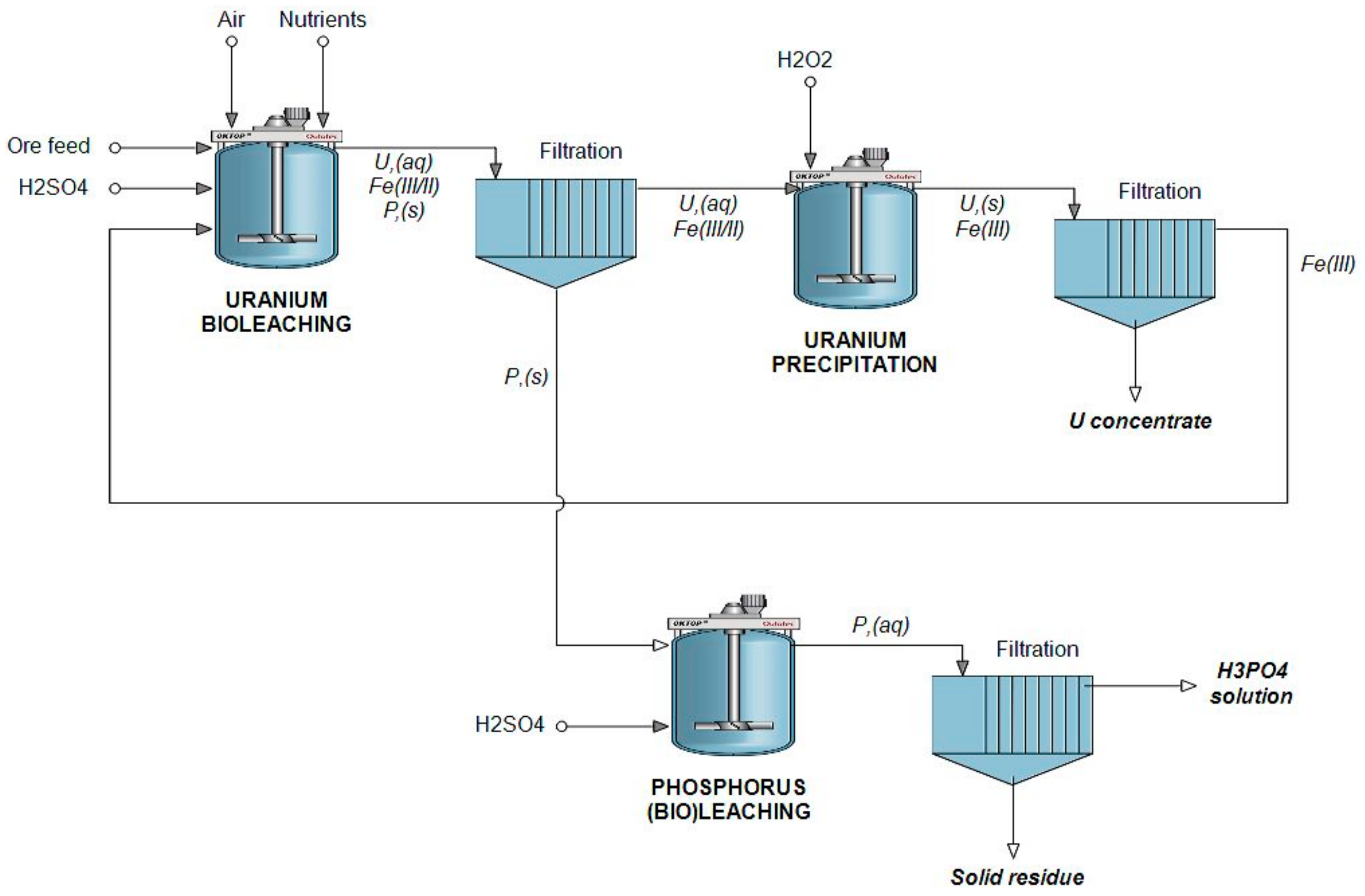
2.2. Leaching Experiments
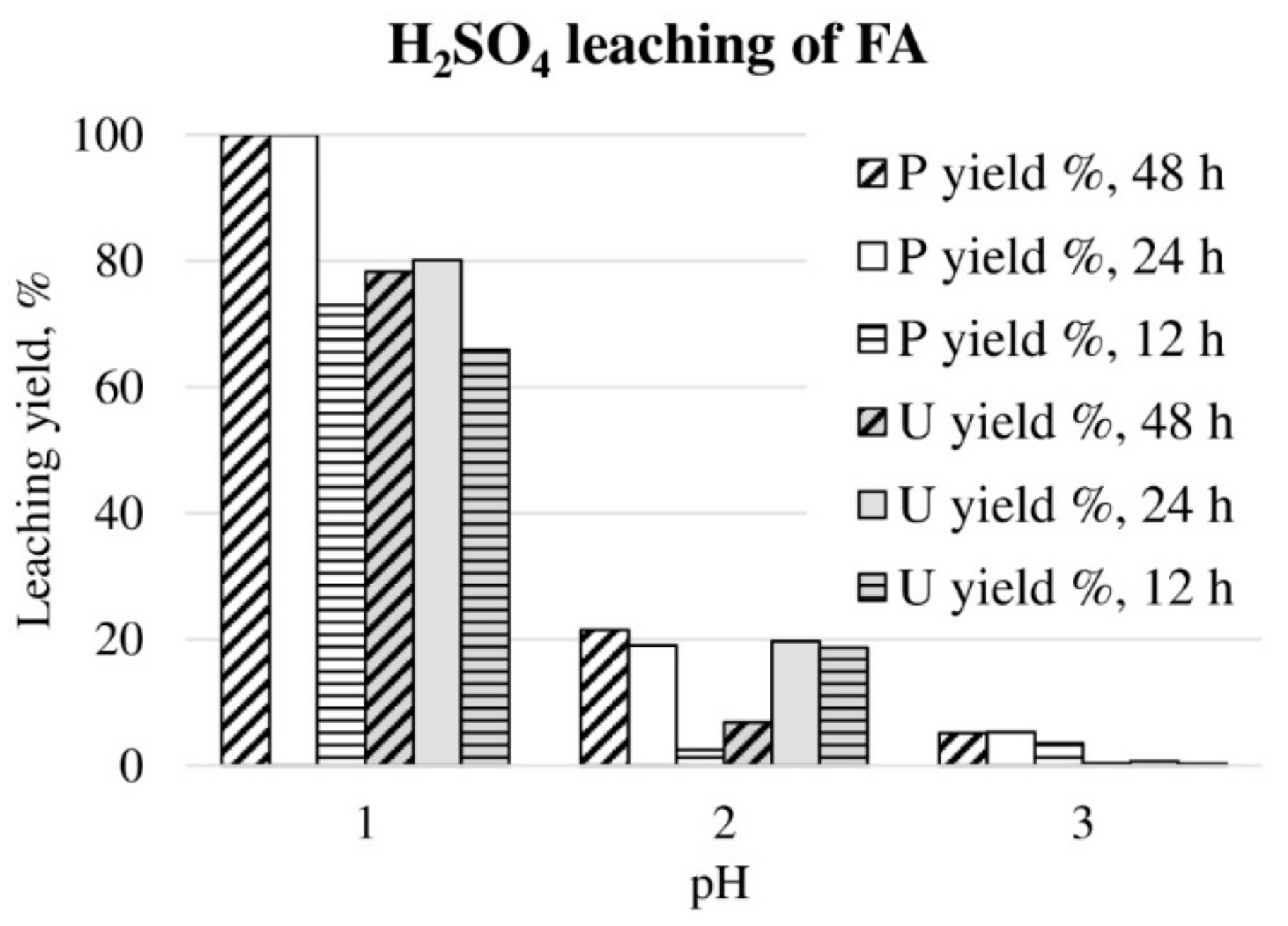
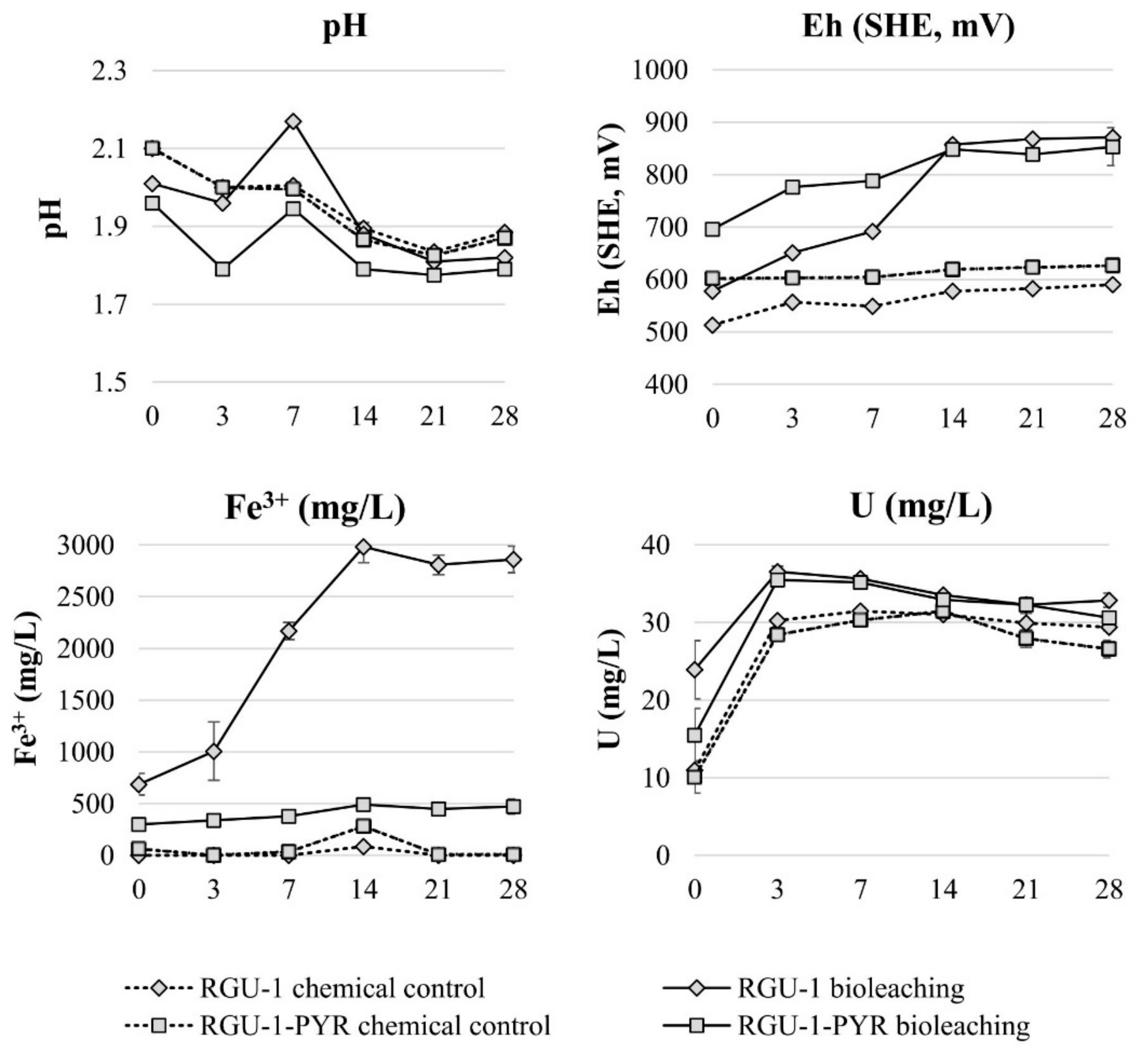
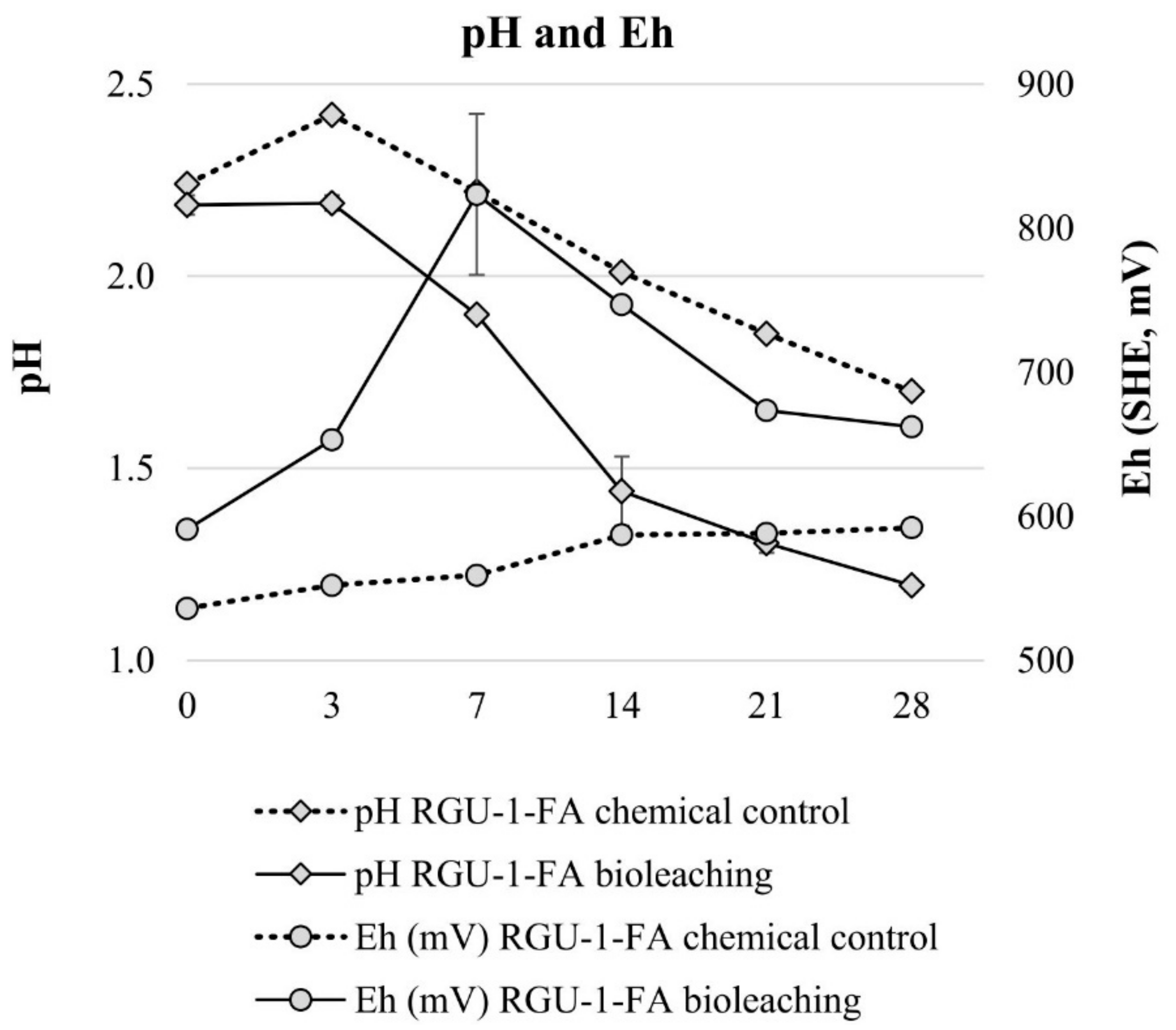
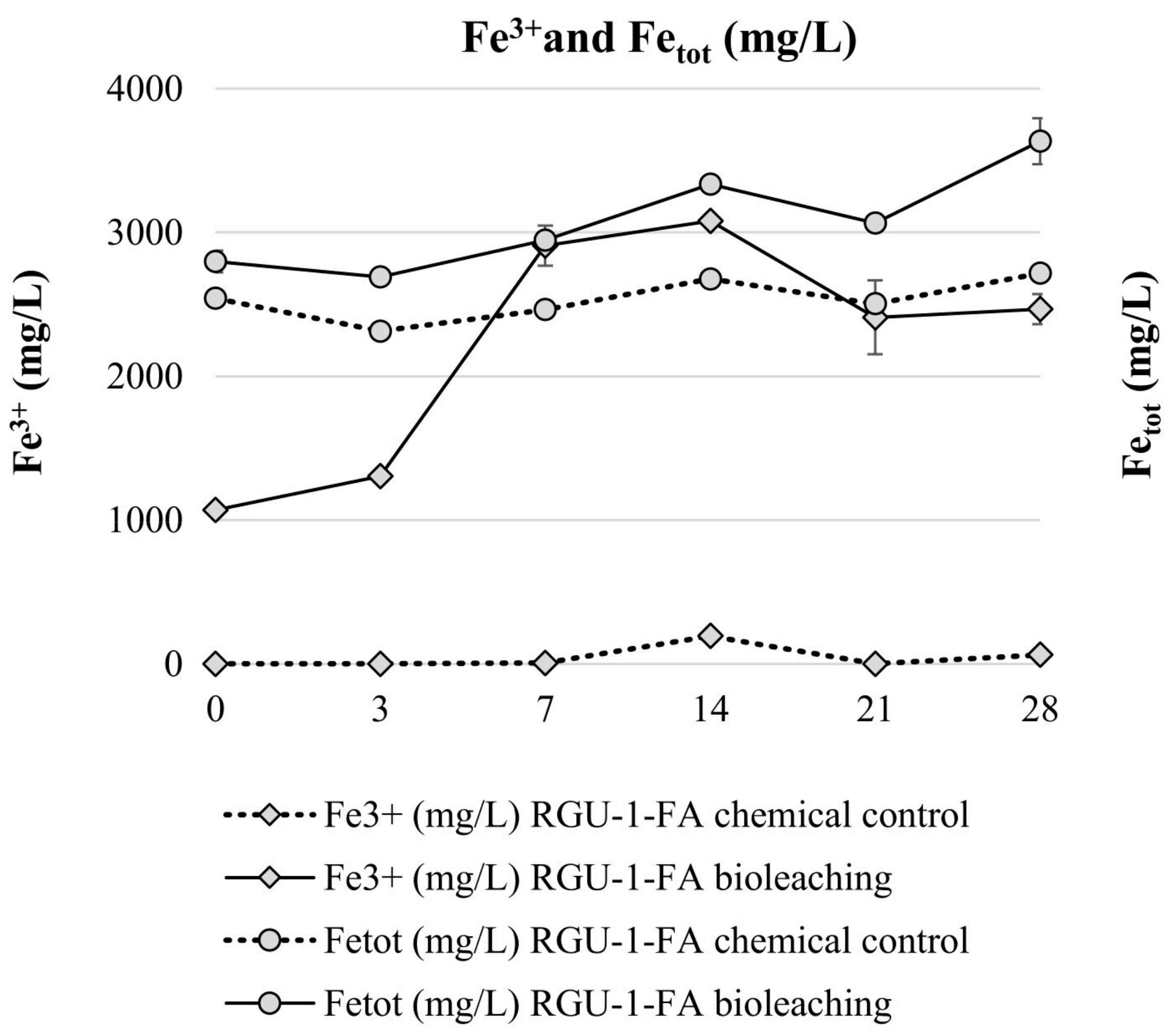
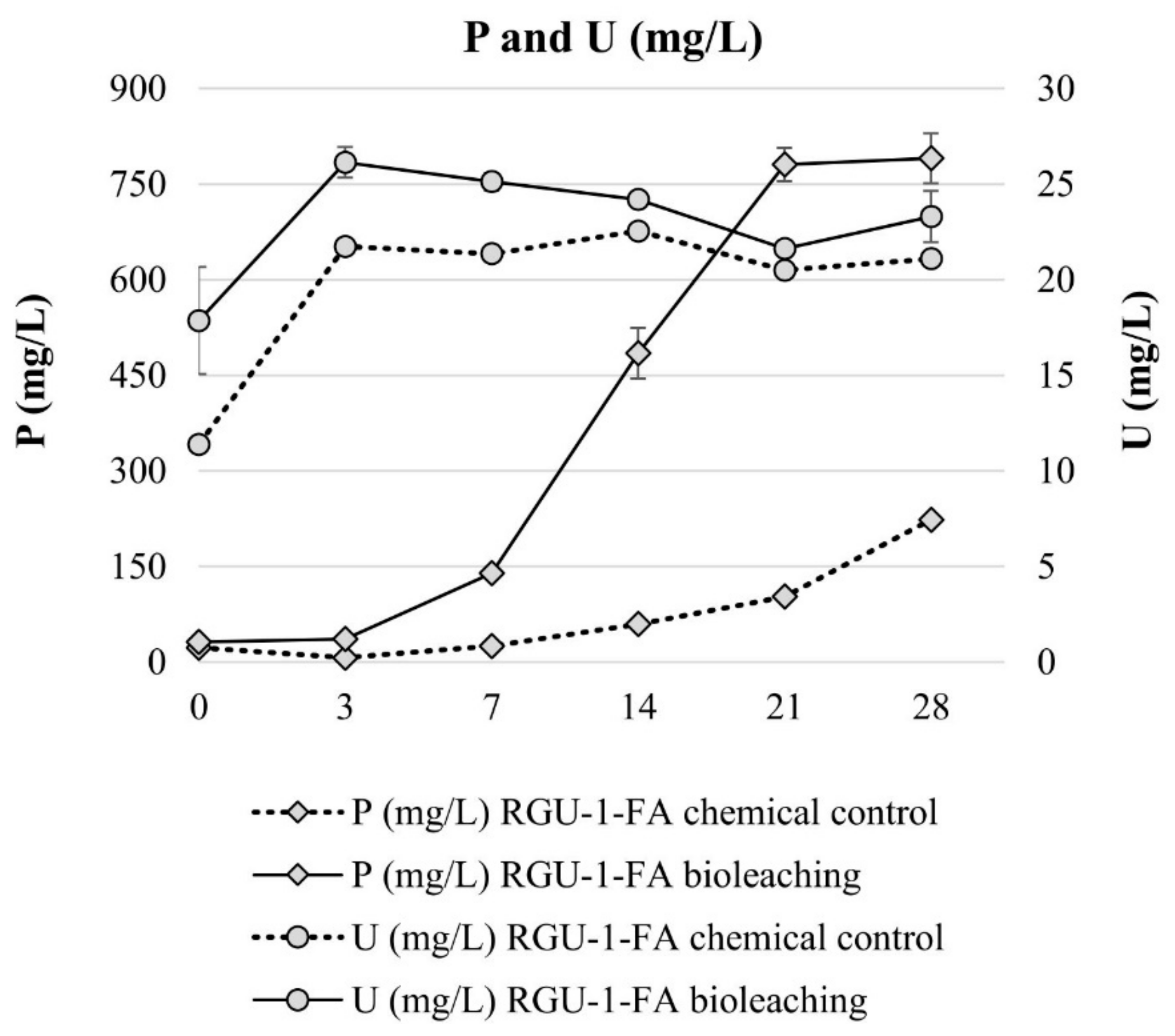
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- U.S. Geological Survey. Mineral Commodity Summaries; U.S. Geological Survey: Reston, VA, USA, 2018; pp. 122–123.
