
Kinetics and Mechanisms of Chalcopyrite Dissolution at Controlled Redox Potential of 750 mV in Sulfuric Acid Solution


Abstract
:
1. Introduction
2. Methodology
2.1. Chalcopyrite Sample
2.2. Leaching Conditions
2.3. Bulk and Surface Analyses
3. Results and Discussion
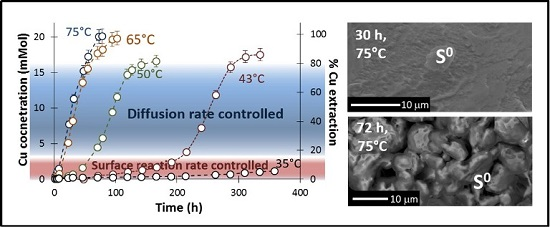
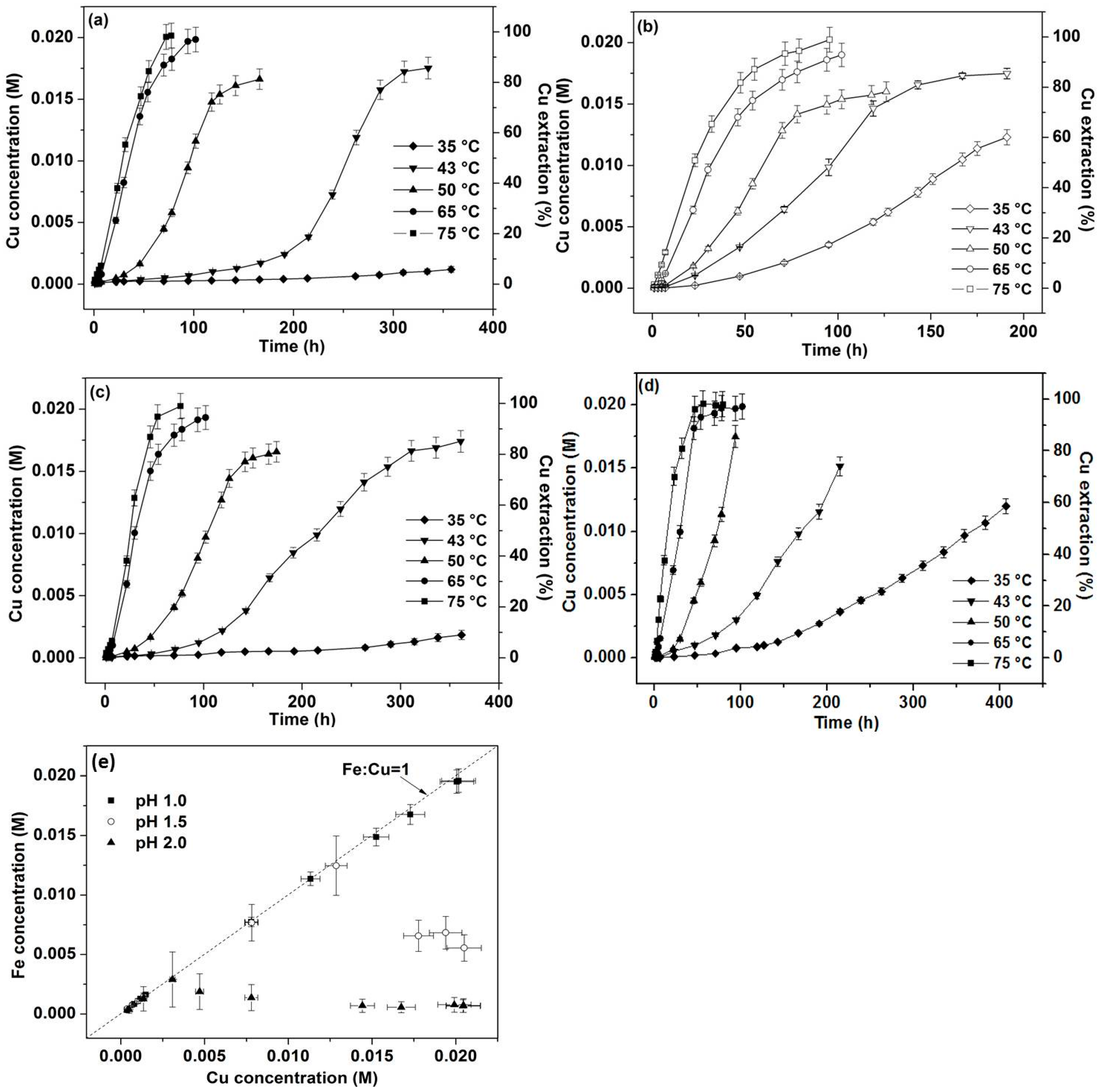
3.1. Effect of Temperature and pH (No Iron Addition)
3.2. Effect of 4 mmol Iron Addition
3.3. Kinetics of Dissolution
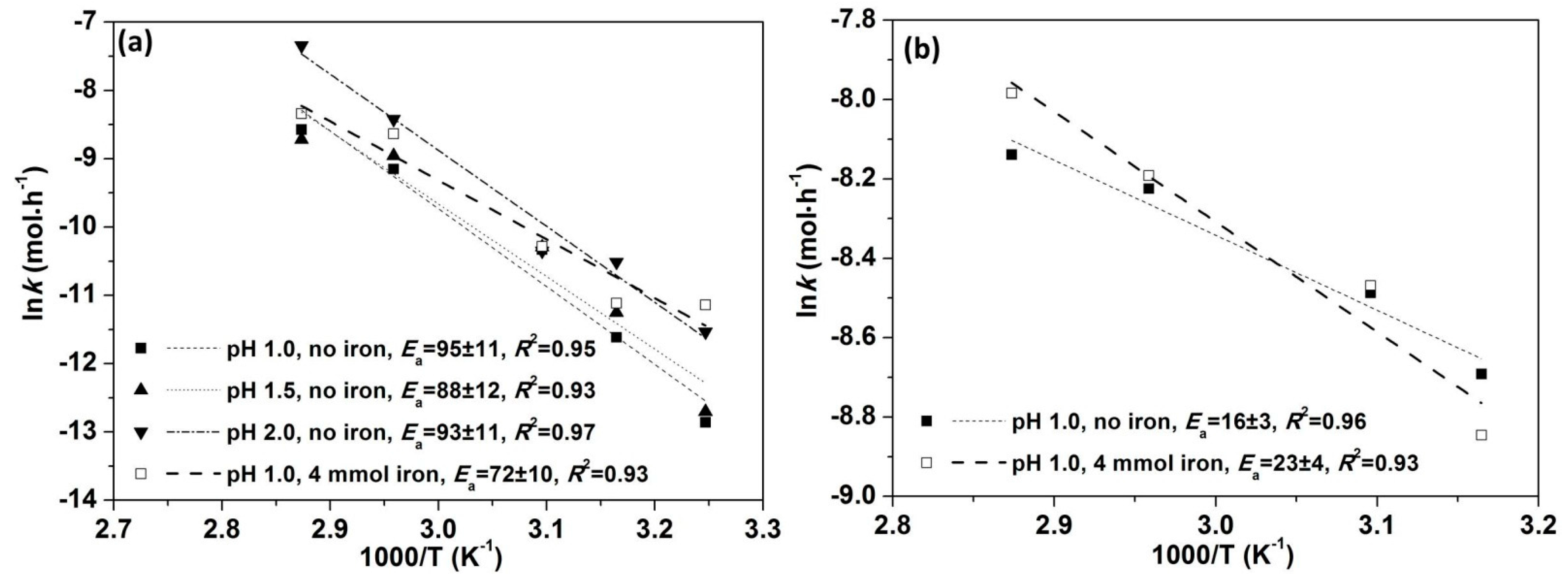
3.3.1. Kinetic Analysis by Apparent Rate (<10% Cu Extraction)
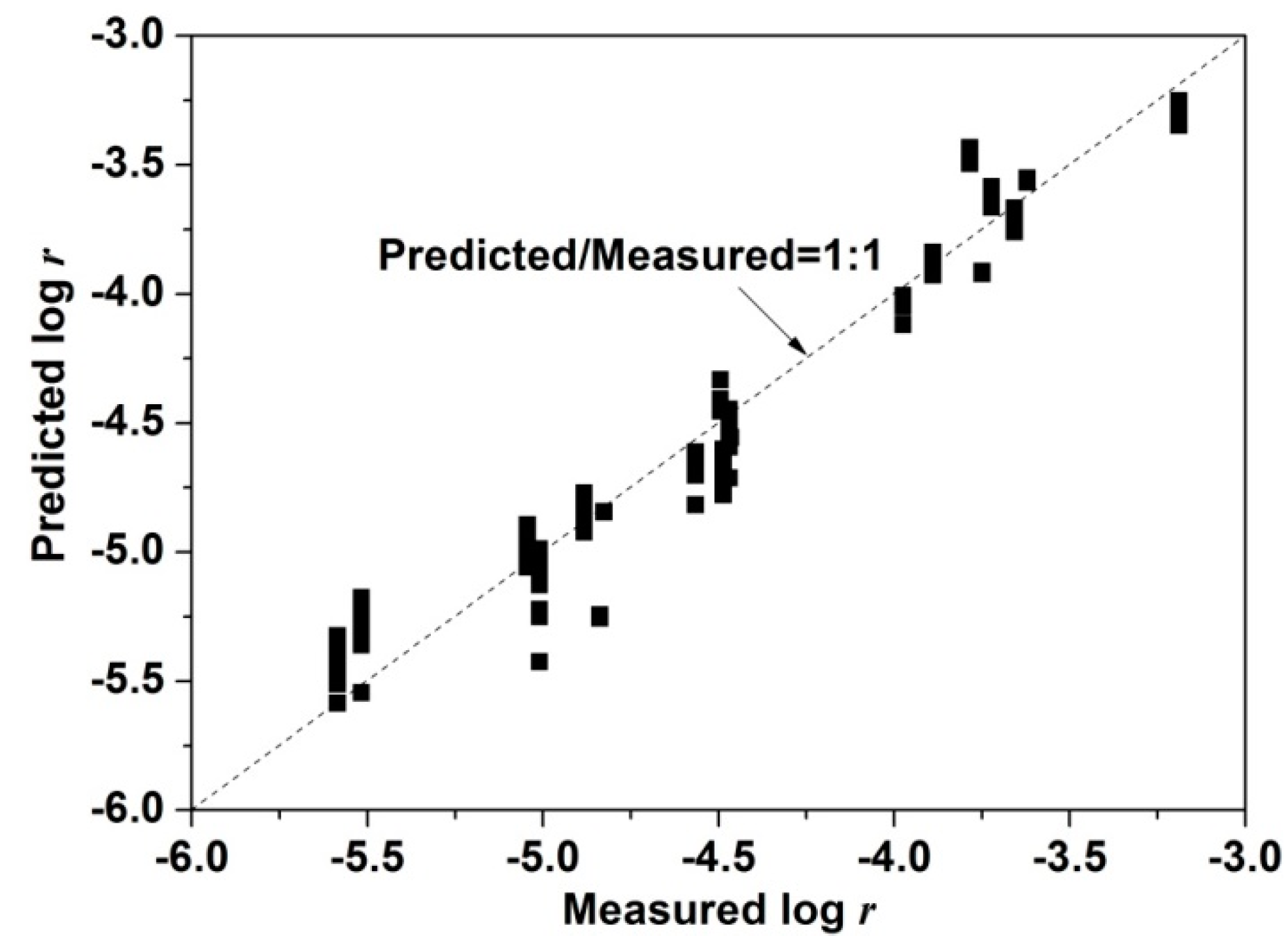
3.3.2. Kinetic Analysis by Multiple Linear Regression (<10% Cu Extraction)
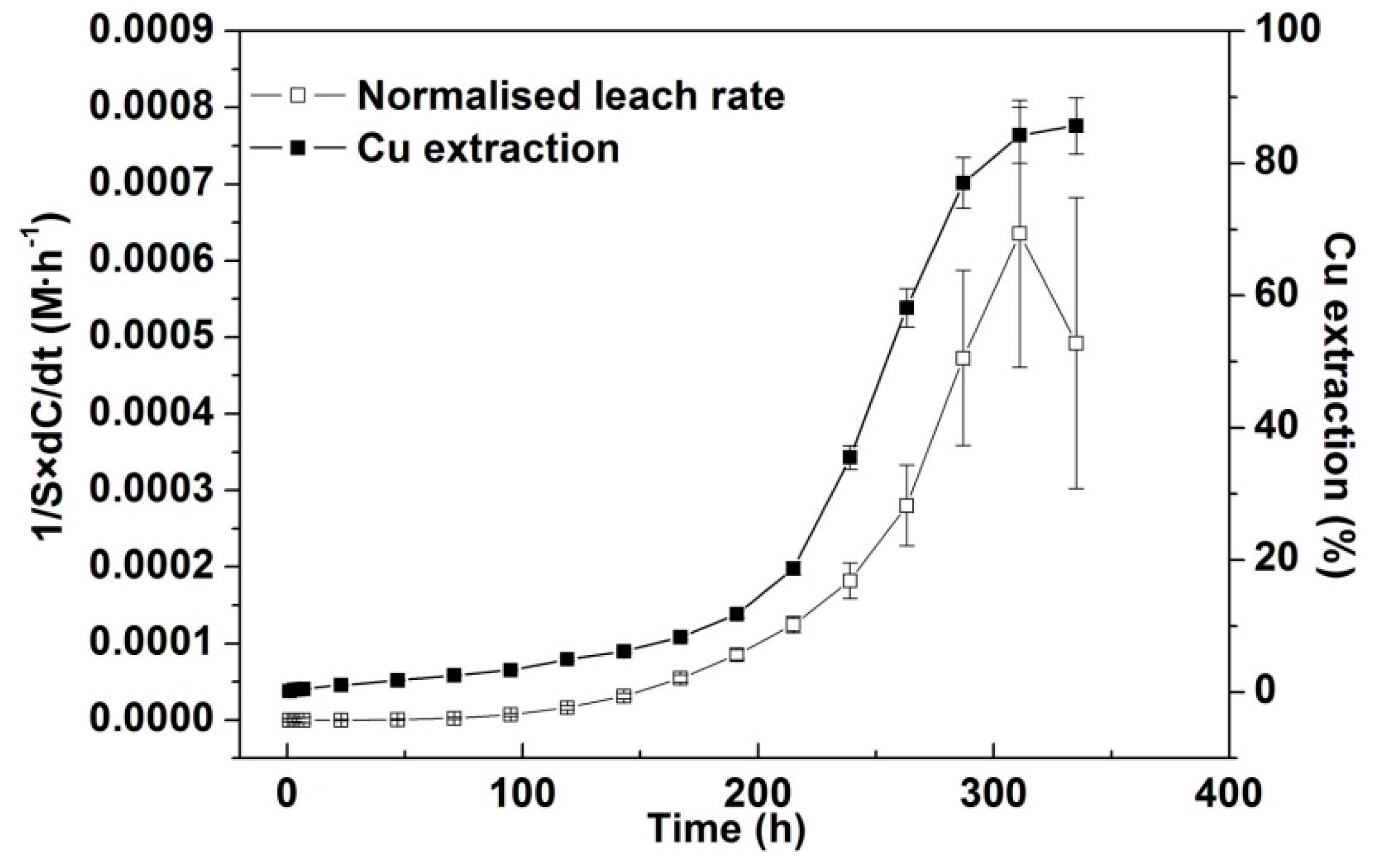
3.3.3. Kinetic Analysis for Cu Extraction > 10%
3.3.4. Unleached Chalcopyrite Particles
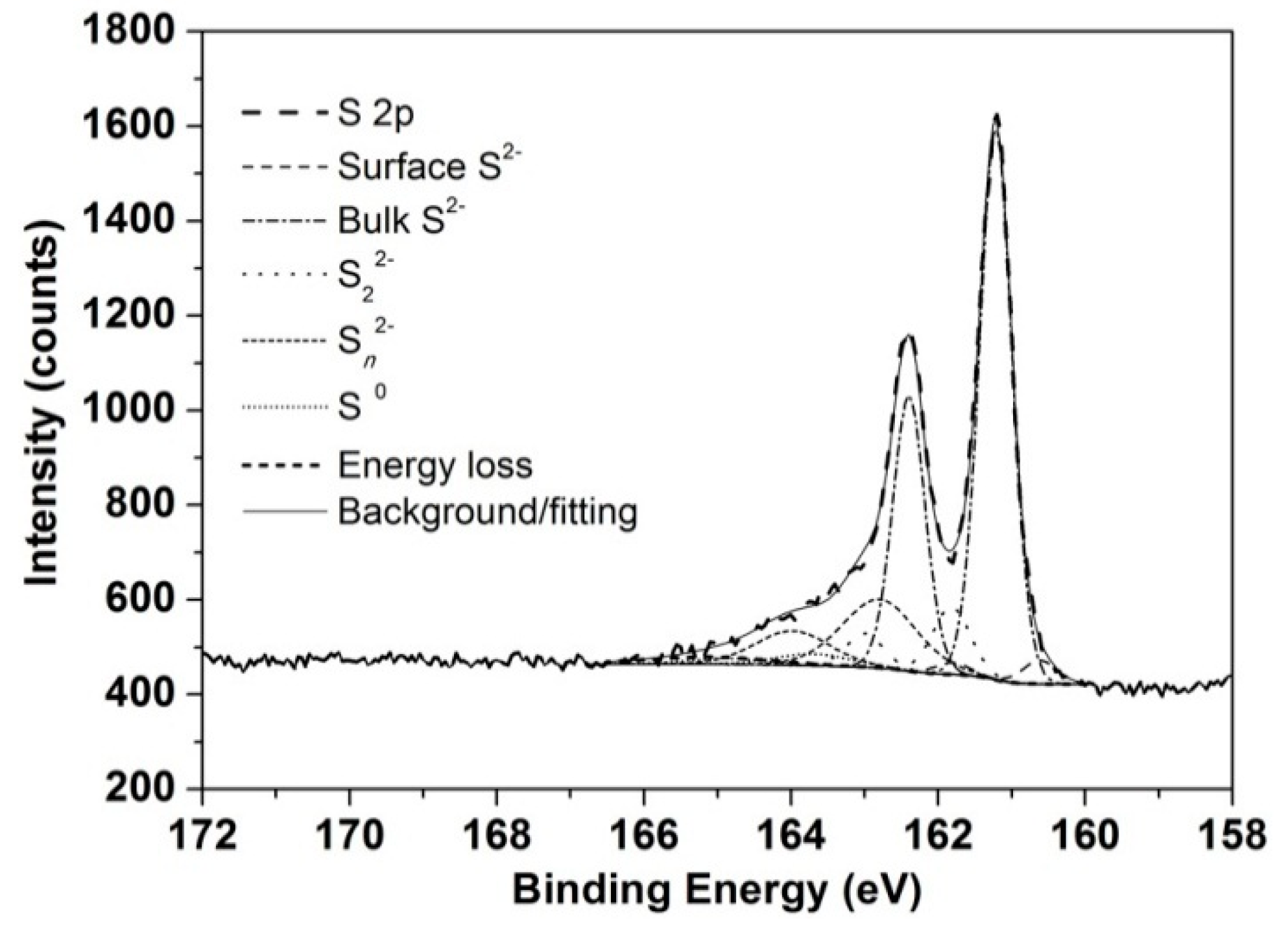
3.4. XPS Analyses for 1 and 30 h Leach Residues from pH 1 Lixiviant
3.5. SEM Analysis
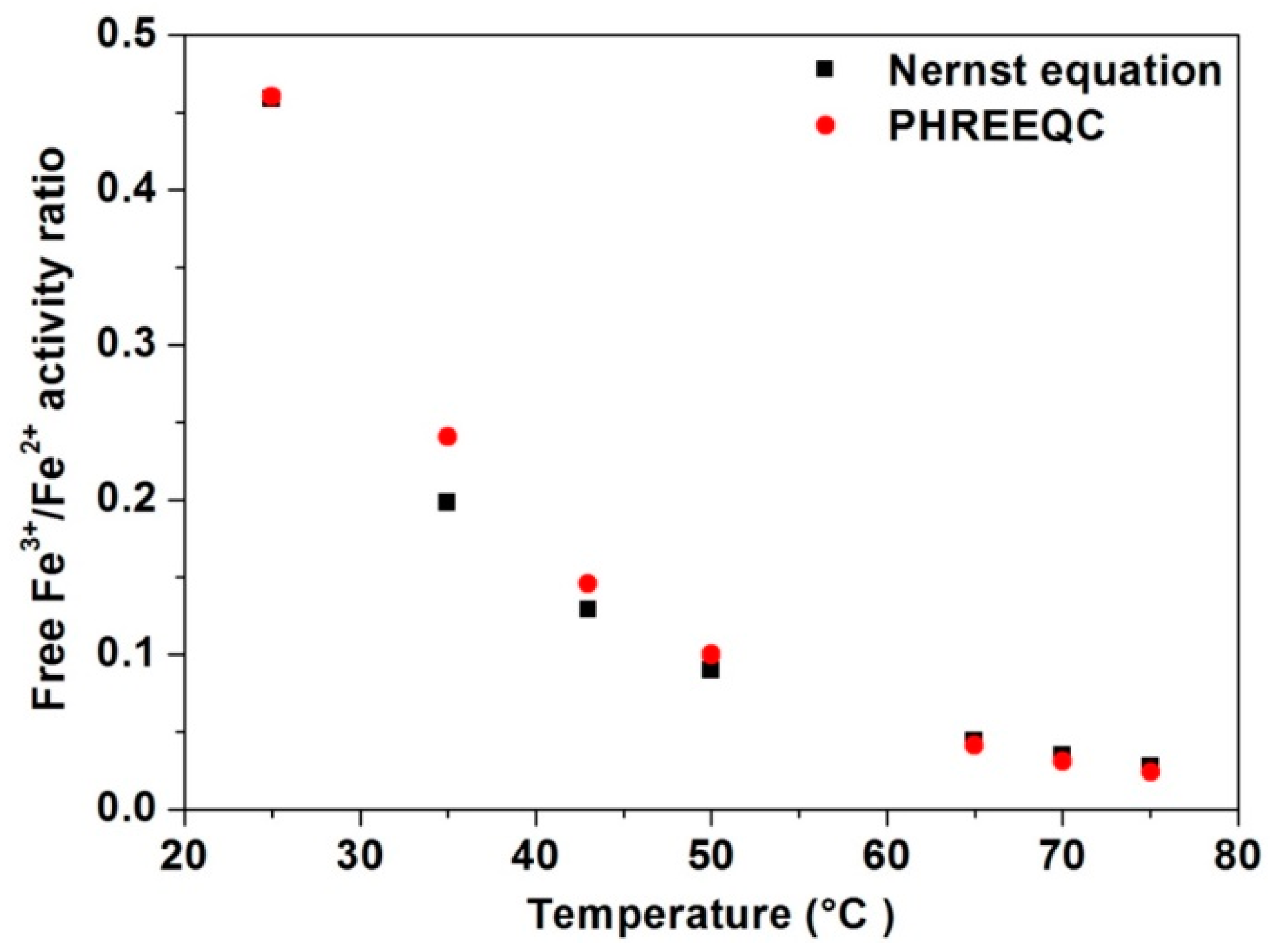
3.6. Identification of Reactions and Mechanisms
4. Conclusions and Future Work
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Habashi, F. Chalcopyrite. In Its Chemistry and Metallurgy; McGraw-Hill: New York, NY, USA, 1978. [Google Scholar]
- Li, J.; Kawashima, N.; Kaplun, K.; Absolon, V.J.; Gerson, A.R. Chalcopyrite leaching: The rate controlling factors. Geochim. Cosmochim. Acta 2010, 74, 2881–2893. [Google Scholar] [CrossRef]
- Kaplun, K.; Li, J.; Kawashima, N.; Gerson, A.R. Cu and Fe chalcopyrite leach activation energies and the effect of added Fe3+. Geochim. Cosmochim. Acta 2011, 75, 5865–5878. [Google Scholar] [CrossRef]
- Kimball, B.E.; Rimstidt, J.D.; Brantley, S.L. Chalcopyrite dissolution rate laws. Appl. Geochem. 2010, 25, 972–983. [Google Scholar] [CrossRef]
