
Effects of Salinity and pH on Clay Colloid Aggregation in Ion-Adsorption-Type Rare Earth Ore Suspensions by Light Scattering Analysis

Abstract
:1. Introduction
2. Experiments
2.1. Materials
2.2. Experimental Methods
2.2.1. Extraction of Clay Colloids from IREO
2.2.2. Mineral Composition and Surface Group Analysis
2.2.3. Aggregation of IREO Colloids
2.2.4. Measurement of ζ Potential on IREO Colloids Surface
2.2.5. Measurement of Acid and Alkali Adsorption on Mineral Surface
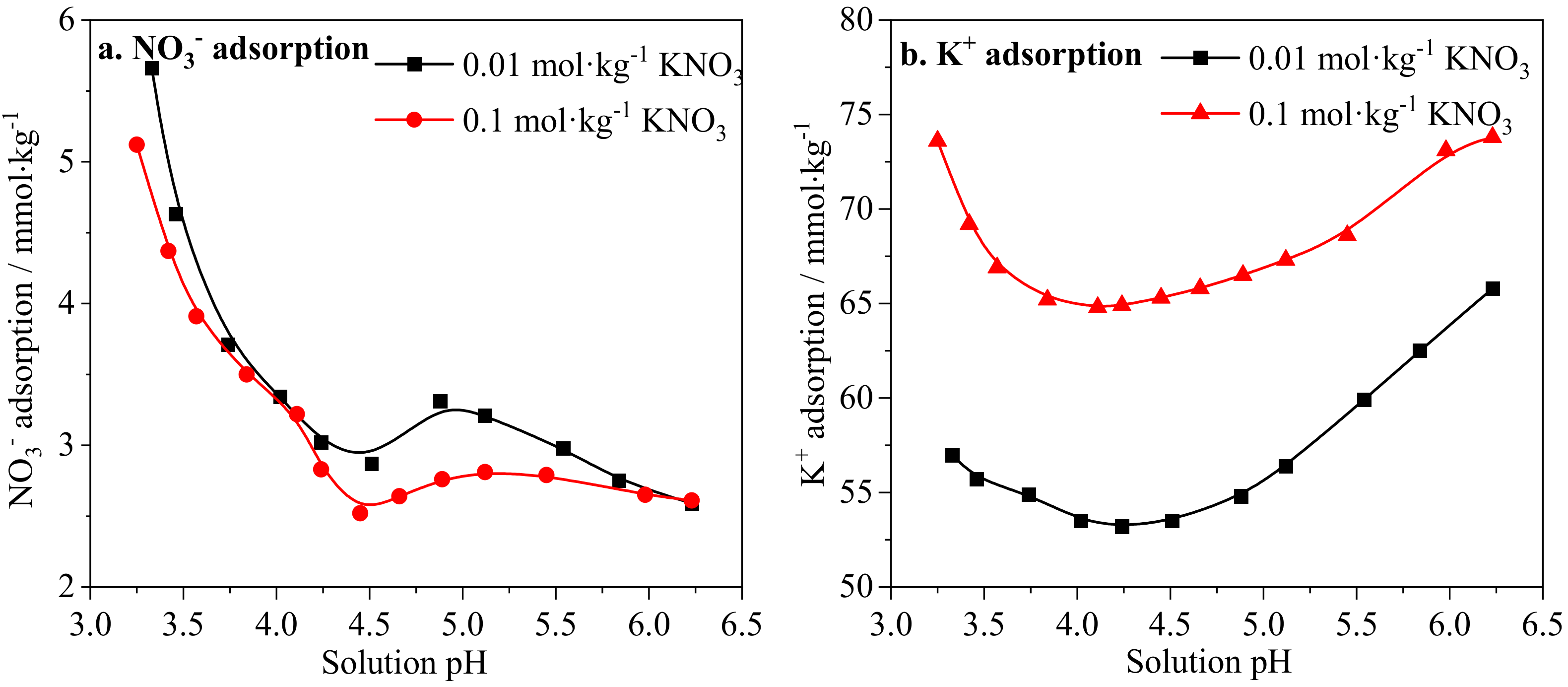
2.2.6. Ion Adsorption on IREOs
3. Results and Discussions
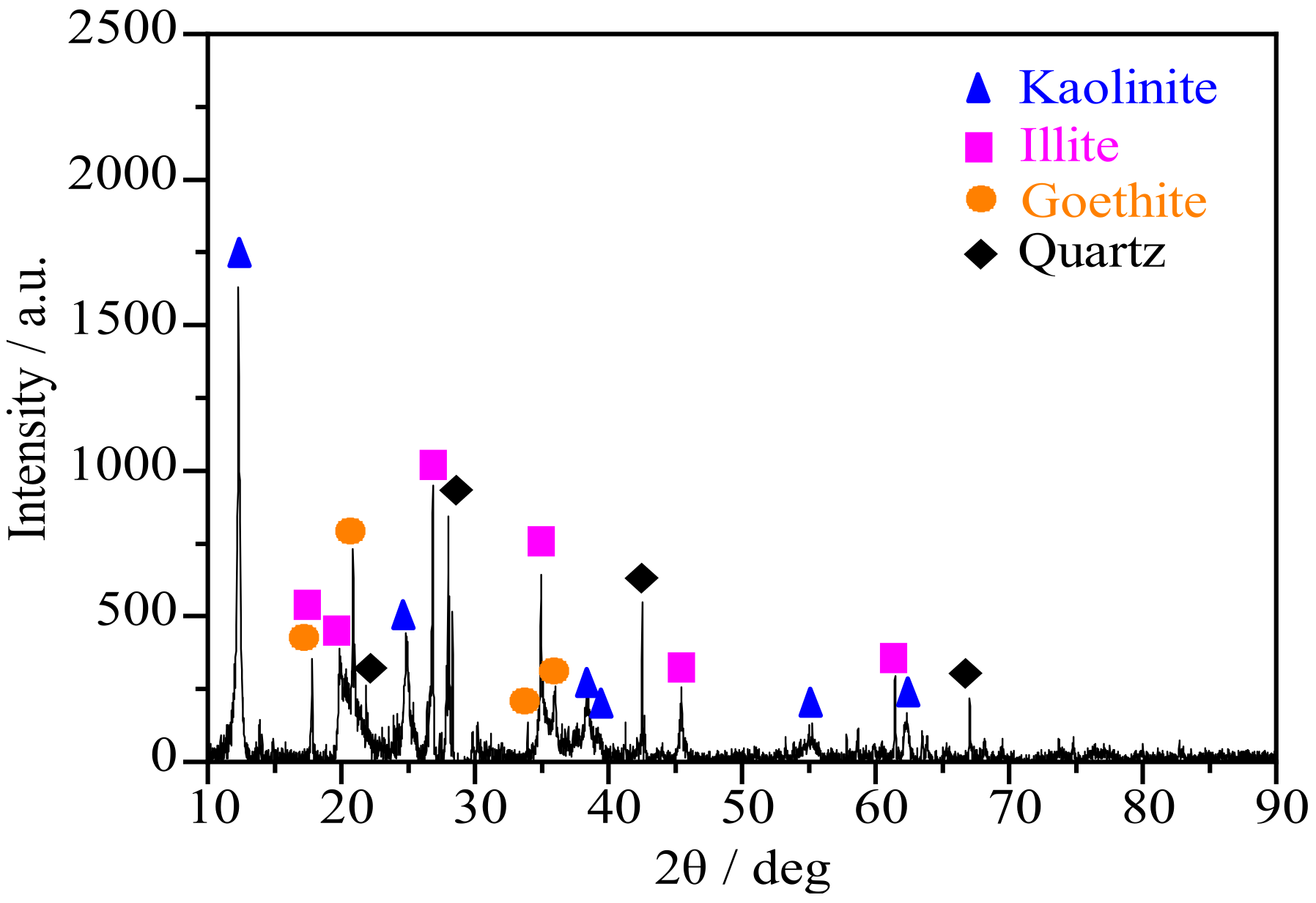
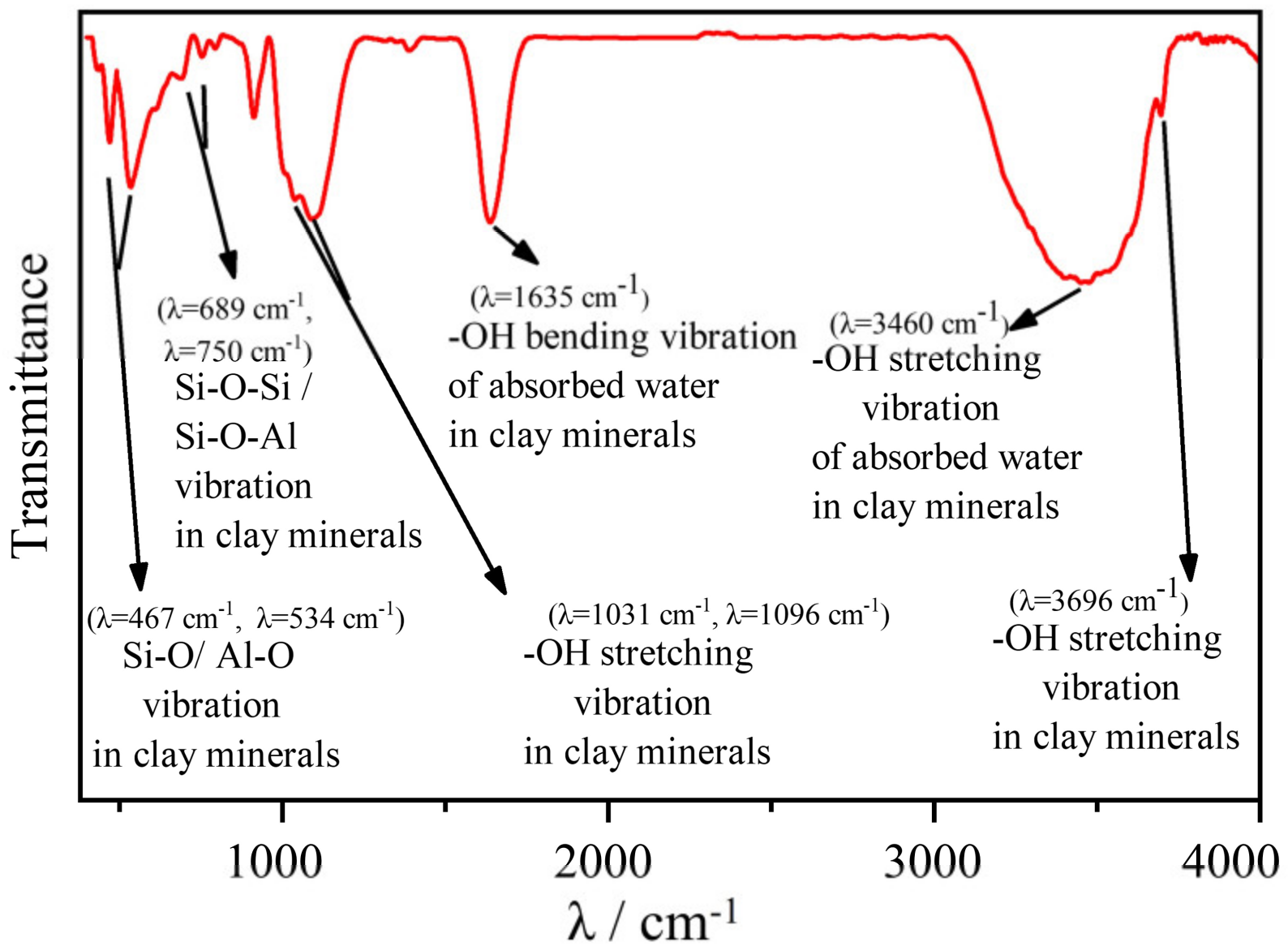
3.1. XRD and FTIR Results
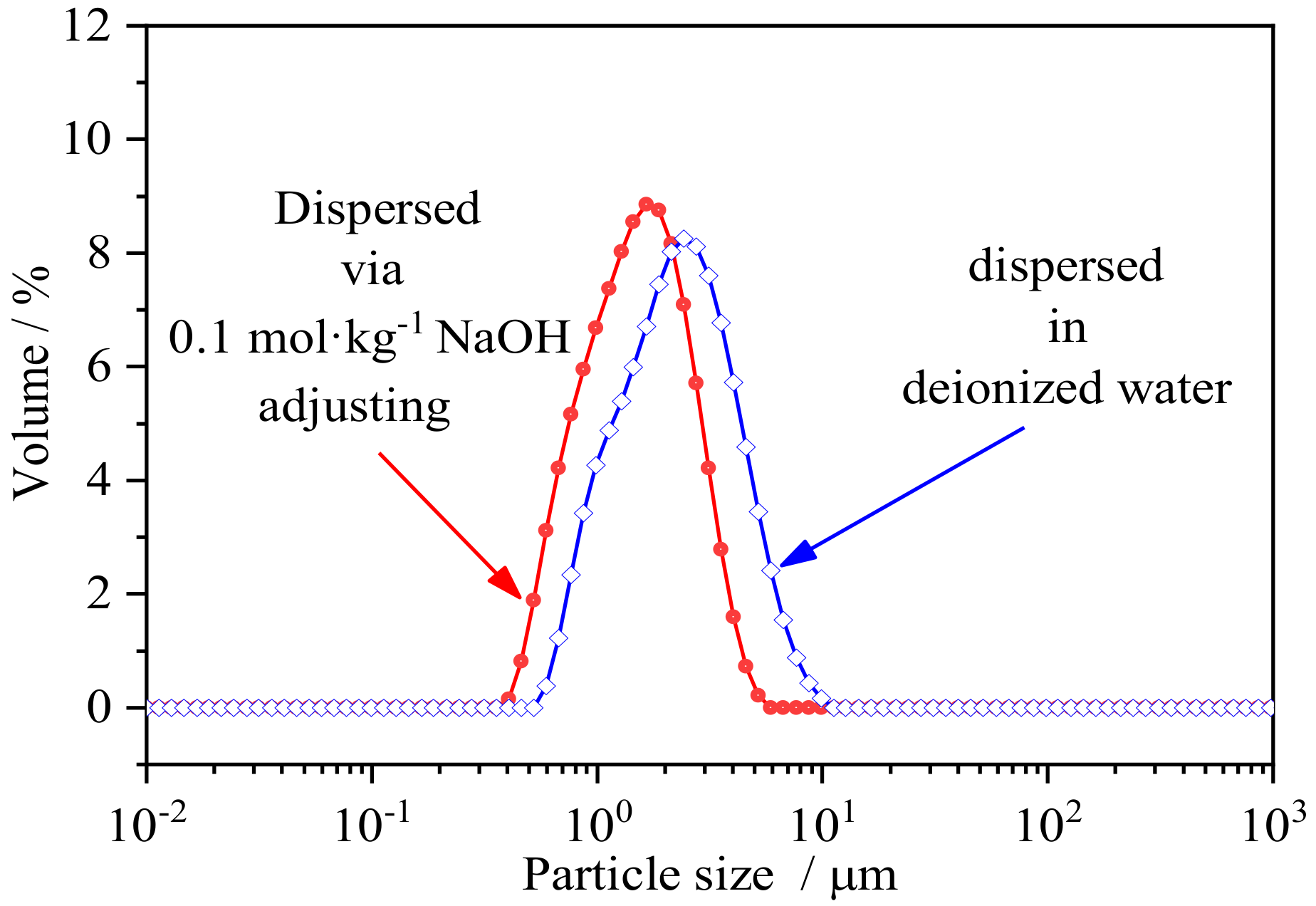
3.2. Stability Analysis on Colloidal Particle Size of IREOs in Aqueous Solution
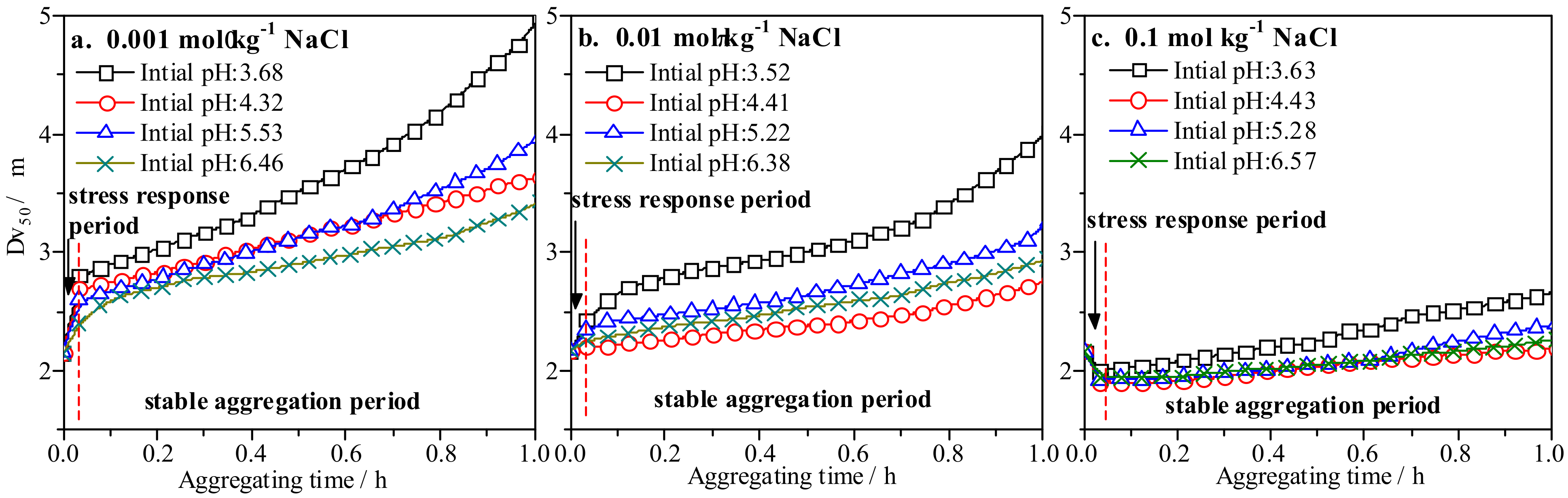
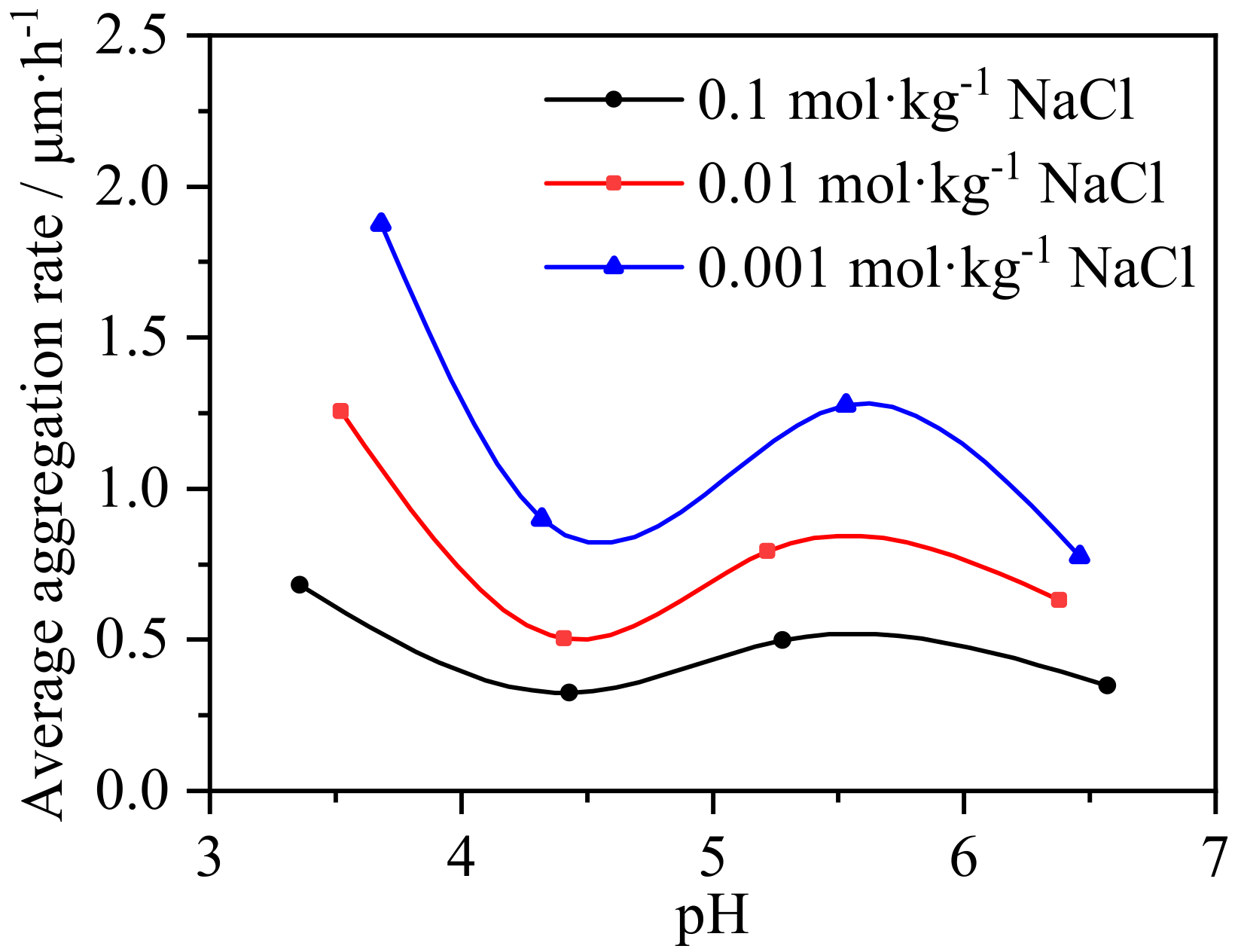
3.3. Impact of pH and Salinity Perturbation on Colloid Aggregation Behavior of IREOs
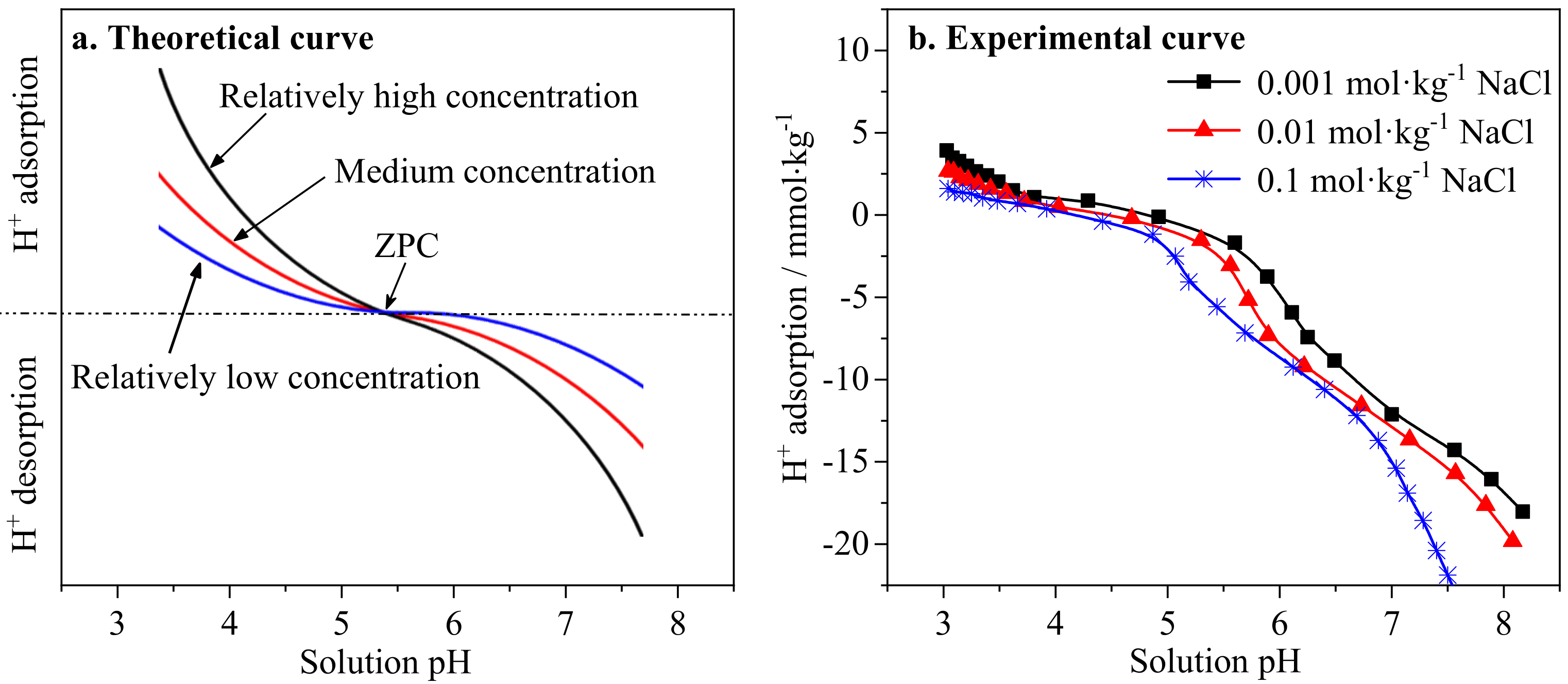
3.4. Effects of pH and NaCl Concentration on the Surface Charge Properties of Ionic Rare Earth Ore Colloids
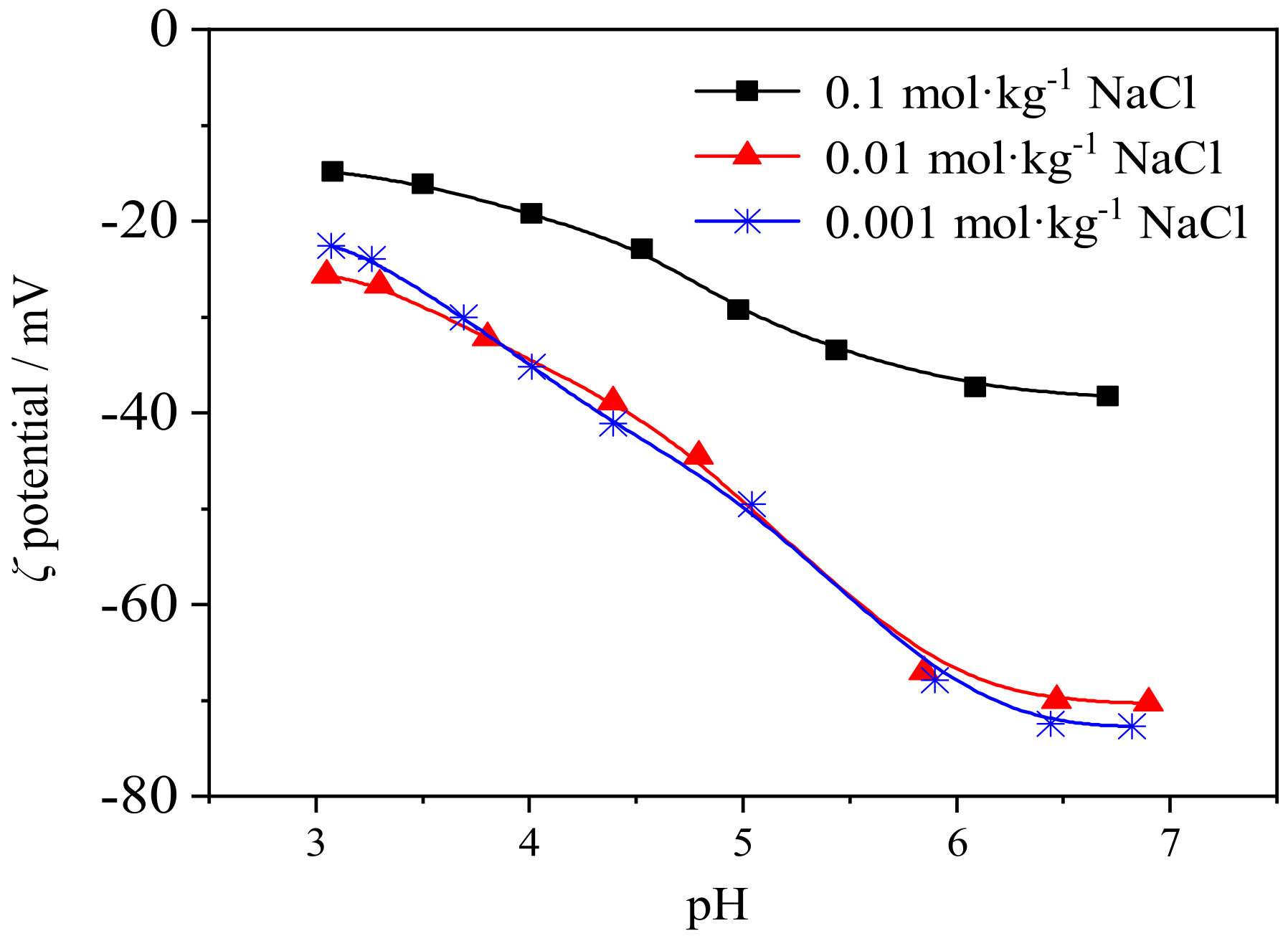
3.5. Effect of pH and Salinity on ζ Potential of the Rare Earth Ore’s Surface
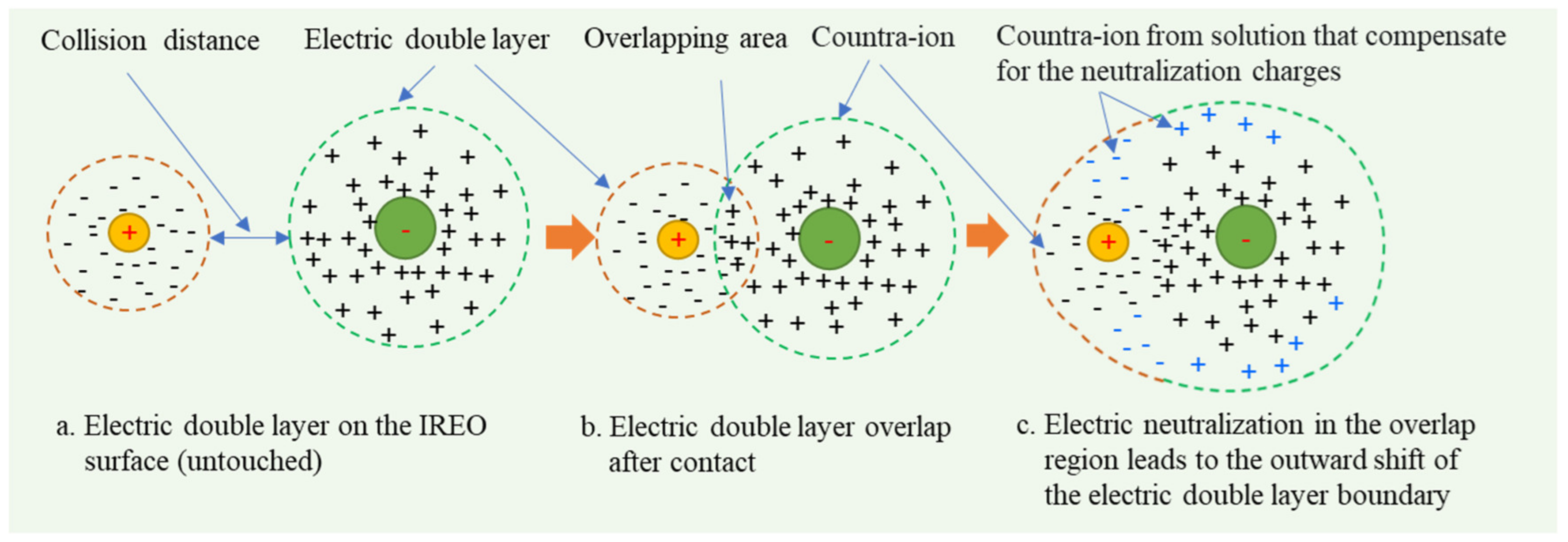