- Van Vuuren, D.P.; Bouwman, A.F.; Beusen, A.H.W. Phosphorus demand for the 1970–2100 period: A scenario analysis of resource depletion. Glob. Environ. Chang. 2010, 20, 428–439. [Google Scholar] [CrossRef]
- Cordell, D.; Drangert, J.-O.; White, S. The story of phosphorus: Global food security and food for thought. Glob. Environ. Chang. 2009, 19, 292–305. [Google Scholar] [CrossRef]
- Van Kauwenbergh, S.J. World Phosphate Rock Reserves and Resources; International Fertilizer Development Center (IFDC): Muscle Shoals, AL, USA, 2010; 50p.
- U.S. Geological Survey. Mineral Commodity Summaries; U.S. Geological Survey: Washington, DC, USA, 2009; 2195p.
- Cooper, J.; Lombardi, R.; Boardman, D.; Carliell-Marquet, C. The distribution and production of global phosphate rock reserves. Resour. Conserv. Recy. 2011, 57, 78–86. [Google Scholar] [CrossRef]
- Beltrami, D.; Cote, G.; Mokhtari, H.; Courtaud, B.; Moyer, B.A.; Chagnes, A. Recovery of Uranium from Wet Phosphoric Acid by Solvent Extraction Processes. Chem. Rev. 2014, 114, 12002–12023. [Google Scholar] [CrossRef] [PubMed]
- Abu-Eishah, S.I.; Abu-Jabal, N.M. Parametric study on the production of phosphoric acid by the dehydrate process. Chem. Eng. J. 2001, 81, 231–250. [Google Scholar] [CrossRef]
- Komnitsas, K.; Kontopoulus, A.; Lazar, I.; Cambridge, M. Risk Assessment and Proposed Remedial Actions in Coastal Tailings Disposal Sites in Romania. Miner. Eng. 1998, 11, 1179–1190. [Google Scholar] [CrossRef]
- Komnitsas, K.; Lazar, I.; Petrisor, I.G. Application of a Vegetative Cover on Phosphogypsum Stacks. Miner. Eng. 1999, 12, 175–185. [Google Scholar] [CrossRef]
- Salo, M.; Mäkinen, J.; Yang, J.; Kurhila, M.; Koukkari, P. Continuous biological sulfate reduction from phosphogypsum waste leachate. Hydrometallurgy 2018, 180, 1–6. [Google Scholar] [CrossRef]
- International Atomic Energy Agency. Extent of Environmental Contamination by Naturally Occurring Radioactive Material (NORM) and Technological Options for Mitigation; International Atomic Energy Agency: Vienna, Austria, 2003; 198p. [Google Scholar]
- International Atomic Energy Agency (IAEA). The Recovery of Uranium from Phosphoric Acid. Report of An Advisory Group Meeting Organized by the International Atomic Energy Agency and Held in Vienna, 16–19 March 1987. Available online: https://www-pub.iaea.org/MTCD/Publications/PDF/te_0533.pdf (accessed on 24 January 2019).
