Comparing Schwertmannite and Hydrobasaluminite Dissolution in Ammonium Oxalate (pH 3.0): Implications for Metal Speciation Studies by Sequential Extraction


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Characterization
2.1.1. Sampling and Chemical Analyses of Waters
2.1.2. Synthesis and Characterization of Mineral Precipitates
2.2. Experimental Setup
2.3. Analytical Methods
3. Results
3.1. Composition of Final Oxalate Solutions
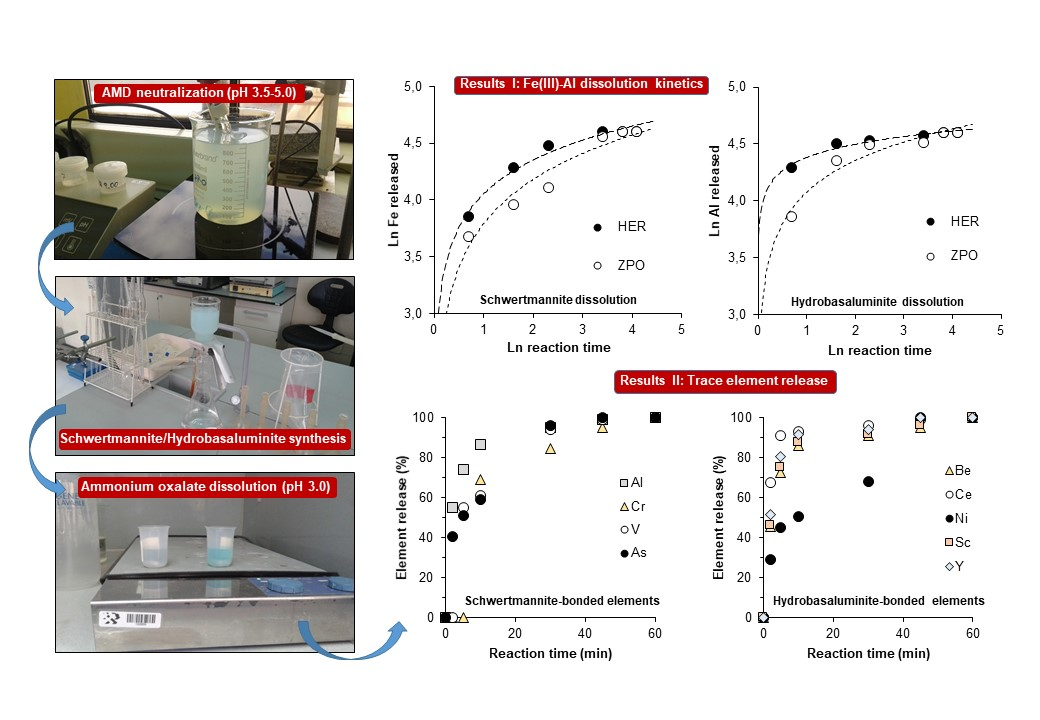
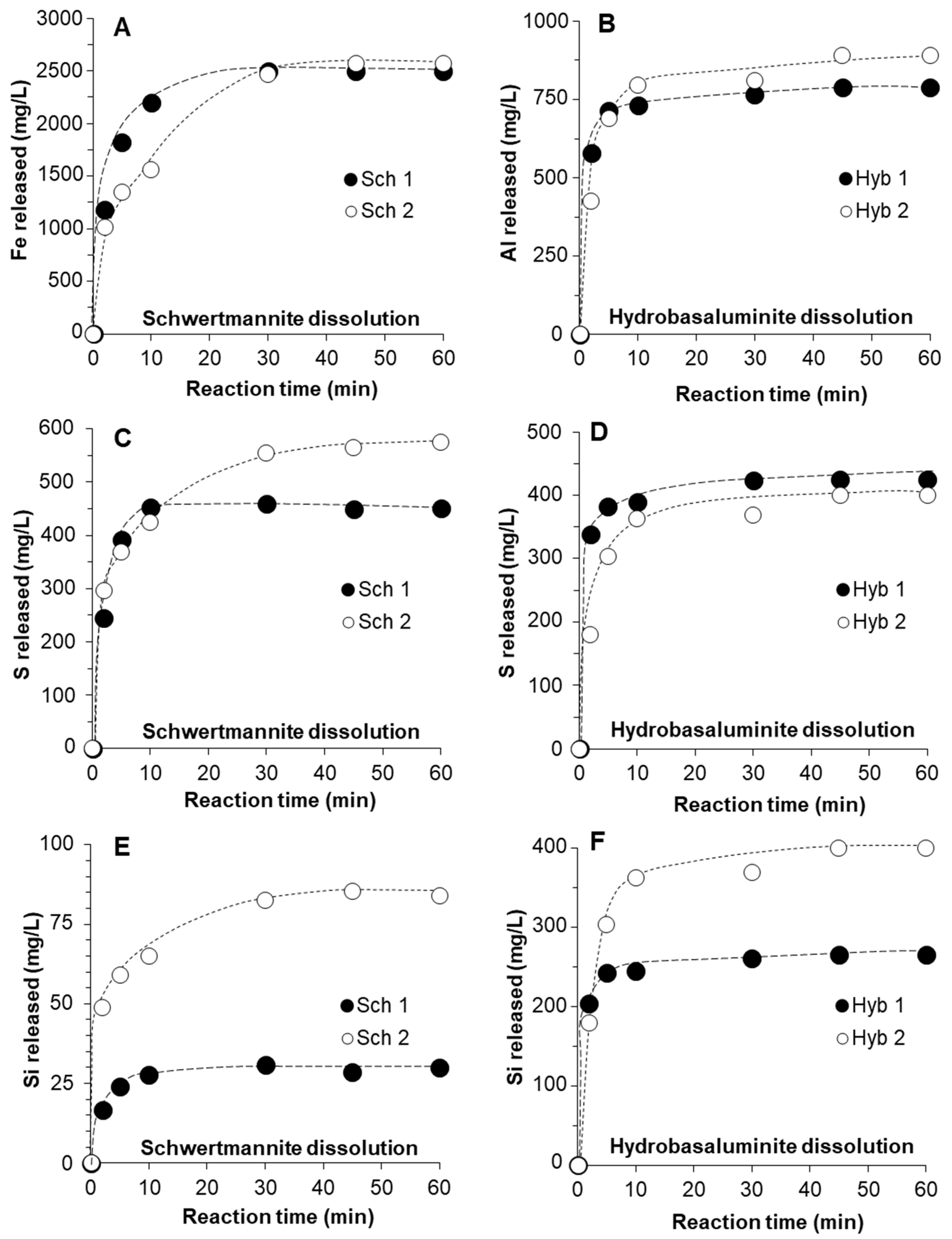
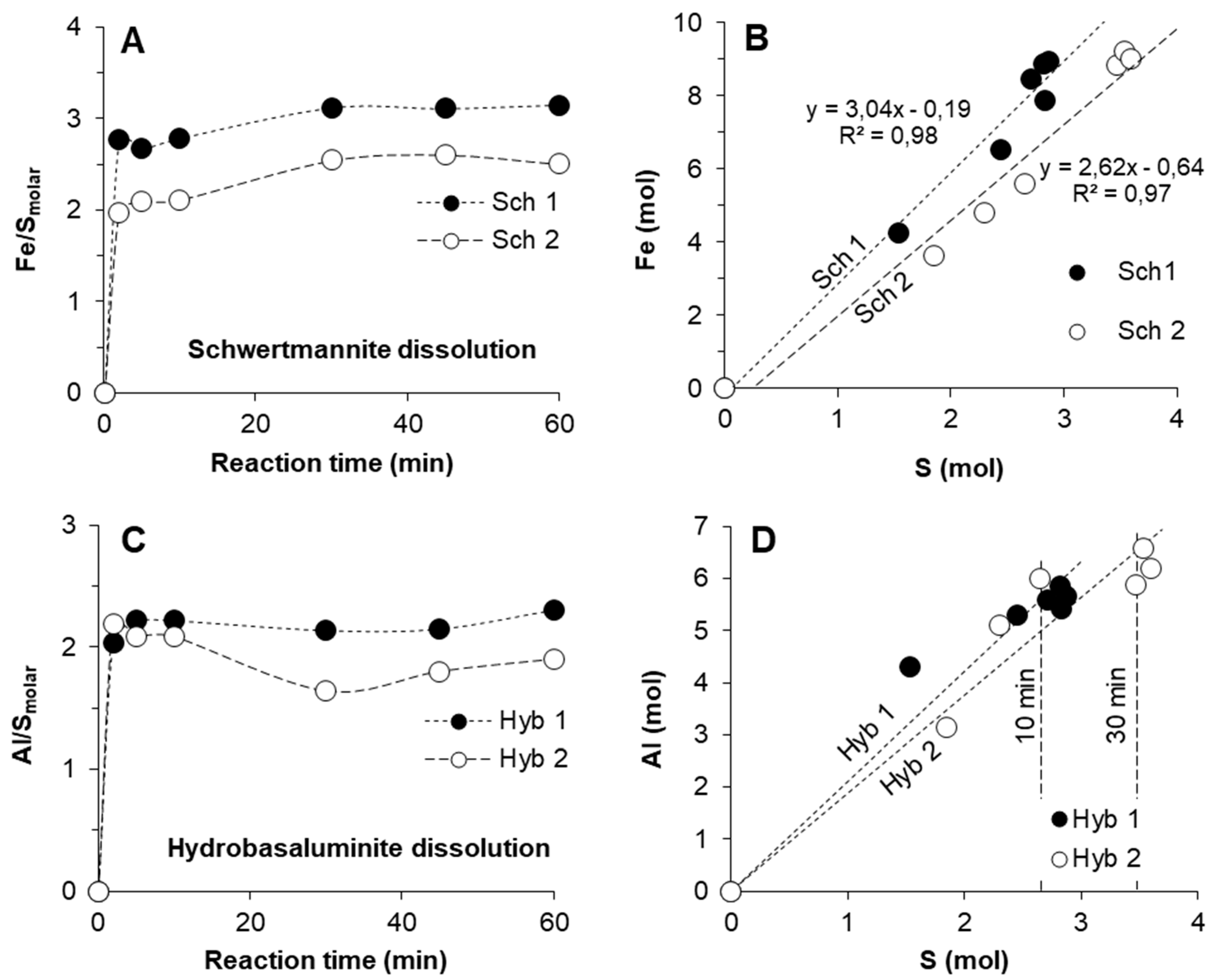
3.2. Kinetics of Schwertmannite and Hydrobasaluminite Dissolution
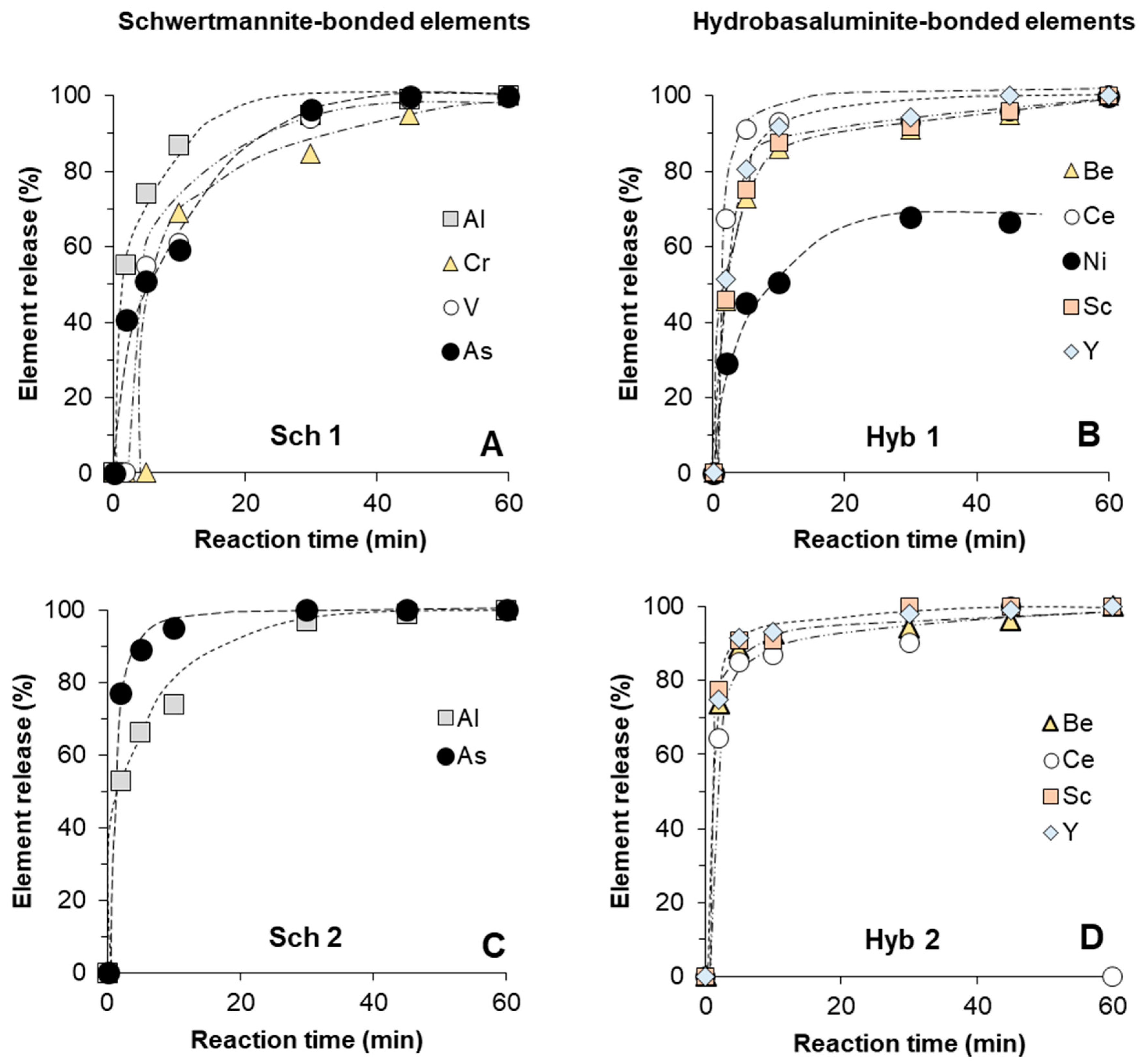
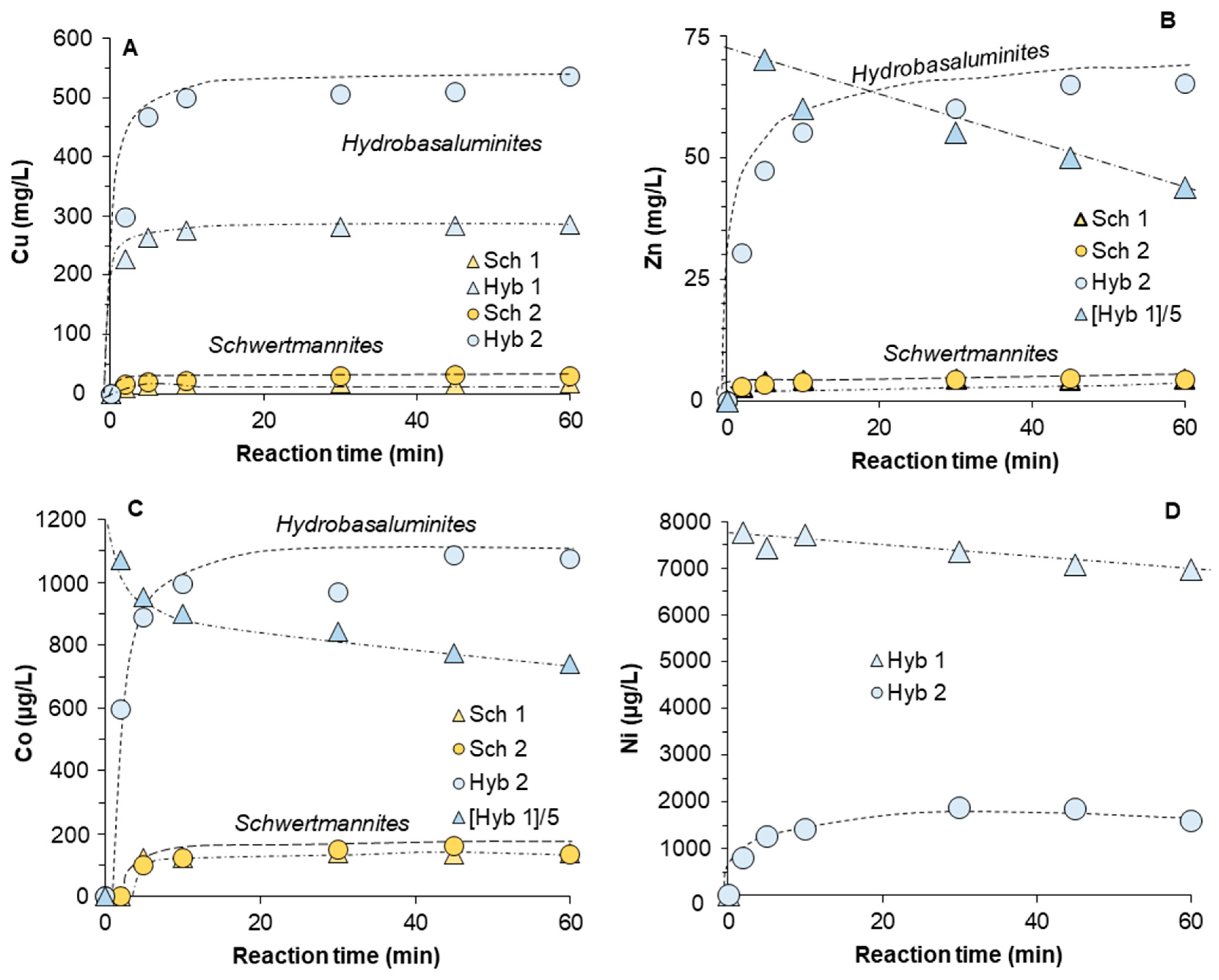
3.3. Release of Trace Elements Associated with Schwertmannite and Hydrobasaluminite
4. Discussion
4.1. Similarities between Schwertmannite and Hydrobasaluminite Dissolution: Implications for Trace Element Speciation Studies
4.2. Control of pH and Ionic Charge on Metal Retention
4.3. Future Research
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Synthesis of Schwertmannite and Hydrobasaluminite Mineral Samples for the Dissolution Experiments
References
- Schwertmann, U.; Schulze, D.G.; Murad, E. Identification of ferrihydrite in soils by dissolution kinetics, differential X-ray diffraction, and Mössbauer spectroscopy. Soil Sci. Soc. Am. J. 1982, 7, 547–552. [Google Scholar] [CrossRef]
- Bigham, J.M.; Schwertmann, U.; Carlson, L.; Murad, E. A poorly crystallized oxyhydroxysulfate of iron formed by bacterial oxidation of Fe(II) in acid mine waters. Geochim. Cosmochim. Acta 1990, 54, 2743–2758. [Google Scholar] [CrossRef]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2003; 664p, ISBN 3-527-30274-3. [Google Scholar]