3.6. Influence Mechanism of Solution pH and Salinity Disturbance on Ionic Rare Earth Ore Aggregation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, L.; Wang, C.; Liao, C.; Yang, Y. Effect of ionic interaction on leaching behavior of ion-adsorption type rare earth ore. Chin. Rare Met. 2018, 42, 1002–1008. [Google Scholar]
- Ni, S.; Chen, Q.; Gao, Y.; Guo, X.; Sun, X. Recovery of rare earths from industrial wastewater using extraction-precipitation strategy for resource and environmental concerns. Miner. Eng. 2020, 151, 106315. [Google Scholar] [CrossRef]
- Lai, A.; Lai, F.; Huang, L.; Qiu, J.; Zhou, X.; Xiao, Y. Non-ammonia enrichment of rare earth elements from rare earth leaching liquor in a magnesium salt system I: Precipitation by calcium oxide. Hydrometallurgy 2020, 193, 105318. [Google Scholar] [CrossRef]
- Wang, L.; Liao, C.; Yang, Y.; Xu, H.; Xiao, Y.; Yan, C. Effects of organic acids on the leaching process of ion-adsorption type rare earth ore. J. Rare Earths 2017, 35, 1233–1238. [Google Scholar] [CrossRef]
- Xiao, Y.; Huang, L.; Long, Z.; Feng, Z.; Wang, L. Adsorption ability of rare earth elements on clay minerals and its practical performance. J. Rare Earths 2016, 34, 543–548. [Google Scholar] [CrossRef]
- Moldoveanu, G.A.; Papangelakis, V.G. Recovery of rare earth elements adsorbed on clay minerals: II. Leaching with ammonium sulfate. Hydrometallurgy 2013, 131, 158–166. [Google Scholar] [CrossRef]
