
CaCl2 as a Mineralizing Agent in Low-Temperature Recycling of Autoclaved Aerated Concrete: Cl-Immobilization by Formation of Chlorellestadite
Abstract
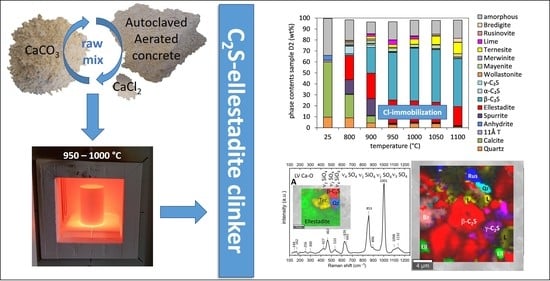
:
1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Oven Experiments
2.2. Chemical Analysis
2.3. X-ray Diffraction
2.4. Raman Spectroscopy and Imaging
2.5. Thermal Analysis
3. Results and Discussion
3.1. Raw Material Composition
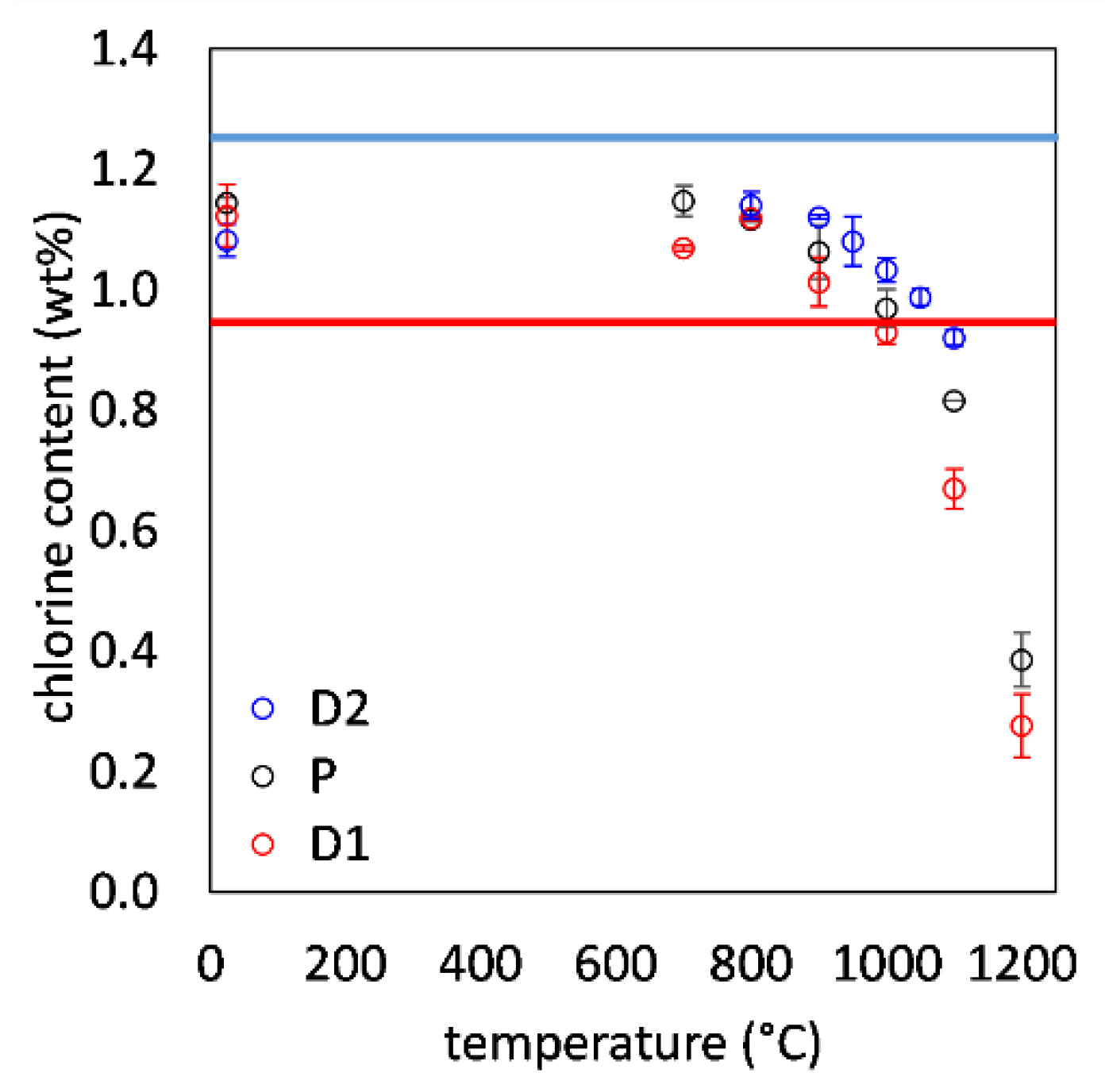
3.2. Variation in Chlorine Content with Synthesis Temperature