- Dold, B. Speciation of the most soluble phases in a sequential extraction procedure adapted for geochemical studies of copper sulfide mine waste. J. Geochem. Explor. 2003, 80, 55–68. [Google Scholar] [CrossRef]
- Dold, B. Dissolution kinetics of schwertmannite and ferrihydrite in oxidized mine samples and their detection by differential X-ray diffraction (DXRD). App. Geochem. 2003, 18, 1531–1540. [Google Scholar] [CrossRef]
- Caraballo, M.A.; Rötting, T.S.; Nieto, J.M.; Ayora, C. Sequential extraction and DXRD applicability to poorly crystalline Fe- and Al-phase characterization from an acid mine water passive remediation system. Am. Mineral. 2009, 94, 1029–1038. [Google Scholar] [CrossRef]
- Alvaro, A. Mineralogía y Geoquímica de Sulfatos Secundarios en Ambientes de Drenaje ácido de Mina. Implicación Ambiental en el Área Minera del Yacimiento de San Miguel (Faja Pirítica Ibérica). Ph.D. Thesis, University of the Basque Country (UPV/EHU), Bilbao, Spain, 2010. [Google Scholar]
- Favas, P.J.C.; Pratas, J.; Goemes, M.E.P.; Cala, V. Selective chemical extraction of heavy metals in tailings and soils contaminated by mining activity: Environmental implications. J. Geochem. Expl. 2011, 111, 160–171. [Google Scholar] [CrossRef]
- Torres, E.; Auleda, M. A sequential extraction procedure for sediments affected by acid mine drainage. J. Geochem. Explor. 2013, 128, 35–41. [Google Scholar] [CrossRef]
- Diez-Ercilla, M. Estudio Hidrogeoquímico del Lago Minero de Cueva de la Mora (FPI, Huelva). Controles Sobre la Concentración de Metales y Modelo de Estratificación. Ph.D. Thesis, University of the Basque Country (UPV/EHU), Bilbao, Spain, 2015. [Google Scholar]