- Wang, L.; Lan, Q.; Li, L.; Yang, Y.; Liao, C. Research status on leach chemistry of ion-adsorption type rare earth ore. Chin. Rare Earths 2018, 39, 135–143. [Google Scholar]
- Zhou, L.; Wang, X.; Zhou, Y.; Hu, K.; Zhong, W.; Huang, G. Dynamic pore structure evolution of the ion adsorbed rare earth ore during the ion exchange process. R. Soc. Open Sci. 2019, 6, 191107. [Google Scholar] [CrossRef]
- Wang, J.; Wang, G.; Hong, G. Effects of Concentration of Pore Solution on Stability of Ion-Absorbed Rare Earth Ore Aggregate. Adv. Civ. Eng. 2021, 2021, 8846605. [Google Scholar] [CrossRef]
- Fan, T.; Wang, Y.; Li, C.; Zhou, D.; Friedman, S. Effects of Soil Organic Matter on Sorption of Metal Ions on Soil Clay Particles. Soil Sci. Soc. Am. J. 2015, 79, 794–802. [Google Scholar] [CrossRef]
- Alonso, U.; Missana, T.; Garcia-Gutierrez, M.; Patelli, A.; Albarran, N.; Rigato, V. Colloid diffusion coefficients in compacted and consolidated clay barriers: Compaction density and colloid size effects. Phys. Chem. Earth 2011, 36, 1700–1707. [Google Scholar] [CrossRef]
- Luckham, P.; Rossi, S. The colloidal and rheological properties of bentonite suspensions. Adv. Colloid Interface Sci. 1999, 82, 43–92. [Google Scholar] [CrossRef]
