Immobilisation of Platinum by Cupriavidus metallidurans


Abstract
:1. Introduction
2. Materials and Methods
2.1. Culturing of Cupriavidus metallidurans
2.2. C. metallidurans and Aqueous Platinum Experiments
2.3. C. metallidurans—Aqueous Pt(II)- and Pt(IV)-Chloride Dose–Response Experiments
2.4. Chemical Analyses of Solutions and Quantification of Pt Associated with Cells
2.5. Transmission Electron Microscopy (TEM)
2.6. X-ray Absorption Spectroscopy (XAS) Data Collection
2.7. XAS Data Analysis
3. Results
3.1. Laboratory-Based C. metallidurans and Aqueous Platinum Experiments
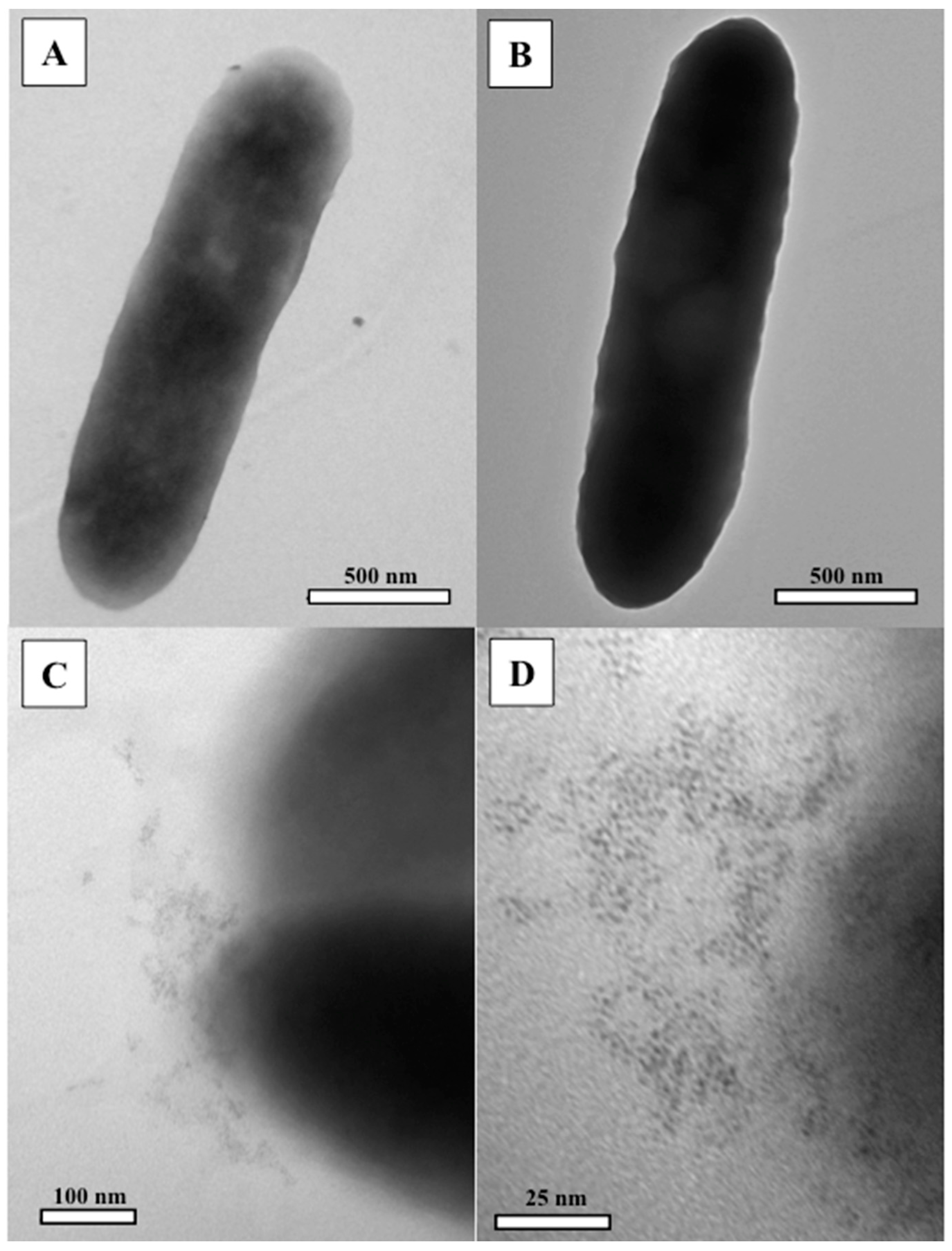
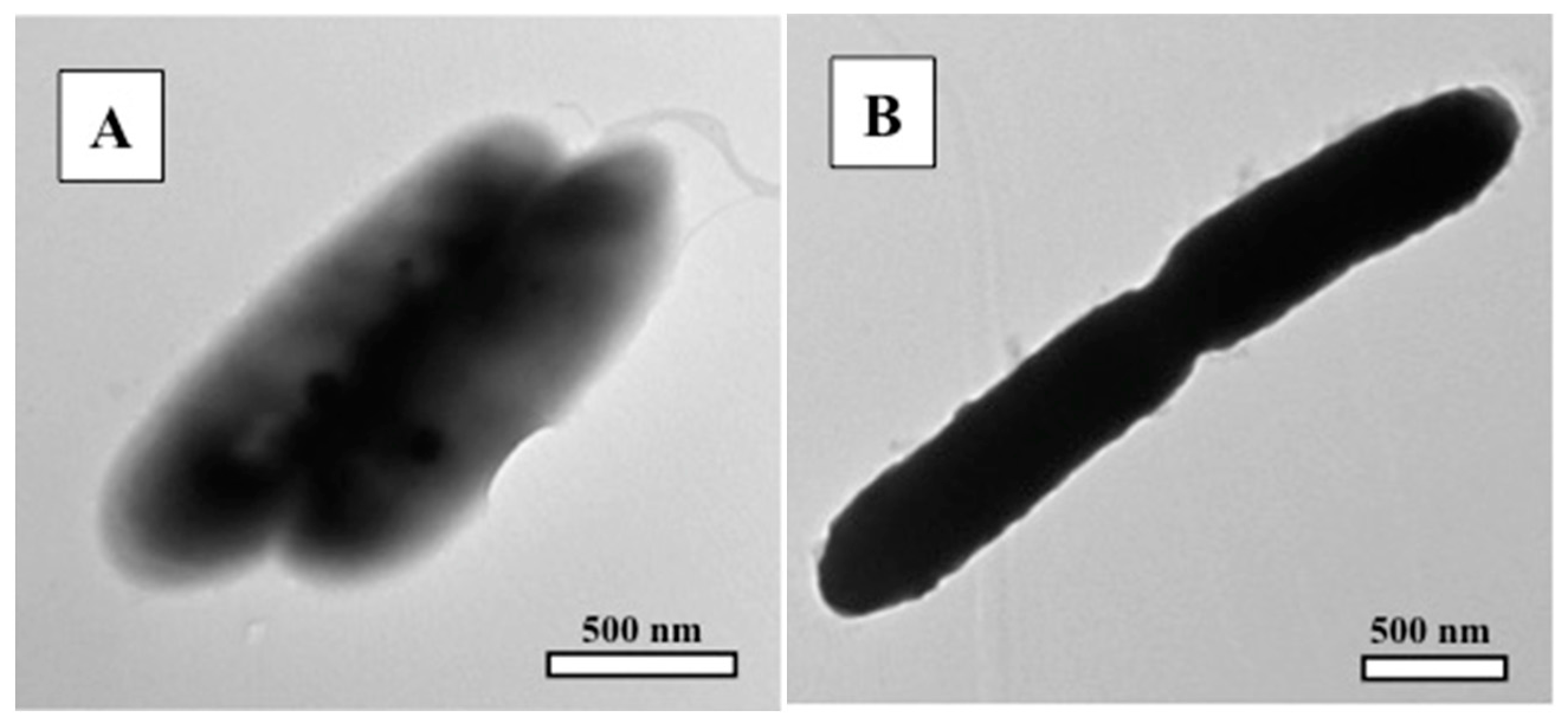
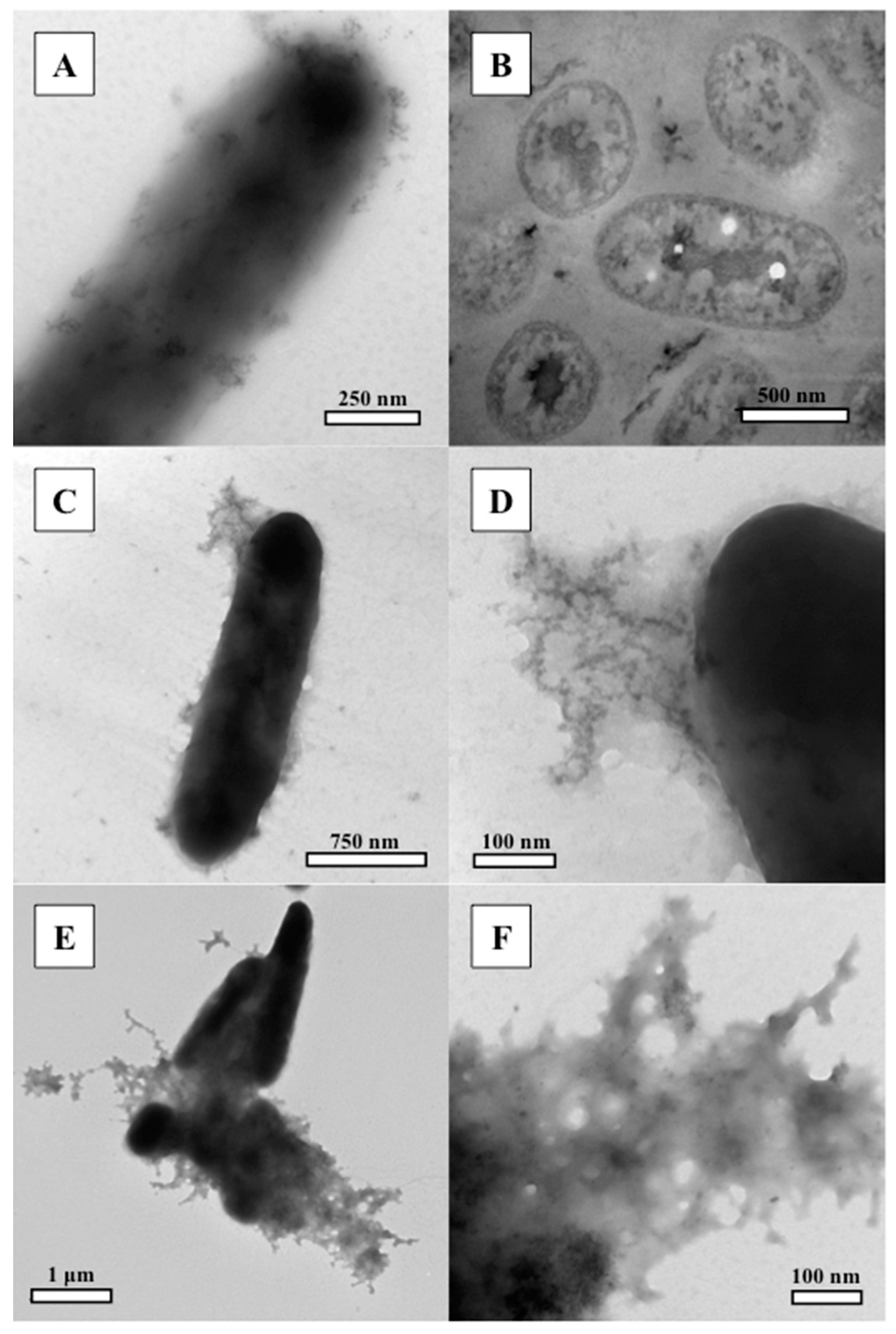
3.2. Transmission Electron Microscopy (TEM)
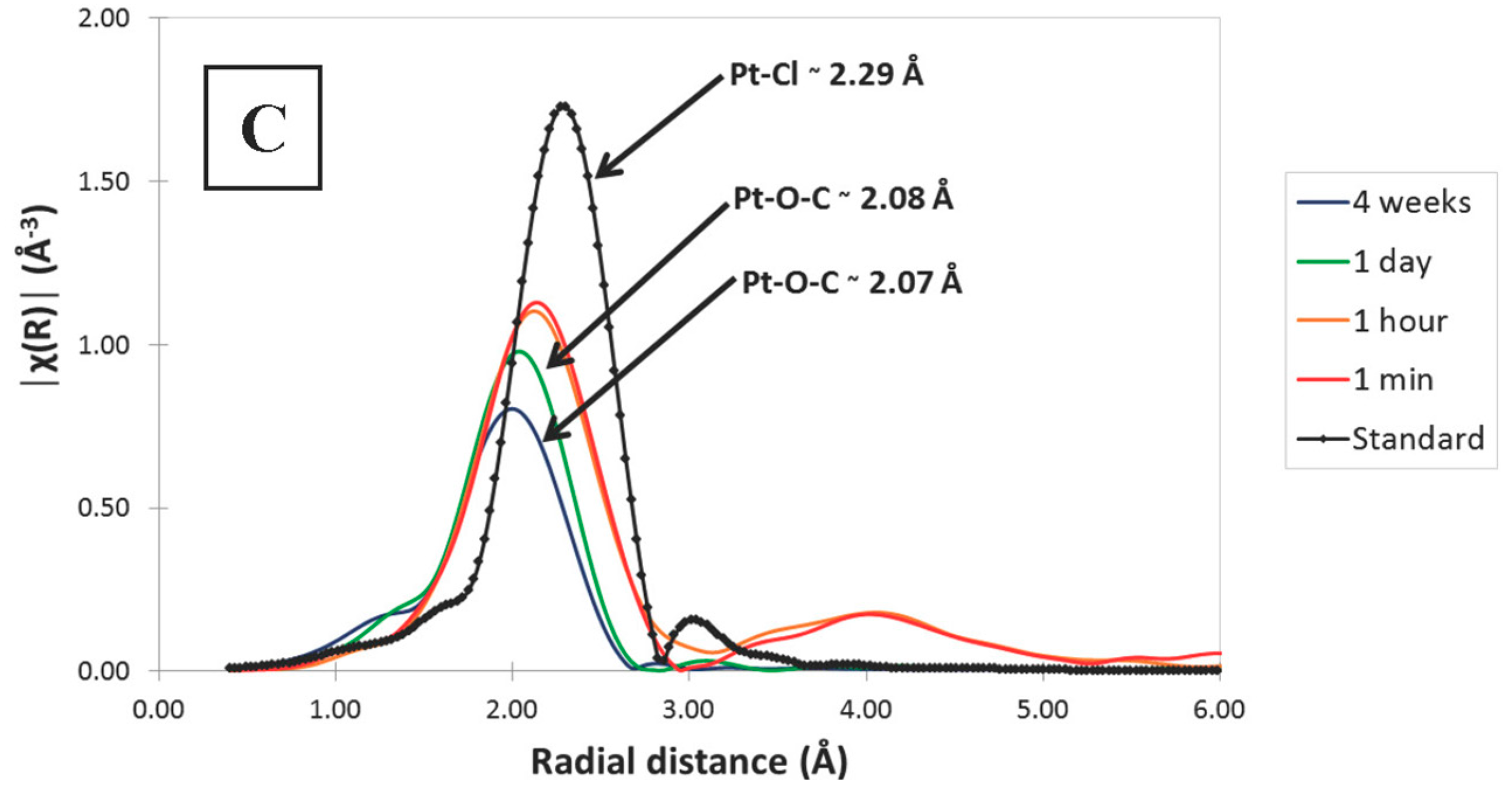
3.3. Synchrotron X-ray Absorption Spectrosocpy (XAS)
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Enders, M.S.; Knickerbocker, C.; Titley, S.R.; Southam, G. The role of bacteria in the supergene environment of the Morenci Porphyry Copper Deposit, Greenlee County, Arizona. Econ. Geol. 2006, 101, 59–70. [Google Scholar] [CrossRef]
- Reith, F.; Brugger, J.; Zammit, C.M.; Gregg, A.L.; Goldfarb, K.C.; Andersen, G.L.; Desantis, T.Z.; Piceno, Y.M.; Brodie, E.L.; Lu, Z.; et al. Influence of geogenic factors on microbial communities in metallogenic Australian soils. ISME J. 2012, 6, 2107–2118. [Google Scholar] [CrossRef] [PubMed]
- Southam, G.; Saunders, J.A. The geomicrobiology of ore deposits. Econ. Geol. 2005, 100, 1067–1084. [Google Scholar] [CrossRef]
- Ehrlich, H.L.; Newman, D.K.; Kappler, A. Ehrlich’s Geomicrobiology, 6th ed.; CRC Press: Boca Raton, FL, USA, 2015; p. 635. [Google Scholar]
- Johnston, C.W.; Wyatt, M.A.; Li, X.; Ibrahim, A.; Shuster, J.; Southam, G.; Magarvey, N. Gold biomineralization by a secondary metabolite from a gold-associated microbe. Nat. Chem. Biol. 2013, 9, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Nies, D.H. Microbial heavy-metal resistance. Appl. Microbiol. Biotechnol. 1999, 51, 730–750. [Google Scholar] [CrossRef] [PubMed]