- Li, Y.; Kawashima, N.; Li, J.; Chandra, A.P.; Gerson, A.R. A review of the structure, and fundamental mechanisms and kinetics of the leaching of chalcopyrite. Adv. Colloid Interface Sci. 2013, 197–198, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Kametani, H.; Aoki, A. Effect of suspension potential on the oxidation rate of copper concentrate in a sulfuric acid solution. Metall. Mater. Trans. B 1985, 16, 695–705. [Google Scholar] [CrossRef]
- Córdoba, E.M.; Muñoz, J.A.; Blázquez, M.L.; González, F.; Ballester, A. Leaching of chalcopyrite with ferric ion. Part III: Effect of redox potential on the silver-catalyzed process. Hydrometallurgy 2008, 93, 97–105. [Google Scholar] [CrossRef]
- Watling, H.R. Chalcopyrite hydrometallurgy at atmospheric pressure: 1. Review of acidic sulfate, sulfate-chloride and sulfate-nitrate process options. Hydrometallurgy 2013, 140, 163–180. [Google Scholar] [CrossRef]
- Jones, D.L.; Peters, E. The leaching of chalcopyrite with ferric sulfate and ferric chloride. Extr. Metall. Copp. 1976, 2, 633–653. [Google Scholar]
- Lin, H.; Sohn, H.; Wadsworth, M. The kinetics of leaching of chalcopyrite and pyrite grains in primary copper ore by dissolved-oxygen. In Hydrometallurgical Reactor Design and Kinetics; TMS-AIME: Warrendale, PA, USA, 1986; pp. 149–168. [Google Scholar]
- Hirato, T.; Majima, H.; Awakura, Y. The leaching of chalcopyrite with ferric sulfate. Metall. Mater. Trans. B 1987, 18, 489–496. [Google Scholar] [CrossRef]
- Hiroyoshi, N.; Miki, H.; Hirajima, T.; Tsunekawa, M. A model for ferrous-promoted chalcopyrite leaching. Hydrometallurgy 2000, 57, 31–38. [Google Scholar] [CrossRef]
- Hiroyoshi, N.; Miki, H.; Hirajima, T.; Tsunekawa, M. Enhancement of chalcopyrite leaching by ferrous ions in acidic ferric sulfate solutions. Hydrometallurgy 2001, 60, 185–197. [Google Scholar] [CrossRef]