- Carstens, J.; Bzchmann, J.; Neuweiler, I. A new approach to determine the relative importance of DLVO and non-DLVO colloid retention mechanisms in porous media. Colloids Surf. A Physicochem. Eng. Asp. 2018, 560, 330–335. [Google Scholar] [CrossRef]
- Nabweteme, R.; Yoo, M.; Kwon, H.; Kim, Y.; Hwang, G.; Lee, C.; Ahn, I. Application of the extended DLVO approach to mechanistically study the algal flocculation. J. Ind. Eng. Chem. 2015, 30, 289–294. [Google Scholar] [CrossRef]
- Soler, J.; Steefel, C.; Gimmi, T.; Leupin, O.; Cloet, V. Modeling the Ionic Strength Effect on Diffusion in Clay. The DR-A Experiment at Mont Terri. ACS Earth Space Chem. 2019, 3, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, M.; Massimo, R. Impact of diffuse layer processes on contaminant forward and back diffusion in heterogeneous sandy-clayey domains. J. Contam. Hydrol. 2021, 237, 103754. [Google Scholar]
- Missana, T.; Alonso, U.; Fernandez, A.; Garcia-Gutierrez, M. Analysis of the stability behaviour of colloids obtained from different smectite clays. Appl. Geochem. 2018, 92, 10–187. [Google Scholar] [CrossRef]
- Kumar, N.; Zhao, C.; Klaassen, A.; van den Ende, D.; Mugele, F.; Siretanu, I. Characterization of the surface charge distribution on kaolinite particles using high resolution atomic force microscopy. Geochim. Cosmochim. Acta 2016, 175, 100–112. [Google Scholar] [CrossRef]