- Silver, S. Bacterial resistances to toxic metal ions—A review. Gene 1996, 179, 9–19. [Google Scholar] [CrossRef]
- Mossman, D.J.; Dyer, B.D. The geochemistry of Witwatersrand-type gold deposits and the possible influence of ancient prokaryotic communities on gold dissolution and precipitation. Precambrian Res. 1985, 30, 303–319. [Google Scholar] [CrossRef]
- Reith, F.; Lengke, M.F.; Falconer, D.; Craw, D.; Southam, G. Winogradsky review: The geomicrobiology of gold. ISME J. 2007, 1, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Shuster, J.; Southam, G. The in-vitro “growth” of gold grains. Geology 2015, 43, 79–82. [Google Scholar] [CrossRef]
- Shuster, J.; Johnston, C.W.; Magarvey, N.A.; Gordon, R.A.; Barron, K.; Banerjee, N.R.; Southam, G. Structural and chemical characterization of placer gold grains: Implications for bacterial contributions to grain formation. Geomicrobiol. J. 2015, 32, 158–169. [Google Scholar] [CrossRef]
- Southam, G.; Lengke, M.F.; Fairbrother, L.; Reith, F. The biogeochemistry of gold. Elements 2009, 5, 303–307. [Google Scholar] [CrossRef]
- Lengke, M.F.; Fleet, M.E.; Southam, G. Morphology of gold nanoparticles synthesized by filamentous cyanobacteria from gold (I)-thiosulphate and gold (III)-chloride complexes. Langmuir 2006, 22, 2780–2787. [Google Scholar] [CrossRef] [PubMed]
- Reith, F.; Rogers, S.L.; McPhail, D.C.; Webb, D. Biomineralization of gold: Biofilms on bacterioform gold. Science 2006, 313, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Reith, F.; Etschmann, B.; Grosse, C.; Moors, H.; Benotmane, M.A.; Monsieurs, P.; Grass, G.; Doonan, C.; Vogt, S.; Lai, B.; et al. Mechanisms of gold biomineralization in the bacterium Cupriavidus metallidurans. Proc. Natl. Acad. Sci. USA 2009, 106, 17757–17762. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, T. Biosorption and recycling of gold using various microorgamisms. J. Gen. Appl. Microbiol. 2004, 50, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Kenney, J.P.L.; Song, Z.; Bunker, B.A.; Fein, J.B. An experimental study of Au removal from solution by non-metabolizing bacterial cells and their exudates. Geochim. Cosmochim. Acta 2012, 87, 51–60. [Google Scholar] [CrossRef]