- Qiu, T.; Nie, G.; Wang, J.; Cui, L. Kinetic process of oxidative leaching of chalcopyrite under low oxygen pressure and low temperature. Trans. Nonferrous Met. Soc. China 2007, 17, 418–422. [Google Scholar] [CrossRef]
- Klauber, C. A critical review of the surface chemistry of acidic ferric sulphate dissolution of chalcopyrite with regards to hindered dissolution. Int. J. Miner. Proc. 2008, 86, 1–17. [Google Scholar] [CrossRef]
- Watling, H.R. The bioleaching of sulphide minerals with emphasis on copper sulphides—A review. Hydrometallurgy 2006, 84, 81–108. [Google Scholar] [CrossRef]
- Munoz, P.B.; Miller, J.D.; Wadsworth, M.E. Reaction mechanism for the acid ferric sulfate leaching of chalcopyrite. Metall. Trans. B 1979, 10, 149–158. [Google Scholar] [CrossRef]
- Pradhan, N.; Nathsarma, K.C.; Srinivasa Rao, K.; Sukla, L.B.; Mishra, B.K. Heap bioleaching of chalcopyrite: A review. Miner. Eng. 2008, 21, 355–365. [Google Scholar] [CrossRef]
- Sokic, M.D.; Markovic, B.; Zivkovic, D. Kinetics of chalcopyrite leaching by sodium nitrate in sulphuric acid. Hydrometallurgy 2009, 95, 273–279. [Google Scholar] [CrossRef]
- Li, Y.; Chandra, A.P.; Gerson, A.R. Scanning photoelectron microscopy studies of freshly fractured chalcopyrite exposed to O2 and H2O. Geochim. Cosmochim. Acta 2014, 133, 372–386. [Google Scholar] [CrossRef]
- Hiroyoshi, N.; Arai, M.; Miki, H.; Tsunekawa, M.; Hirajima, T. A new reaction model for the catalytic effect of silver ions on chalcopyrite leaching in sulfuric acid solutions. Hydrometallurgy 2002, 63, 257–267. [Google Scholar] [CrossRef]
- Koleini, S.M.J.; Jafarian, M.; Abdollahy, M.; Aghazadeh, V. Galvanic leaching of chalcopyrite in atmospheric pressure and sulfate media: Kinetic and surface studies. Ind. Eng. Chem. Res. 2010, 49, 5997–6002. [Google Scholar] [CrossRef]