- Vithana, C.L.; Sullivan, L.A.; Bush, R.; Burton, E.D. Schwertmannite in soil materials: Limits of detection of acidified ammonium oxalate method and differential X-ray diffraction. Geoderma 2015, 249, 51–60. [Google Scholar] [CrossRef]
- Diez-Ercilla, M.; Falagán, C.; Yusta, I.; Sánchez-España, J. Metal mobility and mineral transformations driven by bacterial activity in acidic pit lake sediments: Evidence from column experiments and sequential extraction. J. Soils Sedim. 2018. [Google Scholar] [CrossRef]
- Tessier, A.; Campbell, P.G.C.; Bisson, M. Sequential extraction procedure for the speciation of particulate trace metals. Anal. Chem. 1979, 51, 844–851. [Google Scholar] [CrossRef]
- Nordstrom, D.K. The effect of sulfate on aluminum concentrations in natural waters: Some stability relations in the system Al2O3-SO3-H2O at 298 K. Geochim. Cosmochim. Acta 1982, 46, 681–692. [Google Scholar] [CrossRef]
- Bigham, J.M.; Nordstrom, D.K. Iron and Aluminum Hydroxysulfates from Acid Sulfate Waters. Rev Mineral. Geochem. 2000, 40, 351–403. [Google Scholar] [CrossRef]
- Sánchez-España, J.; López Pamo, E.; Santofimia, E.; Reyes, J.; Martín Rubí, J.A. The Removal of Dissolved Metals by Hydroxysulphate Precipitates during Oxidation and Neutralization of Acid Mine Waters, Iberian Pyrite Belt. Aquat. Geochem. 2006, 12, 269–298. [Google Scholar] [CrossRef]
- Sánchez-España, J. The Behavior of Iron and Aluminum in Acid Mine Drainage: Speciation, Mineralogy, and Environmental Significance. In Thermodynamics, Solubility and Environmental Issues; Letcher, T.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2007; Chapter 7; pp. 137–150. ISBN 978-0-444-52707-3. [Google Scholar]