- Minter, W.E.L.; Goedhart, M.; Knight, J.; Frimmel, H.E. Morphology of Witwatersrand gold grains from the Basal Reef, evidence for their detrital origin. Econ. Geol. 1993, 88, 237–248. [Google Scholar] [CrossRef]
- Wilson, A.F. Origin of quartz-free gold nuggets and supergene gold found in laterites and soils—A review and some new observations. Aust. J. Earth Sci. 1984, 31, 303–316. [Google Scholar]
- Goldschmidt, V.M. Geochemistry; Oxford University Press: Oxford, UK, 1954; p. 730. [Google Scholar]
- Maier, W.D. Pt-group element (PGE) deposits and occurrences: Mineralization styles, genetic concepts, and exploration criteria. J. Afr. Earth Sci. 2005, 41, 165–191. [Google Scholar] [CrossRef]
- Mungall, J.E.; Naldrett, A.J. Ore deposits of the platinum-group elements. Elements 2008, 4, 253–258. [Google Scholar] [CrossRef]
- Cabral, A.R.; Beaudoin, G.; Choquette, M.; Lehmann, B.; Polonia, J.C. Supergene leaching and formation of platinum in alluvium: Evidence from Serro, Minas Gerais, Brazil. Mineral. Petrol. 2007, 90, 141–150. [Google Scholar] [CrossRef]
- Campbell, G.; Reith, F.; Etschmann, B.; Brugger, J.; Gordon, R.A.; Martinez-Criado, G.; Southam, G. Surface transformations of platinum grains from New South Wales, Australia. Am. Mineral. 2015, 100, 1236–1243. [Google Scholar] [CrossRef]
- Reith, F.; Zammit, C.; Shar, S.S.; Etschmann, B.; Bottrill, R.; Southam, G.; Ta, C.; Kilburn, M.; Oberthür, T.; Ball, A.S.; et al. Biological role in the transformation of platinum-group mineral grains. Nat. Geosci. 2016, 9, 294–299. [Google Scholar] [CrossRef]
- Macdonald, A.J. Ore deposit models #12: The platinum group element deposits—Classification and genesis. Geosci. Can. 1987, 14, 155–166. [Google Scholar]