- Antonijevic, M.M.; Jankovic, Z.D.; Dimitrijevic, M.D. Kinetics of chalcopyrite dissolution by hydrogen peroxide in sulphuric acid. Hydrometallurgy 2004, 71, 329–334. [Google Scholar] [CrossRef]
- Adebayo, A.; Ipinmoroti, K.; Ajayi, O. Dissolution kinetics of chalcopyrite with hydrogen peroxide in sulphuric acid medium. Chem. Biochem. Eng. Q. 2003, 17, 213–218. [Google Scholar]
- Yévenes, L.V.; Miki, H.; Nicol, M. The dissolution of chalcopyrite in chloride solutions: Part 2: Effect of various parameters on the rate. Hydrometallurgy 2010, 103, 80–85. [Google Scholar] [CrossRef]
- Aydogan, S.; Ucar, G.; Canbazoglu, M. Dissolution kinetics of chalcopyrite in acidic potassium dichromate solution. Hydrometallurgy 2006, 81, 45–51. [Google Scholar] [CrossRef]
- Saxena, N.N.; Mandre, N.R. Mixed control kinetics of copper dissolution for copper ore using ferric chloride. Hydrometallurgy 1992, 28, 111–117. [Google Scholar] [CrossRef]
- Dutrizac, J.E. Elemental sulphur formation during the ferric sulphate leaching of chalcopyrite. Can. Metall. Q. 1989, 28, 337–344. [Google Scholar] [CrossRef]
- Antonijevic, M.M.; Jankovic, Z.; Dimitrijevic, M. Investigation of the kinetics of chalcopyrite oxidation by potassium dichromate. Hydrometallurgy 1994, 35, 187–201. [Google Scholar] [CrossRef]
- Klauber, C.; Parker, A.; van Bronswijk, W.; Watling, H. Sulphur speciation of leached chalcopyrite surfaces as determined by X-ray photoelectron spectroscopy. Int. J. Miner. Proc. 2001, 62, 65–94. [Google Scholar] [CrossRef]
- Hackl, R.P.; Dreisinger, D.B.; Peters, E.; King, J.A. Passivation of chalcopyrite during oxidative leaching in sulfate media. Hydrometallurgy 1995, 39, 25–48. [Google Scholar] [CrossRef]