- Sánchez-España, J.; Yusta, I.; Diez, M. Schwertmannite and hydrobasaluminite: A re-evaluation of their solubility and control on the iron and aluminum concentration in acidic pit lakes. Appl. Geochem. 2011, 26, 1752–1774. [Google Scholar] [CrossRef]
- Carrero, S.; Pérez-López, R.; Fernandez-Martinez, A.; Cruz-Hernández, P.; Ayora, C.; Poulaine, A. The potential role of aluminium hydroxysulfates in the removal of contaminants in acid mine drainage. Chem. Geol. 2015, 417, 414–423. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Yusta, I.; Burgos, W.D. Geochemistry of dissolved aluminum at low pH: Hydrobasaluminite formation and interaction with trace metals, silica and microbial cells under anoxic conditions. Chem. Geol. 2016, 441, 124–137. [Google Scholar] [CrossRef]
- Acero, P.; Hudson-Edwards, K.A. Influence of pH and temperature on basaluminite dissolution rates. ACS Earth Space Chem. 2018, 2, 203–209. [Google Scholar] [CrossRef]
- Lozano, A.; Fernández-Martínez, A.; Ayora, C.; Poulain, A. Local structure and ageing of basaluminite at different pH values and sulphate concentrations. Chem. Geol. 2018, 496, 25–33. [Google Scholar] [CrossRef]
- McKeague, J.A.; Day, J. Dithionite-and oxalate-extractable Fe and Al as aids in differentiating various classes of soils. Can. J. Soil Sci. 1966, 46, 13–22. [Google Scholar] [CrossRef]
- Tabelin, C.B.; Igarashi, T.; Arima, T.; Sato, D.; Tatsuhara, T.; Tamoto, S. Characterization and evaluation of arsenic and boron adsorption onto natural geologic materials, and their application in the disposal of excavated altered rock. Geoderma 2014, 213, 163–172. [Google Scholar] [CrossRef] [Green Version]
- Francisco, P.C.M.; Sato, T.; Otake, T.; Kasama, T.; Suzuki, S.; Shiwaku, H.; Yaita, T. Mechanisms of Se (IV) Co-precipitation with Ferrihydrite at Acidic and Alkaline Conditions and Its Behavior during Aging. Environ. Sci. Technol. 2018, 52, 4817–4826. [Google Scholar] [CrossRef]