- Nixon, G.T.; Hammack, J.L. Metallogeny of ultramafic-mafic rocks in British Columbia with emphasis on the platinum-group elements. In Ore Deposits, Tectonics, and Metallogeny in the Canadian Cordillera; McMillan, W.J., Ed.; Province of British Columbia-Ministry of Energy, Mines and Petroleum Resources: Vancouver, BC, Canada, 1991; pp. 125–161, Paper 4. [Google Scholar]
- Koek, M.; Kreuzer, O.P.; Maier, W.D.; Porwal, A.K.; Thompson, M.; Guj, P.A. Review of the PGM industry, deposit models and exploration practices: Implications for Australia’s PGM potential. Resour. Policy 2010, 35, 20–35. [Google Scholar] [CrossRef]
- Mountain, B.W.; Wood, S.A. Chemical controls on the solubility, transport and deposition of platinum and palladium in hydrothermal solutions, a thermodynamic approach. Econ. Geol. 1988, 83, 492–510. [Google Scholar] [CrossRef]
- Colombo, C.; Oates, C.J.; Monhemius, A.J.; Plant, J.A. Complexation of platinum, palladium and rhodium with inorganic ligands in the environment. Geochem. Explor. Environ. Anal. 2008, 8, 91–101. [Google Scholar] [CrossRef]
- Reith, F.; Campbell, S.G.; Ball, A.S.; Pring, A.; Southam, G. Platinum in earth surface environments. Earth Sci. Rev. 2014, 131, 1–21. [Google Scholar] [CrossRef]
- Wood, S.A. The interaction of dissolved platinum with fulvic acid and simple organic acid analogues in aqueous solutions. Can. Mineral. 1990, 28, 665–673. [Google Scholar]
- Anthony, E.Y.; Williams, P.A. Thiosulfate complexing of platinum group elements: Implications for supergene chemistry. In Environmental Geochemistry of Sulfide Oxidation; Alpers, C.N., Bowles, D.W., Eds.; American Chemical Society Books: Washington, DC, USA, 1994; pp. 551–560. [Google Scholar]
- Azaroual, M.; Romand, B.; Freyssinet, P.; Disnar, J. Solubility of platinum in aqueous solutions at 25 °C and pHs 4 to 10 under oxidizing conditions. Geochim. Cosmochim. Acta 2001, 65, 4453–4466. [Google Scholar] [CrossRef]
- Hanley, J.J. The aqueous geochemistry of the Platinum-Group Elements (PGE) in surficial, low-T hydrothermal and high-T magmatic-hydrothermal environments. In Exploration for Platinum-Group Element Deposits; Mungall, J.E., Ed.; Mineralogical Association of Canada: Ottawa, ON, Canada, 2005; pp. 35–56. [Google Scholar]