- Tshilombo, A.F.; Petersen, J.; Dixon, D.G. The influence of applied potentials and temperature on the electrochemical response of chalcopyrite during bacterial leaching. Miner. Eng. 2002, 15, 809–813. [Google Scholar] [CrossRef]
- Córdoba, E.M.; Muñoz, J.A.; Blázquez, M.L.; González, F.; Ballester, A. Leaching of chalcopyrite with ferric ion. Part I: General aspects. Hydrometallurgy 2008, 93, 81–87. [Google Scholar] [CrossRef]
- Parker, A.J.; Paul, R.L.; Power, G.P. Electrochemistry of the oxidative leaching of copper from chalcopyrite. J. Electroanal. Chem. Interfacial Electrochem. 1981, 118, 305–316. [Google Scholar] [CrossRef]
- Harmer, S.L.; Thomas, J.E.; Fornasiero, D.; Gerson, A.R. The evolution of surface layers formed during chalcopyrite leaching. Geochim. Cosmochim. Acta 2006, 70, 4392–4402. [Google Scholar] [CrossRef]
- Buckley, A.N.; Woods, R. An X-ray photoelectron spectroscopic study of the oxidation of chalcopyrite. Aust. J. Chem. 1984, 37, 2403–2413. [Google Scholar] [CrossRef]
- Brion, D. Photoelectron spectroscopic study of the surface degradation of pyrite (FeS2), chalcopyrite (CuFeS2), sphalerite (ZnS), and galena (PbS) in air and water. Appl. Surf. Sci. 1980, 5, 133–152. [Google Scholar] [CrossRef]
- Petersen, J.; Dixon, D.G. Thermophilic heap leaching of a chalcopyrite concentrate. Miner. Eng. 2002, 15, 777–785. [Google Scholar] [CrossRef]
- Petersen, J.; Dixon, D.G. Competitive bioleaching of pyrite and chalcopyrite. Hydrometallurgy 2006, 83, 40–49. [Google Scholar] [CrossRef]
- Li, Y.; Qian, G.; Li, J.; Gerson, A. The rate controlling parameters in the hydrometallurgical leaching of chalcopyrite. In Proceedings of the ALTA 2014, Perth, Australia, 24–31 May 2014; pp. 399–410.