- Polizzotto, M.L.; Kocar, B.D.; Benner, S.G.; Sampson, M.; Fendorf, S. Near-surface wetland sediments as a source of arsenic release to ground water in Asia. Nature 2008, 454, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Tabelin, C.B.; Igarashi, T.; Villacorte-Tabelin, M.; Park, I.; Opiso, E.M.; Ito, M.; Hiroyoshi, N. Arsenic, selenium, boron, lead, cadmium, copper, and zinc in naturally contaminated rocks: A review of their sources, modes of enrichment, mechanisms of release, and mitigation strategies. Sci. Total Environ. 2018, 645, 1522–1553. [Google Scholar] [CrossRef] [PubMed]
- Tatsuhara, T.; Arima, T.; Igarashi, T.; Tabelin, C.B. Combined neutralization–adsorption system for the disposal of hydrothermally altered excavated rock producing acidic leachate with hazardous elements. Eng. Geol. 2012, 139, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-España, J.; Yusta, I.; Gray, J.; Burgos, W.D. Geochemistry of dissolved aluminum at low pH: Extent and significance of Al-Fe(III) coprecipitation below pH 4.0. Geochim. Cosmochim. Acta 2016, 175, 128–149. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Yusta, I. Low-crystallinity products of trace-metal precipitation in neutralized pit-lake waters without ferric and aluminous adsorbent: Geochemical modelling and mineralogical analyses. Mineral. Mag. 2015, 79, 781–798. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Yusta, I.; López, G.A. Schwertmannite to jarosite conversion in the water column of an acidic mine pit lake. Mineral. Mag. 2012, 76, 2659–2682. [Google Scholar] [CrossRef]