- Vlassopoulos, D.; Wood, S.A.; Mucci, A. Gold speciation in natural waters: II. The importance of organic complexing-experiments with some simple model ligands. Geochim. Cosmochim. Acta 1990, 54, 1575–1586. [Google Scholar] [CrossRef]
- Bowles, J.F.W. The development of platinum-group minerals in laterites. Econ. Geol. 1986, 81, 1278–1285. [Google Scholar] [CrossRef]
- Lengke, M.F.; Fleet, M.E.; Southam, G. Synthesis of platinum nanoparticles by reaction of filamentous cyanobacteria with platinum(IV)−chloride complex. Langmuir 2006, 22, 7318–7323. [Google Scholar] [CrossRef] [PubMed]
- Konishi, Y.; Ohnoa, K.; Saitoh, N.; Nomura, T.; Nagamine, S.; Hishida, H.; Takahashi, Y.; Uruga, T. Bioreductive deposition of platinum nanoparticles on the bacterium Shewanella algae. J. Biotechnol. 2007, 128, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Cabral, A.R.; Radtke, M.; Munnik, F.; Lehmann, B.; Reinholz, U.; Riesemeier, H.; Tupinambá, M.; Kwitko-Ribeiro, R. Iodine in alluvial platinum–palladium nuggets: Evidence for biogenic precious-metal fixation. Chem. Geol. 2011, 281, 125–132. [Google Scholar] [CrossRef]
- Guiné, V.; Martins, J.M.F.; Causse, B.; Durand, A.; Gaudet, J.P.; Spadini, L. Effect of cultivation and experimental conditions on the surface reactivity of the metal-resistant bacteria Cupriavidus metallidurans CH34 to protons, cadmium and zinc. Chem. Geol. 2007, 236, 266–280. [Google Scholar] [CrossRef]
- Mergeay, M.; Nies, D.; Schlegel, H.G.; Gerits, J.; Charles, P.; Van Gijsegem, F. Alicaligenes eutrophus CH34 is a facultative chemolithotroph with plasmid-bound resistance to heavy metals. J. Bacteriol. 1985, 62, 328–334. [Google Scholar]
- Mergeay, M.; Monchy, S.; Vallaeys, T.; Auquier, V.; Benotmane, A.; Bertin, P.; Taghavi, S.; Dunn, J.; van der Lelie, D.; Wattiez, R. Ralstonia metallidurans, a bacterium specifically adapted to toxic metals: Towards a catalogue of metal-responsive genes. FEMS Microbiol. Rev. 2003, 27, 385–410. [Google Scholar] [CrossRef]