- Sandström, A.; Shchukarev, A.; Paul, J. XPS characterisation of chalcopyrite chemically and bio-leached at high and low redox potential. Miner. Eng. 2005, 18, 505–515. [Google Scholar] [CrossRef]
- Córdoba, E.M.; Muñoz, J.A.; Blázquez, M.L.; González, F.; Ballester, A. Leaching of chalcopyrite with ferric ion. Part II: Effect of redox potential. Hydrometallurgy 2008, 93, 88–96. [Google Scholar] [CrossRef]
- Hiroyoshi, N.; Kitagawa, H.; Tsunekawa, M. Effect of solution composition on the optimum redox potential for chalcopyrite leaching in sulfuric acid solutions. Hydrometallurgy 2008, 91, 144–149. [Google Scholar] [CrossRef]
- Geelhoed, J.S.; Meeussen, J.C.L.; Hillier, S.; Lumsdon, D.G.; Thomas, R.P.; Farmer, J.G.; Paterson, E. Identification and geochemical modeling of processes controlling leaching of CR(VI) and other major elements from chromite ore processing residue. Geochim. Cosmochim. Acta 2002, 66, 3927–3942. [Google Scholar] [CrossRef]
- Schiopu, N.; Tiruta-Barna, L.; Jayr, E.; Méhu, J.; Moszkowicz, P. Modelling and simulation of concrete leaching under outdoor exposure conditions. Sci. Total Environ. 2009, 407, 1613–1630. [Google Scholar] [CrossRef] [PubMed]
- Tiruta-Barna, L. Using phreeqc for modelling and simulation of dynamic leaching tests and scenarios. J. Hazard. Mater. 2008, 157, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Halim, C.E.; Short, S.A.; Scott, J.A.; Amal, R.; Low, G. Modelling the leaching of Pb, Cd, As, and Cr from cementitious waste using phreeqc. J. Hazard. Mater. 2005, 125, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, D.L.; Appelo, C.A.J. User’s Guide to Phreeqc (Version 2): A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; U.S. Geological Survey: Denver, CO, USA, 1999. [Google Scholar]
- Briggs, D.; Seah, M.P. Practical Surface Analysis: By Auger and X-ray Photoelectron Spectroscopy; John Wiley & Sons: Chichester, UK, 1983. [Google Scholar]
- Smart, R.S.C. Surface layers in base metal sulphide flotation. Miner. Eng. 1991, 4, 891–909. [Google Scholar] [CrossRef]
- Mahajan, V.; Misra, M.; Zhong, K.; Fuerstenau, M.C. Enhanced leaching of copper from chalcopyrite in hydrogen peroxide-glycol system. Miner. Eng. 2007, 20, 670–674. [Google Scholar] [CrossRef]
- Yue, G.; Zhao, L.; Olvera, O.G.; Asselin, E. Speciation of the H2SO4–Fe2(SO4)3–FeSO4–H2O system and development of an expression to predict the redox potential of the Fe3+/Fe2+ couple up to 150 °C. Hydrometallurgy 2014, 147–148, 196–209. [Google Scholar] [CrossRef]
- Velardo, A.; Giona, M.; Adrover, A.; Pagnanelli, F.; Toro, L. Two-layer shrinking-core model: Parameter estimation for the reaction order in leaching processes. Chem. Eng. J. 2002, 90, 231–240. [Google Scholar] [CrossRef]
- Córdoba, E.M.; Muñoz, J.A.; Blázquez, M.L.; González, F.; Ballester, A. Leaching of chalcopyrite with ferric ion. Part IV: The role of redox potential in the presence of mesophilic and thermophilic bacteria. Hydrometallurgy 2008, 93, 106–115. [Google Scholar] [CrossRef]
- Dutrizac, J. The dissolution of chalcopyrite in ferric sulfate and ferric chloride media. Metall. Mater. Trans. B 1981, 12, 371–378. [Google Scholar] [CrossRef]
- Nicol, M.; Miki, H.; Velásquez-Yévenes, L. The dissolution of chalcopyrite in chloride solutions: Part 3. Mechanisms. Hydrometallurgy 2010, 103, 86–95. [Google Scholar] [CrossRef]
- Vilcáez, J.; Yamada, R.; Inoue, C. Effect of pH reduction and ferric ion addition on the leaching of chalcopyrite at thermophilic temperatures. Hydrometallurgy 2009, 96, 62–71. [Google Scholar] [CrossRef]
- Vilcáez, J.; Suto, K.; Inoue, C. Bioleaching of chalcopyrite with thermophiles: Temperature-pH-ORP dependence. Int. J. Miner. Proc. 2008, 88, 37–44. [Google Scholar] [CrossRef]
- Vilcáez, J.; Inoue, C. Mathematical modeling of thermophilic bioleaching of chalcopyrite. Miner. Eng. 2009, 22, 951–960. [Google Scholar] [CrossRef]
- He, H.; Xia, J.; Hong, F.; Tao, X.; Leng, Y.; Zhao, Y. Analysis of sulfur speciation on chalcopyrite surface bioleached with acidithiobacillus ferrooxidans. Miner. Eng. 2012, 27–28, 60–64. [Google Scholar] [CrossRef]