3.3. X-ray Diffraction Analysis and Combined Evaluation with RFA, Combustion IC, and TG
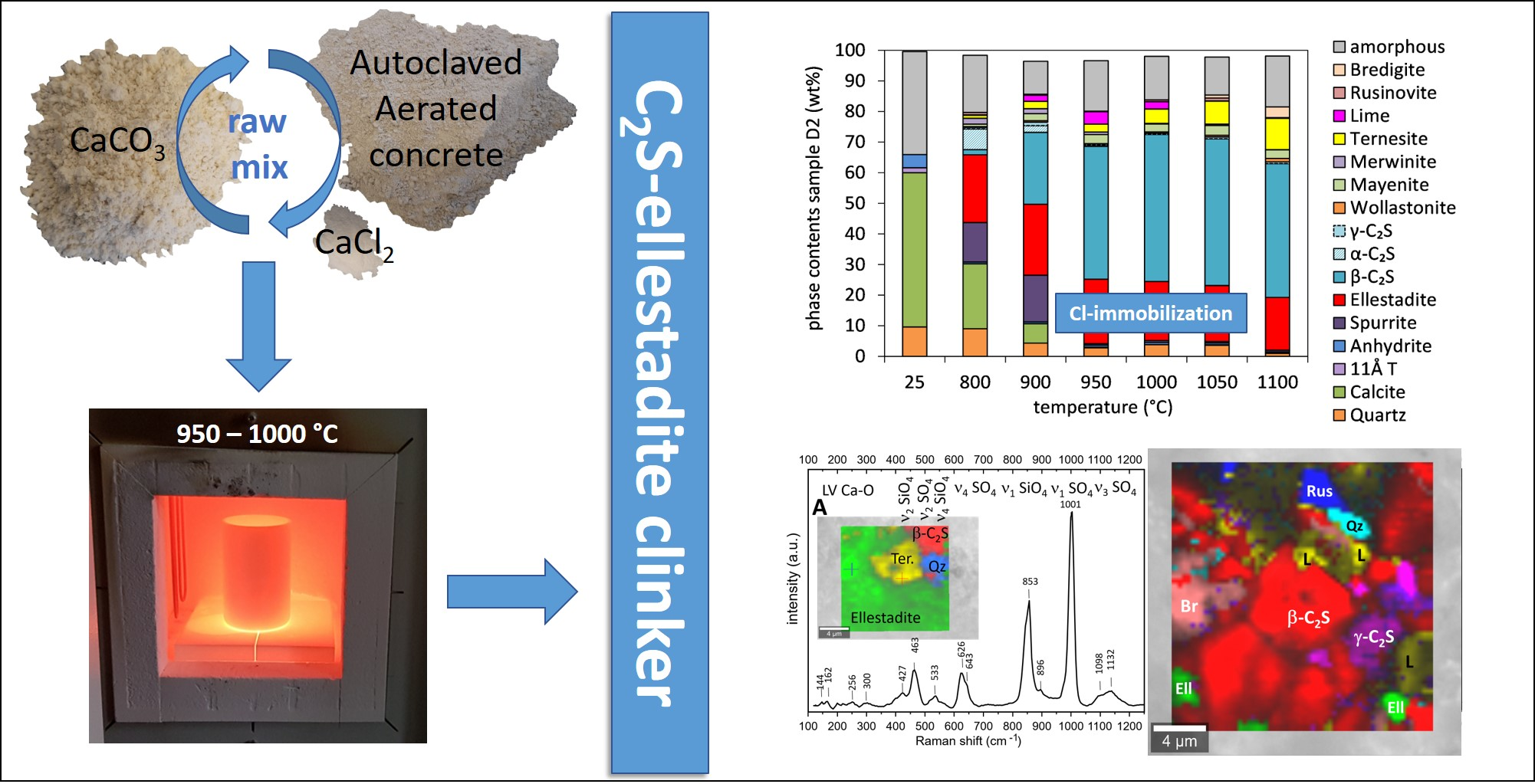
3.3.1. Quantitative Phase Contents
3.3.2. Unit-Cell Volume of β-C2S and Ellestadite
3.3.3. Yield of C2S and Ellestadite
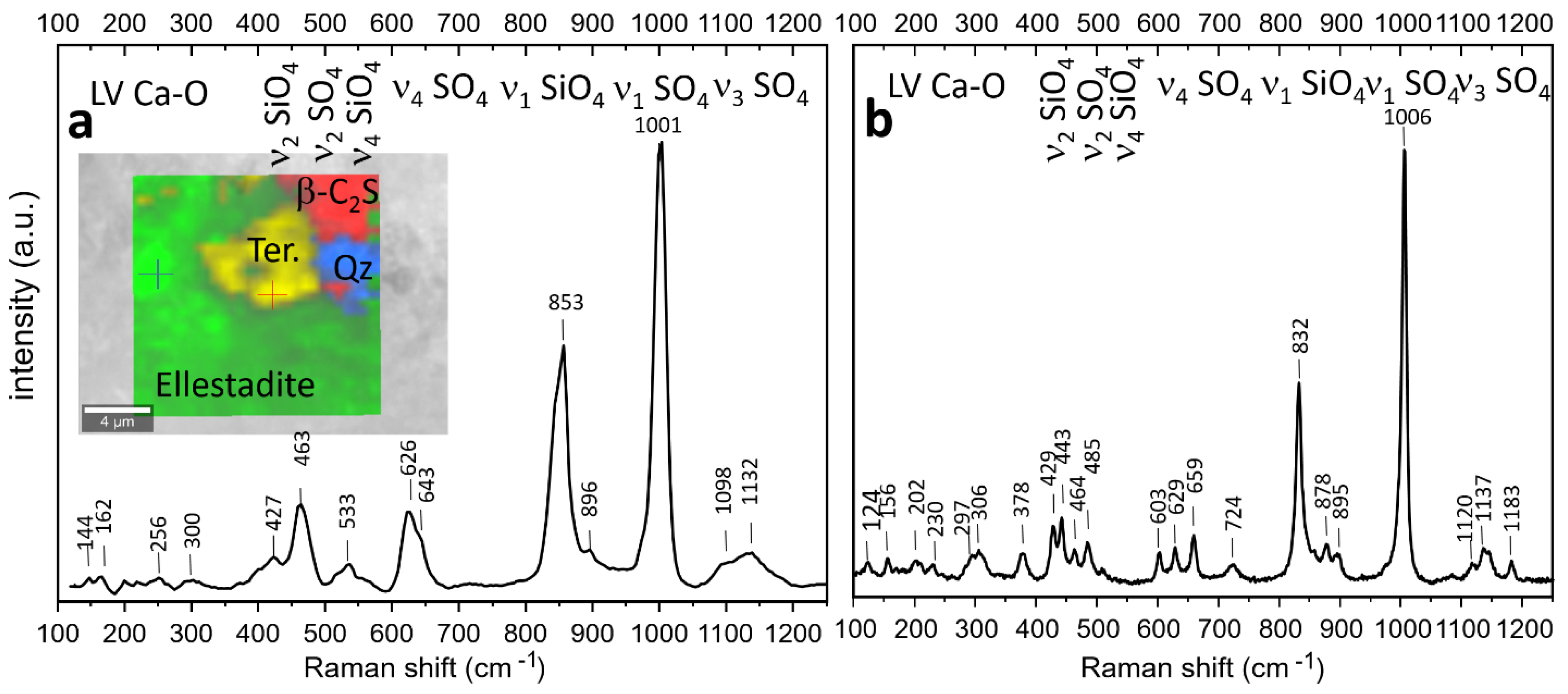