- Zhu, L.; Tian, R.; Liu, X.; Xiong, H.; Li, H. A general theory for describing coagulation kinetics of variably charged nanoparticles. Colloid Surface A 2017, 527, 158–163. [Google Scholar] [CrossRef]
- Alagha, L.; Wang, S.; Yan, L.; Xu, Z.; Masliyah, J. Probing adsorption of polyacrylamide-based polymers on anisotropic basal planes of kaolinite using quartz crystal microbalance. Langmuir 2013, 29, 3989–3998. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, L.; Min, F.; Zhang, M.; Song, S. Hydrophobic agglomeration of colloidal kaolinite in aqueous suspensions with dodecylamine. Colloids Surf. A Physicochem. Eng. Asp. 2013, 434, 281–286. [Google Scholar] [CrossRef]
- Shao, H.; Chang, J.; Lu, Z.; Luo, B.; Grundy, J.; Xie, G.; Xu, Z.; Liu, Q. Probing Anisotropic Surface Properties of illite by atomic force microscopy. Langmuir 2019, 35, 6532–6539. [Google Scholar] [CrossRef]
- Baalousha, M. Aggregation and disaggregation of iron oxide nanoparticles: Influence of particle concentration, pH and natural organic matter. Sci. Total Environ. 2009, 407, 2093–2101. [Google Scholar] [CrossRef]
- Baalousha, M.; Manciulea, A.; Cumberland, S.; Kendall, K.; Lead, J. Aggregation and surface properties of iron oxide nanoparticles: Influence of pH and natural organic matter. Environ. Toxicol. Chem. 2008, 27, 1875–1882. [Google Scholar] [CrossRef]