- Gabriel, S.; Baschwitz, A.; Mathonnière, G.; Eleouet, T.; Fizaine, F. A critical assessment of global uranium resources, including uranium in phosphate rocks, and the possible impact of uranium shortages on nuclear power fleets. Ann. Nucl. Energy 2013, 58, 213–220. [Google Scholar] [CrossRef]
- Nuclear Energy Agency. Uranium 2016: Resources, Production and Demand; Nuclear Energy Agency: Boulogne-Billancourt, France, 2016; 548p. [Google Scholar]
- Venter, R.; Boylett, M. The Evaluation of Various Oxidants Used in Acid Leaching of Uranium. Hydrometallurgy Conference 2009, The Southern African Institute of Mining and Metallurgy, 2009. Available online: https://www.saimm.co.za/Conferences/Hydro2009/445-456_Venter.pdf (accessed on 22 March 2019).
- Ram, R.; Charalambous, F.A.; McMaster, S.; Pownceby, M.I.; Tardio, J.; Bhargava, S.K. An investigation on the dissolution of natural uraninite ores. Miner. Eng. 2013, 50–51, 83–92. [Google Scholar] [CrossRef]
- Muñoz, J.A.; Gonzáles, F.; Blázquez, M.L.; Ballester, A. A study of the bioleaching of Spanish uranium ore. Part I: A review of the bacterial leaching in the treatment of uranium ores. Hydrometallurgy 1995, 38, 39–57. [Google Scholar] [CrossRef]
- Bosecker, K. Bioleaching: Metal solubilisation by microorganisms. FEMS Microbiol. Rev. 1997, 20, 591–604. [Google Scholar] [CrossRef]
- Rawlings, D.E.; Silver, S. Mining with Microbes. Biotechnology 1995, 13, 773–778. [Google Scholar] [CrossRef]
- Rawlings, D.E. Heavy Metal Mining Using Microbes. Annu. Rev. Microbiol. 2002, 56, 65–91. [Google Scholar] [CrossRef]
- Abhilash, S.S.; Mehta, K.D.; Kumar, V.; Pandey, B.D.; Pandey, V.M. Dissolution of uranium from silicate-apatite ore by Acidithiobacillus ferrooxidans. Hydrometallurgy 2009, 95, 70–75. [Google Scholar] [CrossRef]
- Schippers, A.; Sand, W. Bacterial leaching of metal sulfides proceeds by two indirect mechanisms via thiosulfate or via polysulfides and sulfur. Appl. Environ. Microbiol. 1999, 65, 319–321. [Google Scholar]
- Tuovinen, O.H.; Hiltunen, P.; Vuorinen, A. Solubilization of phosphorus, uranium and iron from apatite- and uranium-containing rock samples in synthetic and microbiologically produced acid leach solutions. Eur. J. Appl. Microbiol. Biotechnol. 1983, 17, 327–333. [Google Scholar] [CrossRef]
- Umanskii, A.B.; Klyushnikov, A.M. Bioleaching of low grade uranium ore containing pyrite using A. ferrooxidans and A. thiooxidans. J. Radioanal. Nucl. Chem. 2013, 295, 151–156. [Google Scholar] [CrossRef]
- Mäkinen, J.; Kinnunen, P.; Arnold, M.; Priha, O.; Sarlin, T. Fluoride Toxicity in Bioleaching of Fluorapatite. Adv. Mater. Res. 2015, 1130, 406–409. [Google Scholar] [CrossRef]
- Bhatti, T.M.; Yawar, W. Bacterial solubilisation of phosphorus from phosphate rock containing sulfur-mud. Hydrometallurgy 2010, 103, 54–59. [Google Scholar] [CrossRef]
- Priha, O.; Sarlin, T.; Blomberg, P.; Wendling, L.; Mäkinen, J.; Arnold, M.; Kinnunen, P. Bioleaching phosphorus from fluorapatites with acidophilic bacteria. Hydrometallurgy 2014, 150, 269–275. [Google Scholar] [CrossRef]