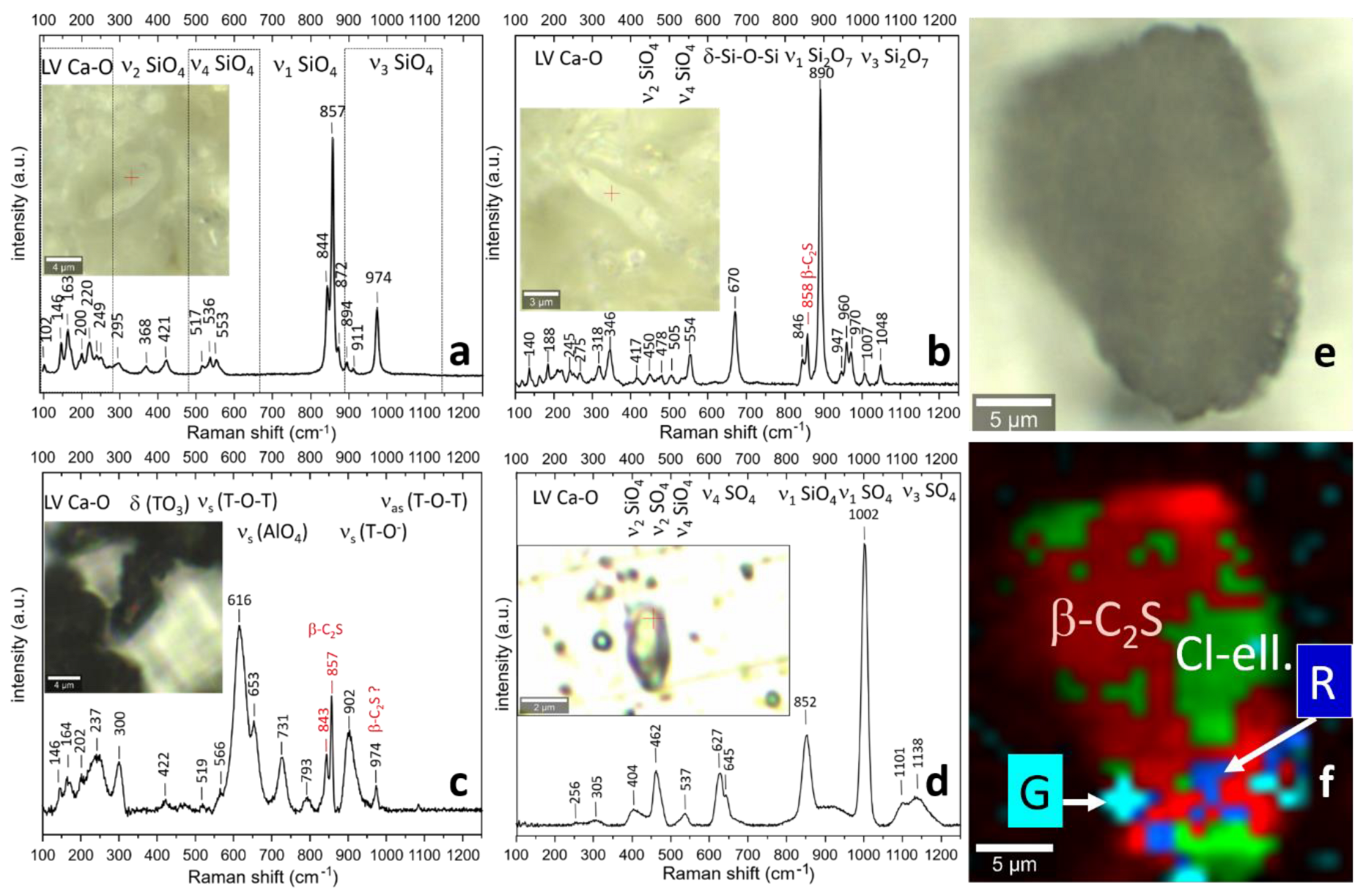
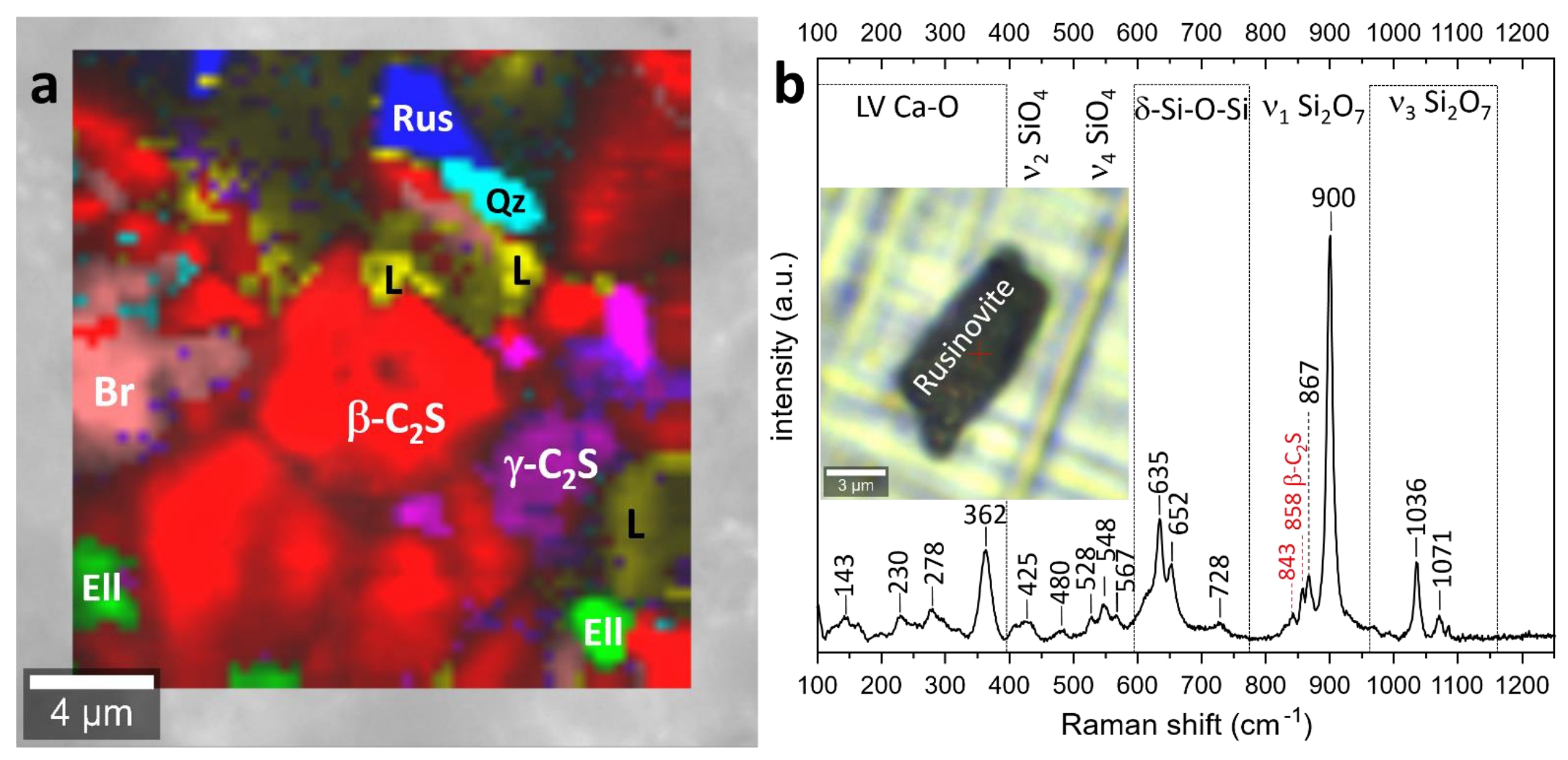
3.4. Raman Spectroscopy
3.4.1. Sample D2 1100 °C
3.4.2. Sample P 1200 °C
3.4.3. Sample D1 1200 °C
4. Summary and Conclusions
- The amount of ellestadite depends strongly on the sulfate content and to a lesser extent on the synthesis temperature.
- The optimal temperature for the formation of ellestadite beside large amounts of C2S is 950–1000 °C where the decomposition of carbonate is accomplished resulting in low spurrite contents. In this range, still no loss of chlorine takes place.