- Goetz, A.J.; Ziegler, A.; Gaebel, J.; Wiacek, C.; Schlömann, M.; Schmahl, W.W. Structural variations in biogenic and synthetic schwertmannite. Macla 2010, 13, 118. [Google Scholar]
- Carrero, S.; Fernandez-Martinez, A.; Pérez-López, R.; Lee, D.; Aquilanti, G.; Poulain, A.; Lozano, A.; Nieto, J.M. The nanocrystalline structure of basaluminite, an aluminum hydroxide sulfate from acid mine drainage. Am. Mineral. 2017, 102, 2381–2389. [Google Scholar] [CrossRef]
- Fukushi, K.; Sato, T.; Yanase, N.; Minato, J.; Yamada, H. Arsenate sorption on schwertmannite. Am. Mineral. 2004, 89, 1728–1734. [Google Scholar] [CrossRef]
- Reggenspurg, S.; Peiffer, S. Arsenate and chromate incorporation in schwertmannite. App. Geochem. 2005, 20, 1226–1239. [Google Scholar] [CrossRef]
- Burton, E.D.; Bush, R.T.; Johnston, S.G.; Watling, K.M.; Hocking, R.K.; Sullivan, L.A.; Parker, G.K. Sorption of Arsenic(V) and Arsenic(III) to Schwertmannite. Environ. Sci. Technol. 2009, 43, 9202–9207. [Google Scholar] [CrossRef] [PubMed]
- Doua, X.; Mohan, D.; Pittman, C.U. Arsenate adsorption on three types of granular schwertmannite. Water Res. 2013, 47, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Ayora, C.; Macías, F.; Torres, E.; Lozano, A.; Carrero, S.; Nieto, J.M.; Pérez-López, R.; Fernández-Martínez, A.; Castillo-Michel, H. Recovery of Rare Earth Elements and Yttrium from Passive-Remediation Systems of Acid Mine Drainage. Environ. Sci. Technol. 2016, 50, 8255–8262. [Google Scholar] [CrossRef]
- Lozano, A.; Ayora, C.; Fernández-Martínez, A.; Macías, F. Rare Earth Element adsorption onto basaluminite. Macla 2017, 22, 81–82. [Google Scholar]
- Karbouj, R. Aluminium leaching using chelating agents as compositions of food. Food Chem. Toxicol. 2007, 45, 1688–1693. [Google Scholar] [CrossRef]