- Tschapek, M.; Tcheichvili, L.; Wasowski, C. The point of zero charge (pzc) of kaolinite and SiO2+AI2O3 mixtures. Clay Miner. 1974, 10, 219–229. [Google Scholar] [CrossRef]
- Zhu, L.; Li, Z.; Tian, R.; Li, H. Specific ion effects of divalent cations on the aggregation of positively charged goethite nanoparticles in aqueous suspension. Colloids Surf. A Physicochem. Eng. Asp. 2019, 3, 442–451. [Google Scholar] [CrossRef]
- Sakurai, K.; Teshima, A.; Kyuma, K. Changes in zero point of charge (ZPC), specific surface area (SSA), and cation exchange capacity (CEC) of kaolinite and montmorillonite, and strongly weathered soils caused by Fe and Al coatings. Soil Sci. Plant Nutr. 1990, 36, 73–81. [Google Scholar] [CrossRef]
- Balan, E.; Delattre, S.; Guillaumet, M.; Salje, E. Low-temperature infrared spectroscopic study of OH-stretching modes in kaolinite and dickite. Am. Mineral. 2010, 95, 1257–1266. [Google Scholar] [CrossRef]
- Noneman, H.; Hollingsworth, M.; Singh, J.; White, R. A high sensitivity variable temperature infrared spectroscopy investigation of kaolinite structure changes. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2021, 247, 119113. [Google Scholar] [CrossRef]