- Careful adaption of both the amount of CaCl2 addition to the sulfate content of the sample and the C/S ratio to the expected ellestadite content must be considered.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chatterjee, A.K. High belite cements—Present status and future technological options: Part I. Cem. Concr. Res. 1996, 26, 1213–1225. [Google Scholar] [CrossRef]
- Chatterjee, A.K. Future technological options: Part II. Cem. Concr. Res. 1996, 26, 1227–1237. [Google Scholar] [CrossRef]
- Dienemann, W.; Schmitt, D.; Bullerjahn, F.; Haha, M.B. Belite-Calciumsulfoaluminate-Ternesite (BCT)-A new low-carbon clinker Technology. Cem. Int. 2013, 11, 100–109. [Google Scholar]
- Stanĕk, T.; Sulovský, P. Active low-energy belite cement. Cem. Concr. Res. 2015, 68, 203–210. [Google Scholar] [CrossRef]
- Cuesta, A.; Ayuela, A.; Aranda, M.A.G. Belite cements and their activation. Cem. Concr. Res. 2021, 140, 106319. [Google Scholar] [CrossRef]
- Ullrich, A.; Garbev, K.; Bergfeld, B. In-Situ X-ray Diffraction at High Temperatures: Formation of Ca2SiO4 and Ternesite in Recycled Autoclaved Aerated Concrete. Minerals 2021, 11, 789. [Google Scholar] [CrossRef]
- Taylor, H.F.W. Cement Chemistry, 2nd ed.; Thomas Telford Ltd.: London, UK, 1997. [Google Scholar]
- Serpell, R.; Zunino, F. Recycling of hydrated cement pastes by synthesis of α’H-C2S. Cem. Concr. Res. 2017, 100, 398–412. [Google Scholar] [CrossRef]
- Serpell, R.; Lopez, M. Properties of mortars with reactivated cementitious materials. Cem. Concr. Res. 2015, 64, 16–26. [Google Scholar] [CrossRef]
- Hunsinger, H.; Beuchle, G.; Stemmermann, P.; Schweike, U.; Giziewicz, K.; Garbev, K. Method for Producing Dicalcium Silicate. U.S. Patent 9751771B2, 5 September 2017. [Google Scholar]
- Yamashita, M.; Tanaka, H. Low-temperature burnt portland cement clinker using mineralizer. Cem. Sci. Concr. Technol. 2011, 65, 82–87. [Google Scholar] [CrossRef]
- Engelsen, C.J. Effect of Mineralizers in Cement Production; SINTEF Report: Trondheim, Norway, 2007; pp. 1–25. ISBN 9788253609843. [Google Scholar]
- Böhme, N.; Hauke, K.; Neuroth, M.; Geisler, T. In situ Raman imaging of high-temperature solid-state reactions in the CaSO4-SiO2 system. Int. J. Coal Sci. Technol. 2019, 6, 247–259. [Google Scholar] [CrossRef]
- Böhme, N.; Hauke, K.; Neuroth, M.; Geisler, T. In situ Hyperspectral Raman Imaging of Ternesite Formation and Decomposition at High Temperatures. Minerals 2020, 10, 287. [Google Scholar] [CrossRef] [Green Version]
- Garbev, K.; Beuchle, G.; Schweike, U.; Merz, D.; Dregert, O.; Stemmermann, P. Preparation of a Novel Cementitious Material from Hydrothermally Synthesized C-S-H Phases. J. Am. Ceram. Soc. 2014, 97, 2298–2307. [Google Scholar] [CrossRef]
- Stemmermann, P.; Schweike, U.; Garbev, K.; Beuchle, G.; Möller, H. Celitement-A sustainable prospect fort the cement industry. Cem. Int. 2010, 8, 52–67. [Google Scholar]
- Beuchle, G.; Stemmermann, P.; Schweike, U.; Garbev, K. Single-Phase Hydraulic Binder, Methods for the Production Thereof and Building/Structural Material Produced Therewith. U.S. Patent 8382892, 26 February 2013. [Google Scholar]
- Saint-Jean, S.J.; Jøns, E.; Lundgaard, N.; Hansen, S. Chlorellestadite in the preheater system of cement kilns as an indicator of HCl formation. Cem. Concr. Res. 2005, 35, 431–437. [Google Scholar] [CrossRef]
- Gerassimidou, S.; Velis, C.A.; Williams, P.T.; Castaldi, M.J.; Black, L.; Komilis, D. Chlorine in waste-derived solid recovered fuel (SRF), co-combusted in cement kilns: A systematic review of sources, reactions, fate and implications. Crit. Rev. Environ. Sci. Technol. 2020, 51, 140–186. [Google Scholar] [CrossRef]
- Martauz, P.; Strigáč, J.; Jamnický, M. Decreasing chloride levels in cement rotary kiln atmosphere by sorption into hydroxylapatite structure. Mat. Sci. 2007, 60, 75–86. [Google Scholar]