- Ledrich, M.; Stemmler, S.; Laval-Gilly, P.; Foucaud, L.; Falla, J. Precipitation of silver-thiosulphate complex and immobilization of silver by Cupriavidus metallidurans CH34. BioMetals 2005, 18, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Sanders, E.R. Aseptic laboratory techniques: Plating methods. J. Vis. Exp. 2012, 63, e3064. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, M.; Bankston, P. Measurement of live bacteria by Nomarski interference microscopy and steriologic methods as tested by macroscopic rod-shaped models. Appl. Environ. Microbiol. 1988, 54, 105–109. [Google Scholar] [PubMed]
- Williams, G.P. Electron binding energies. In X-ray Data Booklet; Thompson, A.C., Vaughan, D., Eds.; Lawrence Berkeley National Laboratory, University of California: Berkeley, CA, USA, 2001; Section 1.1. [Google Scholar]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.; Doyle, J.; Rosenberg, M. Mechanism of enhancement of microbial cell hydrophobicity by cationic polymers. J. Bacteriol. 1990, 172, 5650–5654. [Google Scholar] [CrossRef] [PubMed]
- Ohba, S.; Sato, S.; Saito, Y. Electron-density distribution in crystals of potassium tetrachloroplatinate (II) and influence of X-ray diffuse scattering. Acta Crystallogr. Sect. B 1983, 39, 49–53. [Google Scholar] [CrossRef]
- Moret, M.E.; Keller, S.F.; Slootweg, J.C.; Chen, P. Mononuclear platinum (II) complexes incorporating κ2-carboxylate ligands: Synthesis, structure, and reactivity. Inorg. Chem. 2009, 48, 6972–6978. [Google Scholar] [CrossRef] [PubMed]
- Ankudinov, A.L.; Rehr, J.J.; Bare, S.R. Hybridization peaks in Pt-Cl XANES. Chem. Phys. Lett. 2000, 316, 495–500. [Google Scholar] [CrossRef]
- Wood, S.A.; Mountain, B.W.; Pan, P. The aqueous geochemistry of platinum, palladium and gold: Recent experimental constraints and a re-evaluation of theoretical predictions. Can. Mineral. 1992, 30, 955–982. [Google Scholar]
- Beveridge, T.J.; Murray, R.G.E. Sites of metal deposition in the cell wall of Bacillus subtilis. J. Bacteriol. 1980, 141, 876–887. [Google Scholar] [PubMed]