- Labille, J.; Thomas, F.; Bihannic, I.; Santaella, C. Destabilization of montmorillonite suspensions by Ca2+ and succinoglycan. Clay Miner. 2003, 38, 173–185. [Google Scholar] [CrossRef]
- Xiong, Y. Soil Colloids: The Characteristic Basis of Soil Colloids; Science Press: Beijing, China, 1983. [Google Scholar]
- Baik, M.; Lee, S. Colloidal stability of bentonite clay considering surface charge properties as a function of pH and ionic strength. J. Ind. Eng. Chem. 2010, 16, 837–841. [Google Scholar] [CrossRef]
- Li, Z.; Xu, R.; Li, J. Effect of clay colloids on the zeta potential of Fe/Al oxide-coated quartz: A streaming potential study. J. Soil Sediments 2016, 16, 2676–2686. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TREO | Main Component Elements/wt.% | |||||||
|---|---|---|---|---|---|---|---|---|
| Na | Mg | Ca | Si | Fe | Al | K | O | |
| 0.16% | 0.09 | 0.044 | 0.019 | 15.22 | 0.76 | 14.38 | 1.99 | 45.22 |
| Solution Medium | Dv50/μm | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No.1 | No.2 | No.3 | No.4 | No.5 | No.6 | No.7 | No.8 | No.9 | No.10 | |
| Deionized Water | 2.14 | 2.15 | 2.13 | 2.15 | 2.14 | 2.14 | 2.14 | 2.14 | 2.15 | 2.15 |
| 0.1 mol·kg−1 NaCl Disturbed | 1.93 | 1.94 | 1.93 | 1.92 | 1.93 | 1.93 | 1.93 | 1.93 | 1.92 | 1.92 |
| CNaCl/mol·kg−1 | Initial pH | Fitting Equation | R-Squared |