- Hashem, M. Using Eco-Cement Made from Municipal Solid Waste Incineration Residues as a Mineral Additive for Concrete. Master’s Thesis, McGill University, Montreal, QC, Canada, 2021. [Google Scholar]
- Ghouleh, Z.; Shao, Y. Turning municipal solid waste incineration into a cleaner cement production. J. Clean. Prod. 2018, 195, 268–279. [Google Scholar] [CrossRef]
- Pasero, M.; Kampf, A.R.; Ferraris, C.; Pekov, I.V.; Rakovan, J.; White, T. Nomenclature of the apatite supergroup minerals. Eur. J. Mineral. 2010, 22, 163–179. [Google Scholar] [CrossRef]
- Saint-Jean, S.J.; Hansen, S. Nonstoichiometry in chlorellestadite. Solid State Sci. 2005, 7, 97–102. [Google Scholar] [CrossRef]
- Środek, D.; Galuskina, I.O.; Galuskin, E.; Dulski, M.; Książek, M.; Kusz, J.; Gazeev, V. Chlorellestadite, Ca5(SiO4)1.5(SO4)1.5Cl, a new ellestadite-group mineral from Shadil-Khokh volcano, South Ossetia. Mineral. Petrol. 2018, 112, 743–752. [Google Scholar] [CrossRef]
- Sudarsanan, K. Structure of Hydroxylellestadite. Acta Cryst. 1980, B36, 1636–1639. [Google Scholar] [CrossRef]
- Organova, N.I.; Rastsvetaeva, R.K.; Kuz’mina, O.V. Crystal Structure of Low-Symmetry Ellestadite in Comparison with other Apatite-Like Structures. Kristallografiya 1994, 39, 278–282. [Google Scholar]
- Fang, Y.N.; Ritter, C.; White, T.J. Crystal chemical characteristics of ellestadite-type apatite: Implications for toxic metal immobilization. Dalton Trans. 2014, 43, 16031–16043. [Google Scholar] [CrossRef]
- Fang, Y.; Ritter, C.; White, T. The Crystal Chemistry of Ca10-y(SiO4)3(SO4)3Cl2-x−2yFx Ellestadite. Inorg. Chem. 2011, 50, 12641–12650. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Fang, Y. The Chemical Composition and Crystal Parameters of Calcium Chlorosulfatosilicate. Cem. Concr. Res. 1989, 19, 184–188. [Google Scholar] [CrossRef]
- Stemmermann, P.; Pöllmann, H. The system CaO-SiO2-CaCl2-phase equilibria and polymorphs below 1000 °C. An interpretation on garbage combustion ashes. Neues Jahrb. Mineral. Monatsh. 1992, 9, 409–431. [Google Scholar]
- Chesnokov, B.V.; Vilisov, V.; Bushmakin, A.; Kotlyarov, V.; Belogub, E.V. New minerals from a fired dump of the Chelyabinsk coal basin. Ural Mineral. Zbor. 1994, 3, 3–34. [Google Scholar]
- Hermoneit, B.; Ziemer, B.; Malewski, G. Single crystal growth and some properties of the new compound Ca3Si2O7∙13CaCl2. J. Cryst. Growth 1981, 52, 660–664. [Google Scholar] [CrossRef]
- Galuskin, E.V.; Galuskina, I.O.; Lazic, B.; Armbruster, T.; Zadov, A.E.; Krzykawski, T.; Banasik, K.; Gazeev, V.M.; Pertsev, N.N. Rusinovite, Ca10(Si2O7)3Cl2: A new skarn mineral from the Upper Chegem caldera, Kabardino-Balkaria, Northern Caucasus, Russia. Eur. J. Mineral. 2011, 23, 837–844. [Google Scholar] [CrossRef]
- Środek, D.; Juroszek, R.; Krüger, H.; Krüger, B.; Galuskina, I.; Gazeev, V. New Occurrence of Rusinovite, Ca10(Si2O7)3Cl2: Composition, Structure and Raman Data of Rusinovite from Shadil-Khokh volcano, south Ossetia and Bellerberg Volcano, Germany. Minerals 2018, 8, 399. [Google Scholar] [CrossRef]
- Barreneche, C.; Fernandez, A.I.; Cabeza, L.F.; Cuypers, R. Thermophysical Characterization of Sorption TCM. Energy Procedia 2014, 48, 273–279. [Google Scholar] [CrossRef]
- National Institute of Standards and Technology. Certificate of Analysis Standard Reference Material® 640e, Line Position and Line Shape Standard for Powder Diffraction (Silicon Powder); Department of Commerce: Gaithersburg, MD, USA, 2015.
- National Institute of Standards and Technology. Certificate of Analysis Standard Reference Material® 674b, X-ray Powder Diffraction Intensity Set (Quantitative Powder Diffraction Standard); Department of Commerce: Gaithersburg, MD, USA, 2017.