- Pan, H.; Yang, H.; Tong, L.; Zhong, C.; Zhao, Y. Control method of chalcopyrite passivation in bioleaching. Trans. Nonferrous Met. Soc.China 2012, 22, 2255–2260. [Google Scholar] [CrossRef]
- Gómez, E.; Blázquez, M.L.; Ballester, A.; González, F. Study by SEM and EDS of chalcopyrite bioleaching using a new thermophilic bacteria. Miner. Eng. 1996, 9, 985–999. [Google Scholar] [CrossRef]
- Third, K.A.; Cord-Ruwisch, R.; Watling, H.R. The role of iron-oxidizing bacteria in stimulation or inhibition of chalcopyrite bioleaching. Hydrometallurgy 2000, 57, 225–233. [Google Scholar] [CrossRef]
- Gericke, M.; Pinches, A.; van Rooyen, J.V. Bioleaching of a chalcopyrite concentrate using an extremely thermophilic culture. Int. J. Miner. Proc. 2001, 62, 243–255. [Google Scholar] [CrossRef]
- Keeling, S.E.; Palmer, M.L.; Caracatsanis, F.C.; Johnson, J.A.; Watling, H.R. Leaching of chalcopyrite and sphalerite using bacteria enriched from a spent chalcocite heap. Miner. Eng. 2005, 18, 1289–1296. [Google Scholar] [CrossRef]
- Ahmadi, A.; Schaffie, M.; Petersen, J.; Schippers, A.; Ranjbar, M. Conventional and electrochemical bioleaching of chalcopyrite concentrates by moderately thermophilic bacteria at high pulp density. Hydrometallurgy 2011, 106, 84–92. [Google Scholar] [CrossRef]
- Tshilombo, A.F. Mechanism and Kinetics of Chalcopyrite Passivation and Depassivation During Ferric and Microbial Leaching. Ph.D. Thesis, University of Brithish Columbia, Vancouver, BC, Canada, 2004. [Google Scholar]
- Wang, J.; Qin, W.; Zhang, Y.; Yang, C.; Zhang, J.; Nai, S.; Shang, H.; Qiu, G. Bacterial leaching of chalcopyrite and bornite with native bioleaching microorganism. Trans. Nonferrous Met. Soc. China 2008, 18, 1468–1472. [Google Scholar] [CrossRef]
- Mahajan, V.K. Study of Chalcopyrite Oxidation in Hydrogen Peroxide-Ethylene Glycol System. Master’s Thesis, University of Nevada, Reno, NV, USA, 2005. [Google Scholar]
- Dreisinger, D.; Abed, N. A fundamental study of the reductive leaching of chalcopyrite using metallic iron part I: Kinetic analysis. Hydrometallurgy 2002, 66, 37–57. [Google Scholar] [CrossRef]
- Li, Y.; Qian, G.; Li, J.; Gerson, A.R. Kinetics and roles of solution and surface species of chalcopyrite dissolution at 650 mV. Geochim. Cosmochim. Acta 2015, 161, 188–202. [Google Scholar] [CrossRef]
- Dong, T.; Hua, Y.; Zhang, Q.; Zhou, D. Leaching of chalcopyrite with brønsted acidic ionic liquid. Hydrometallurgy 2009, 99, 33–38. [Google Scholar] [CrossRef]
- Brümmer, G.W.; Barrow, N.J.; Fischer, L. Effect of porosity of goethite on the sorption of six heavy metal ions. Eur. J. Soil Sci. 2013, 64, 805–813. [Google Scholar] [CrossRef]
- Nesbitt, H.W.; Muir, I.J. X-ray photoelectron spectroscopic study of a pristine pyrite surface reacted with water vapour and air. Geochim. Cosmochim. Acta 1994, 58, 4667–4679. [Google Scholar] [CrossRef]
- McIntyre, N.S.; Zetaruk, D.G. X-ray photoelectron spectroscopic studies of iron oxides. Anal. Chem. 1977, 49, 1521–1529. [Google Scholar] [CrossRef]
- Nesbitt, H.W.; Muir, I.J. Oxidation states and speciation of secondary products on pyrite and arsenopyrite reacted with mine waste waters and air. Mineral. Petrol. 1998, 62, 123–144. [Google Scholar] [CrossRef]
- Acres, R.G.; Harmer, S.L.; Beattie, D.A. Synchrotron xps, nexafs, and tof-sims studies of solution exposed chalcopyrite and heterogeneous chalcopyrite with pyrite. Miner. Eng. 2010, 23, 928–936. [Google Scholar] [CrossRef]
- Mikhlin, Y.L.; Tomashevich, Y.V.; Asanov, I.P.; Okotrub, A.V.; Varnek, V.A.; Vyalikh, D.V. Spectroscopic and electrochemical characterization of the surface layers of chalcopyrite (CuFeS2) reacted in acidic solutions. Appl. Surface Sci. 2004, 225, 395–409. [Google Scholar] [CrossRef]
- Ghahremaninezhad, A.; Dixon, D.G.; Asselin, E. Electrochemical and xps analysis of chalcopyrite (CuFeS2) dissolution in sulfuric acid solution. Electrochim. Acta 2013, 87, 97–112. [Google Scholar] [CrossRef]