- Dzombak, D.A.; Morel, F.M.M. Surface Complexation Modeling: Hydrous Ferric Oxide; John Wiley: New York, NY, USA, 1990; 416p, ISBN 978-0-471-63731-8. [Google Scholar]
- Crisponi, G.; Nurchi, V.M.; Bertolasi, V.; Remelli, M.; Faa, G. Chelating agents for human diseases related to aluminium overload. Coordin. Chem. Rev. 2012, 256, 89–104. [Google Scholar] [CrossRef]
- Exley, C.; Korchazhkina, O.; Job, D.; Strekopytov, S.; Polwart, A.; Crome, P. Non-invasive therapy to reduce the body burden of aluminium in Alzheimer’s disease. J. Alzheimers’ Dis. 2006, 10, 17–24. [Google Scholar] [CrossRef]
- Beardmore, J.; Lopez, X.; Mujika, J.I.; Exley, C. What is the mechanism of formation of hydroxyaluminosilicates? Sci. Rep. 2016, 6, 30913. [Google Scholar] [CrossRef] [Green Version]
- Prietzel, J.; Hirsch, C. Extractability and dissolution kinetics of pure and soil-added synthesized aluminium hydroxy sulphate minerals. Eur. J. Soil. Sci. 1998, 49, 669–681. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Major Elements (mg/L) | |||||||||||
| Water sample | K | Na | Mg | Ca | SO4 | SiO2 | Fe | Mn | Cu | Zn | Al |
| HER | 1.4 | 48 | 666 | 530 | 5900 | 66 | 342 | 158 | 21 | 130 | 76 |
| ZP | 0.6 | 37 | 756 | 552 | 14,712 | 150 | 3901 | 317 | 184 | 182 | 876 |
| Trace Elements (µg/L) | |||||||||||
| Water sample | As | Be | Ni | Cd | Co | Cr | Pb | Se | Tl | U | V |
| HER | 44 | 37 | 2494 | 186 | 3379 | 34 | 26 | 58 | 10 | 20 | 2 |
| ZP | 5688 | 23 | 3994 | 355 | 4126 | 156 | 408 | 167 | 12 | 118 | 177 |
| Sample | Locality | Fe | S | Si | Al | Cu | Zn | As | Cr | [Fe/S] |
| wt % | wt % | wt % | wt % | ppm | ppm | ppm | ppm | |||
| Sch 1 | Herrerías | 41.25 | 6.10 | 0.25 | 0.40 | 46 | 1862 | 8 | 3 | 3.9 |