|---|---|---|---|
| 0.1 | 3.63 | Dv50 = 1.81 + 0.22e1.39t | 0.977 |
| 4.43 | Dv50 = 1.71 + 0.19e0.98t | 0.962 | |
| 5.28 | Dv50 = 1.87 + 0.06e2.20t | 0.984 | |
| 6.57 | Dv50 = 1.76 + 0.16e1.14t | 0.993 | |
| 0.01 | 3.52 | Dv50 = 2.46 + 0.19e2.04t | 0.988 |
| 4.41 | Dv50 = 2.24 + 0.03e2.84t | 0.976 | |
| 5.22 | Dv50 = 2.20 + 0.19e1.62t | 0.994 | |
| 6.38 | Dv50 = 2.29 + 0.03e3.05t | 0.955 | |
| 0.001 | 3.68 | Dv50 = 2.66 + 0.28e2.01t | 0.993 |
| 4.32 | Dv50 = 2.60 + 0.21e1.64t | 0.958 | |
| 5.53 | Dv50 = 2.58 + 0.18e2.06t | 0.980 | |
| 6.46 | Dv50 = 2.50 + 0.19e1.60t | 0.976 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Wang, L.; Liu, Q. Effects of Salinity and pH on Clay Colloid Aggregation in Ion-Adsorption-Type Rare Earth Ore Suspensions by Light Scattering Analysis. Minerals 2023, 13, 38. https://doi.org/10.3390/min13010038
Li L, Wang L, Liu Q. Effects of Salinity and pH on Clay Colloid Aggregation in Ion-Adsorption-Type Rare Earth Ore Suspensions by Light Scattering Analysis. Minerals. 2023; 13(1):38. https://doi.org/10.3390/min13010038
Chicago/Turabian StyleLi, Liang, Li Wang, and Qiaochu Liu. 2023. "Effects of Salinity and pH on Clay Colloid Aggregation in Ion-Adsorption-Type Rare Earth Ore Suspensions by Light Scattering Analysis" Minerals 13, no. 1: 38. https://doi.org/10.3390/min13010038