- Guiné, V.; Spadini, L.; Sarret, G.; Muris, M.; Delolme, C.; Gaudet, J.P.; Martins, J.M.F. Zinc sorption to three gram-negative bacteria: Combined titration, modeling, and EXAFS study. Environ. Sci. Technol. 2006, 40, 1806–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fein, J.; Daughney, C.; Yee, N.; Davis, T.A. Chemical equilibrium model for metal adsorption onto bacterial surfaces. Geochim. Cosmochim. Acta 1997, 61, 3319–3328. [Google Scholar] [CrossRef]
- Kubrakova, I.V.; Fortygin, A.V.; Lobov, S.G.; Koshcheeva, I.Y.; Tyutyunnik, O.A.; Mironenko, M.V. Migration of platinum, palladium, and gold in the water systems of platinum deposits. Geochem. Int. 2011, 49, 1072–1084. [Google Scholar] [CrossRef]
- Janssen, P.J.; Van Houdt, R.; Moors, H.; Monsieurs, P.; Morin, N.; Michaux, A.; Benotmane, M.A.; Leys, N.; Vallaeys, T.; Lapidus, A.; et al. The complete genome sequence of Cupriavidus metallidurans strain CH34, a master survivalist in harsh and anthropogenic environments. PLoS ONE 2010, 5, e10433. [Google Scholar] [CrossRef] [PubMed]
- Urrutia, M.; Kemper, M.; Doyle, R.; Beveridge, T.J. The membrane-induced proton motive force influences the metal binding ability of Bacillus subtilis cell walls. Appl. Environ. Microbiol. 1992, 58, 3837–3844. [Google Scholar]
- Etschmann, B.; Brugger, J.; Fairbrother, L.; Grosse, C.; Nies, D.H.; Martinez-Criado, G.; Reith, F. Applying the Midas touch: Differing toxicity of mobile gold and platinum complexes drives biomineralization in the bacterium Cupriavidus metallidurans. Chem. Geol. 2016, 438, 103–111. [Google Scholar] [CrossRef]
- Fuchs, W.A.; Rose, A.W. The geochemical behavior of platinum and palladium in the weathering cycle in the Stillwater Complex, Montana. Econ. Geol. 1974, 69, 332–346. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [Pt(II)] | cfu/mL after 1 min Exposure | cfu/mL after 1 h Exposure | cfu/mL after 1 day Exposure |
|---|---|---|---|
| 0 µM | 5.0 ± 0.2 × 108 | 5.0 ± 0.2 × 108 | 5.0 ± 0.2 × 108 |
| 0.5 µM | 4.9 ± 0.1 × 108 | 2.7 ± 0.1 × 108 | 4.6 ± 0.1 × 108 |