| Sch 2 | La Zarza | 32.70 | 6.50 | 0.46 | 2.86 | 1805 | 387 | 25 | 29 | 3.0 |
| Sample | Locality | Al | S | Si | Fe | Cu | Zn | Co | Ni | [Al/S] |
| wt % | wt % | wt % | wt % | wt % | wt % | ppm | ppm | |||
| Hyb 1 | Herrerías | 10.80 | 5.05 | 1.61 | 0.12 | 3.7 | 15.2 | 1662 | 1821 | 2.6 |
| Hyb 2 | La Zarza | 11.52 | 5.80 | 2.17 | 0.25 | 7.1 | 0.8 | 160 | 209 | 2.4 |
| Major Metals (mg/L) | Trace Elements (µg/L) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | S | Si | Fe | Al | Cu | Zn | As | Be | Ce | Cr | Co | Ni | Sc | V | Y |
| Sch 1 | 460 | 30 | 2500 | 240 | 15 | 4.5 | <100 | 60 | <100 | 195 | 140 | <100 | <10 | <25 | <10 |
| Sch 2 | 575 | 85 | 2570 | 440 | 30 | 4.5 | 7580 | <10 | <100 | 125 | 160 | <100 | <10 | 230 | 70 |
| Hyb 1 | 425 | 265 | <1 | 790 | 285 | 425 | <100 | 265 | 980 | <25 | 5355 | 7765 | 110 | <25 | 3525 |
| Hyb 2 | 575 | 405 | <1 | 890 | 535 | 65 | <100 | 105 | 1010 | <25 | 1085 | 2780 | 115 | <25 | 2890 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-España, J.; Reyes, J. Comparing Schwertmannite and Hydrobasaluminite Dissolution in Ammonium Oxalate (pH 3.0): Implications for Metal Speciation Studies by Sequential Extraction. Minerals 2019, 9, 57. https://doi.org/10.3390/min9010057
Sánchez-España J, Reyes J. Comparing Schwertmannite and Hydrobasaluminite Dissolution in Ammonium Oxalate (pH 3.0): Implications for Metal Speciation Studies by Sequential Extraction. Minerals. 2019; 9(1):57. https://doi.org/10.3390/min9010057
Chicago/Turabian StyleSánchez-España, Javier, and Jesús Reyes. 2019. "Comparing Schwertmannite and Hydrobasaluminite Dissolution in Ammonium Oxalate (pH 3.0): Implications for Metal Speciation Studies by Sequential Extraction" Minerals 9, no. 1: 57. https://doi.org/10.3390/min9010057