- Fairthorne, G.; Fornasiero, D.; Ralston, J. Effect of oxidation on the collectorless flotation of chalcopyrite. Int. J. Miner. Proc. 1997, 49, 31–48. [Google Scholar] [CrossRef]
- Acres, R.G.; Harmer, S.L.; Shui, H.W.; Chen, C.H.; Beattie, D.A. Synchrotron scanning photoemission microscopy of homogeneous and heterogeneous metal sulfide minerals. J. Synchrotron Radiat. 2011, 18, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.G.; Mayne, D.D.; Baxter, K.G. Galvanox™—A novel galvanically-assisted atmospheric leaching technology for copper concentrates. Can. Metall. Q. 2008, 47, 327–336. [Google Scholar] [CrossRef]
- Cancho, L.; Blázquez, M.L.; Ballester, A.; González, F.; Muñoz, J.A. Bioleaching of a chalcopyrite concentrate with moderate thermophilic microorganisms in a continuous reactor system. Hydrometallurgy 2007, 87, 100–111. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, L.; Gu, G.; Hu, Y.; Su, L. Effects of microorganisms on surface properties of chalcopyrite and bioleaching. Trans. Nonferrous Met. Soc. China 2008, 18, 1421–1426. [Google Scholar] [CrossRef]
- Fu, B.; Zhou, H.; Zhang, R.; Qiu, G. Bioleaching of chalcopyrite by pure and mixed cultures of acidithiobacillus spp. And leptospirillum ferriphilum. Int. Biodeterior. Biodegrad. 2008, 62, 109–115. [Google Scholar] [CrossRef]
- Antonijevic, M.M.; Bogdanovic, G.D. Investigation of the leaching of chalcopyritic ore in acidic solutions. Hydrometallurgy 2004, 73, 245–256. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
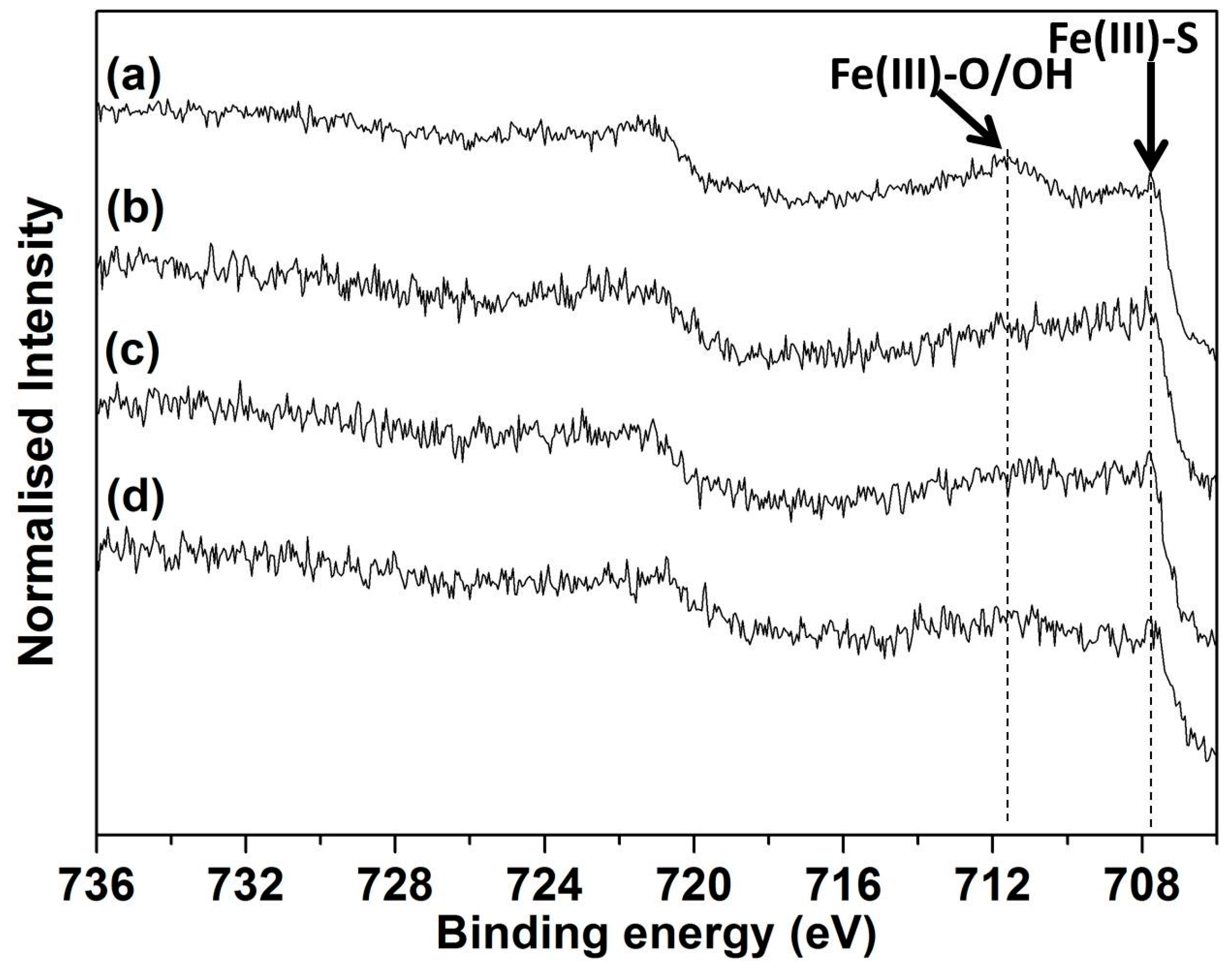{kind=link}
{kind=link}
{kind=link}
| Element | S | Cu | Fe | Si | Ca | Bi | Zn | Pb | As | Al | Co | K | Mg |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| wt % | 34 | 33 | 29 | 0.89 | 0.76 | 0.29 | 0.54 | 0.30 | 0.06 | 0.04 | 0.10 | 0.02 | 0.02 |
| Solution Species | Activity (10−4) | |||
|---|---|---|---|---|
| No Iron Addition | 4 mmol Iron Addition | |||
| 1 h | 30 h | 1 h | 30 h | |
| Cu2+ | 1.07 | 29.20 | 0.85 | 33.06 |
| CuSO4 | 0.46 | 15.78 | 0.38 | 19.01 |
| Fe2+ | 0.97 | 28.45 | 12.75 | 39.76 |
| FeSO4 | 0.29 | 10.64 | 3.99 | 15.82 |
| Fe3+ | 0.023 | 0.67 | 0.30 | 0.94 |
| FeSO4+ | 0.034 | 1.27 | 0.48 | 1.89 |
| FeOH2+ | 1.47 × 10−3 | 0.04 | 0.02 | 0.06 |
| Fe(SO4)2− | 1.31 × 10−4 | 6.12 × 10−3 | 1.92 × 10−3 | 9.67 × 10−3 |
| Fe(OH)2+ | 4.86 × 10−6 | 1.42 × 10−4 | 6.38 × 10−5 | 1.99 × 10−4 |
| HSO4− | 823.5 | 1037 | 867.2 | 1103 |
| SO42− | 18.73 | 23.6 | 19.73 | 25.10 |
| Element Core-Level | Average Binding Energy (eV) | Feed | No Iron Addition | 4 mmol Iron Addition | ||
|---|---|---|---|---|---|---|
| 1 h | 30 h | 1 h | 30 h | |||
| Cu 2p3/2 | 932.4 | 12 | 22 | 13 | 14 | 8 |
| Fe 2p3/2 | 710.3 | 8 | 12 | 9 | 10 | 3 |
| S 2p3/2 | 162.6 | 80 | 66 | 78 | 76 | 89 |
| S Species | Binding Energy (eV) | FWHM (eV) | Feed | No Iron | 4 mmol Iron Addition | ||
|---|---|---|---|---|---|---|---|
| 1 h | 30 h | 1 h | 30 h | ||||
| Surface S2− | 160.5–160.8 | 0.5–0.6 | 0 | 3 | 2 | 0 | 0 |
| Bulk S2− | 161.1 | 0.5–0.6 | 41 | 64 | 54 | 53 | 3 |
| S22− | 162.0–162.2 | 0.6–0.7 | 17 | 9 | 6 | 9 | 14 |
| Sn2− | 162.9–163.2 | 1.0–1.2 | 30 | 19 | 33 | 33 | 71 |
| S0 | 163.6–163.8 | 1.0–1.2 | 0 | 3 | 2 | 3 | 9 |
| SO42− | 168.2–169.2 | 1.2–1.5 | 8 | 0 | 0 | 0 | 0 |
| Energy loss | 164.8–165.2 | 1.2–1.5 | 4 | 2 | 3 | 2 | 3 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Wei, Z.; Qian, G.; Li, J.; Gerson, A.R. Kinetics and Mechanisms of Chalcopyrite Dissolution at Controlled Redox Potential of 750 mV in Sulfuric Acid Solution. Minerals 2016, 6, 83. https://doi.org/10.3390/min6030083
Li Y, Wei Z, Qian G, Li J, Gerson AR. Kinetics and Mechanisms of Chalcopyrite Dissolution at Controlled Redox Potential of 750 mV in Sulfuric Acid Solution. Minerals. 2016; 6(3):83. https://doi.org/10.3390/min6030083
Chicago/Turabian StyleLi, Yubiao, Zhenlun Wei, Gujie Qian, Jun Li, and Andrea R. Gerson. 2016. "Kinetics and Mechanisms of Chalcopyrite Dissolution at Controlled Redox Potential of 750 mV in Sulfuric Acid Solution" Minerals 6, no. 3: 83. https://doi.org/10.3390/min6030083