| 5 µM | 3.7 ± 0.2 × 108 | 3.2 ± 0.2 × 108 | 4.3 ± 0.2 × 106 |
| 50 µM | 4.1 ± 0.3 × 108 | 1.3 ± 0.1 × 107 | 2.1 ± 0.1 × 103 |
| 500 µM | 9.9 ± 0.3 × 106 | 0 | 0 |
| 5000 µM | 0 | 0 | 0 |
| [Pt(IV)] | cfu/mL after 1 min Exposure | cfu/mL after 1 h Exposure | cfu/mL after 1 day Exposure |
|---|---|---|---|
| 0 µM | 5.0 ± 0.2 × 108 | 5.0 ± 0.2 × 108 | 5.0 ± 0.2 × 108 |
| 0.5 µM | 4.5 ± 0.1 × 108 | 3.2 ± 0.1 × 108 | 3.3 ± 0.1 × 108 |
| 5 µM | 4.9 ± 0.3 × 108 | 2.5 ± 0.1 × 108 | 7.6 ± 0.4 × 107 |
| 50 µM | 1.1 ± 0.1 × 108 | 6.4 ± 0.4 × 106 | 4.8 ± 0.3 × 103 |
| 500 µM | 6.6 ± 0.4 × 102 | 0 | 0 |
| 5000 µM | 0 | 0 | 0 |
| [Pt(II)] | Intracellular Concentration after 1 min Exposure (mM) | Intracellular Concentration after 1 h Exposure (mM) | Intracellular Concentration after 1 day Exposure (mM) |
|---|---|---|---|
| 5 µM | 1.8 ± 0.01 | 1.6 ± 0.02 | 15 ± 0.3 |
| 50 µM | 51 ± 0.3 | 44 ± 0.5 | 67 ± 1 |
| 500 µM | 120 ± 0.70 | 120 ± 1.5 | 410 ± 7.3 |
| 5000 µM | 850 ± 12 | 800 ± 12 | 850 ± 6.5 |
| [Pt(IV)] | Intracellular Concentration after 1 min Exposure (mM) | Intracellular Concentration after 1 h Exposure (mM) | Intracellular Concentration after 1 day Exposure (mM) |
|---|---|---|---|
| 5 µM | 1.6 ± 0.01 | 2.6 ± 0.02 | 9.6 ± 0.05 |
| 50 µM | 11 ± 0.05 | 13 ± 0.1 | 40 ± 0.2 |
| 500 µM | 45 ± 0.22 | 160 ± 1.3 | 210 ± 1.2 |
| 5000 µM | 310 ± 5.8 | 350 ± 0.45 | 870 ± 0.75 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campbell, G.; MacLean, L.; Reith, F.; Brewe, D.; Gordon, R.A.; Southam, G. Immobilisation of Platinum by Cupriavidus metallidurans. Minerals 2018, 8, 10. https://doi.org/10.3390/min8010010
Campbell G, MacLean L, Reith F, Brewe D, Gordon RA, Southam G. Immobilisation of Platinum by Cupriavidus metallidurans. Minerals. 2018; 8(1):10. https://doi.org/10.3390/min8010010
Chicago/Turabian StyleCampbell, Gordon, Lachlan MacLean, Frank Reith, Dale Brewe, Robert A. Gordon, and Gordon Southam. 2018. "Immobilisation of Platinum by Cupriavidus metallidurans" Minerals 8, no. 1: 10. https://doi.org/10.3390/min8010010