- Xiao, C.Q.; Chi, R.A.; Li, W.S.; Zheng, Y. Biosolubilization of phosphorus from rock phosphate by moderately thermophilic and mesophilic bacteria. Miner. Eng. 2011, 24, 956–958. [Google Scholar] [CrossRef]
- Faye, G.H.; Bowman, W.S.; Sutarno, R. Uranium ore BL-5—A Certified Reference Material; CANMET report 1979, 79–4; CANMET, Energy, Mines and Resources Canada: Ottawa, ON, Canada, 1979. [Google Scholar]
- IAEA. Preparation of Gamma-ray Spectrometry Reference Materials RGU-1 RGTh-1 and RGK-1 Report; International Atomic Energy Agency: Vienna, Austria, 1987. [Google Scholar]
- CEN. Characterization of Waste—Leaching Behaviour Tests—Influence of pH on Leaching with Continuous pH-Control; CEN/TS 14997; CEN: Brussels, Belgium, 2006. [Google Scholar]
- Halinen, A.-K.; Rahunen, N.; Kaksonen, A.H.; Puhakka, J.A. Heap bioleaching of a complex sulfide ore Part I: Effect of pH on metal extraction and microbial composition in pH controlled columns. Hydrometallurgy 2009, 98, 92–100. [Google Scholar] [CrossRef]
- Silverman, M.P.; Lundgren, D.G. Studies on the chemoautotrophic iron bacterium Ferrobacillus ferrooxidans. I. An improved medium and a harvesting procedure for securing high cell yields. J. Bacteriol. 1959, 77, 642–647. [Google Scholar]
- Anwar-ul-Haq, M.; Ahmad, I.; Niazi, M.T.; Arif, M.; Mahmood, K. Evaluation of problematic indigenous phosphate deposits for phosphoric acid manufacture. Fert. Res. 1990, 24, 53–56. [Google Scholar] [CrossRef]
- Chi, R.; Xiao, C.; Gao, H. Bioleaching of phosphorus from rock phosphate containing pyrites by Acidithiobacillus ferrooxidans. Miner. Eng. 2006, 19, 979–981. [Google Scholar] [CrossRef]
- Gupta, R.; Pandey, V.M.; Pranesh, A.B.; Chakravarty, A.B. Study of an improved technique for precipitation of uranium from eluated solution. Hydrometallurgy 2004, 71, 429–434. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element (wt%) | FA | RGU-1 | RGU-1-PYR | RGU-1-FA |
|---|---|---|---|---|
| P | 3.6 | - | - | 0.95 |
| U | 0.0007 | 0.040 | 0.039 | 0.029 |
| Fe | 14.0 | 0.030 | 1.2 | 3.7 |
| S | - | 0.0020 | 1.3 | 0.0015 |
| Si | 14.0 | 46.4 | 45.3 | 37.9 |
| Ca | 8.7 | 0.030 | 0.029 | 2.3 |
| Mg | 7.0 | 0.010 | 0.0098 | 1.8 |
| Al | 4.4 | 0.10 | 0.10 | 1.2 |
| K | 1.1 | 0.0020 | 0.0020 | 0.29 |
| F | 0.71 | - | - | 0.19 |
| Ore and Test | Nutrient Media | Inoculum | Iron Source | Sulfur Source | pH Control |
|---|---|---|---|---|---|
| RGU-1 (CC) | Yes | No | Yes | No | Yes |
| RGU-1 (BL) | Yes | Yes | Yes | No | Yes |
| RGU-1-PYR (CC) | Yes | No | No | No | Yes |
| RGU-1-PYR (BL) | Yes | Yes | No | No | Yes |
| RGU-1-FA (CC) | Yes | No | Yes | Yes | Yes |
| RGU-1-FA (BL) | Yes | Yes | Yes | Yes | Yes |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mäkinen, J.; Wendling, L.; Lavonen, T.; Kinnunen, P. Sequential Bioleaching of Phosphorus and Uranium. Minerals 2019, 9, 331. https://doi.org/10.3390/min9060331
Mäkinen J, Wendling L, Lavonen T, Kinnunen P. Sequential Bioleaching of Phosphorus and Uranium. Minerals. 2019; 9(6):331. https://doi.org/10.3390/min9060331
Chicago/Turabian StyleMäkinen, Jarno, Laura Wendling, Tiina Lavonen, and Päivi Kinnunen. 2019. "Sequential Bioleaching of Phosphorus and Uranium" Minerals 9, no. 6: 331. https://doi.org/10.3390/min9060331