- Marchbank, H.R.; Clark, A.H.; Hyde, T.I.; Playford, H.Y.; Tucker, M.G.; Thompsett, D.; Fisher, J.; Chapman, K.W.; Beyer, K.A.; Monte, M.; et al. Structure of nano-sized CeO2 material: A combined scattering and spectroscopic investigation. ChemPhysChem 2016, 17, 3494–3503. [Google Scholar] [CrossRef] [PubMed]
- Pederson, B.M.; Schaible, K.J.; Winburn, R.S. Minimization of errors due to microabsorption or Absorption contrast. Adv. X-ray Anal. 2004, 47, 200–205. [Google Scholar]
- Le Page, Y.; Donnay, G. Refinement of the crystal structure of low-quartz. Acta Cryst. 1976, 32, 2456–2459. [Google Scholar] [CrossRef]
- Liu, S.; Li, H.; Liu, J. Obtaining optimal structural data from X-ray powder diffraction using the Rietveld method. Powder Diffr. 2014, 29, 396–403. [Google Scholar] [CrossRef]
- Merlino, S.; Bonaccorsi, E.; Armbruster, T. The real structure of tobermorite 11 A: Normal and anomalous forms, OD character and polytypic modifications. Eur. J. Mineral. 2001, 13, 577–590. [Google Scholar] [CrossRef]
- De Villiers, J.P.R. Crystal structures of aragonite, strontianite, and witherite. Am. Mineral. 1971, 56, 758–766. [Google Scholar]
- Kamhi, S.R. On the structure of vaterite, CaCO3. Acta Cryst. 1963, 16, 770–772. [Google Scholar] [CrossRef]
- Hawthorne, F.C.; Ferguson, R.B. Anhydrous Sulfates II. Refinement of the Crystal Structure of Anhydrite. Can. Mineral. 1975, 13, 289–292. [Google Scholar]
- Ballirano, P.; Maras, A.; Meloni, S.; Caminiti, R. The monoclinic I2 structure of bassanite, calcium sulphate hemihydrate (CaSO4*0.5(H2O)). Eur. J. Mineral. 2001, 13, 985–993. [Google Scholar] [CrossRef]
- Allan, D.R.; Angel, R.J. A high-pressure structural study of microcline (KAlSi3O8) to 7 GPa. Eur. J. Mineral. 1997, 9, 263–275. [Google Scholar] [CrossRef]
- Winter, J.K.; Okamura, F.P.; Ghose, S. A high temperature structural study of high albite, monalbite, and the analbite-monalbite phase transition. Am. Mineral. 1979, 64, 409–423. [Google Scholar]
- Leclaire, A.; Borel, M.M. Le dichlorure de calcium dehydrate. Acta Cryst. 1977, 33, 1608–1610. [Google Scholar] [CrossRef]
- Mumme, W.G.; Hill, R.J.; Bushnell, G.W.; Segnit, E.R. Rietveld crystal structure refinements, crystal chemistry and calculated powder diffraction data for the polymorphs of dicalcium silicate and related phases. Neues Jahrb. Mineral. 1995, 169, 35–68. [Google Scholar]
- Mumme, W.G.; Cranswick, L.; Chakoumakos, B. Rietveld crystal structure refinement from high temperature neutron powder diffraction data for the polymorphs of dicalcium silicate. Neues Jahrb. Mineral. Abh. 1996, 170, 171–188. [Google Scholar]
- Galuskin, E.V.; Kusz, J.; Armbruster, T.; Bailau, R.; Galuskina, I.O.; Ternes, B.; Murashko, M. A reinvestigation of mayenite from the type locality, the Ettringer Bellerberg volcano near Mayen, Eifel district, Germany. Mineral. Mag. 2012, 76, 707–716. [Google Scholar] [CrossRef]
- Ruilun, Y.; Wang, X.; Zhang, Z. Crystal structure of the new compound Ca8Mg(SiO4)4Cl2. Guisuanyan Xuebao 1987, 15, 309–314. [Google Scholar]
- Anselment, B. Die Dynamik der Phasenumwandlung vom Rutil-in den CaCl2-Typ am Beispiel des CaBr2 and zur Polymorphie des CaCl2; Dissertation Universität Karlsruhe: Karlsruhe, Germany, 1985; pp. 1–195. [Google Scholar]
- Golovastikov, N.I.; Kazak, V.F. The crystal structure of calcium chlorosilicate Ca2SiO3Cl2. Kristallografiya 1977, 22, 962–965. [Google Scholar]
- Irran, E.; Tillmanns, E.; Hentschel, G. Ternesite, Ca5(SiO4)2SO4, a new mineral from the Ettringer Bellerberg/Eifel, Germany. Mineral. Petrol. 1997, 60, 121–132. [Google Scholar] [CrossRef]
- Huang, Q.; Chmaissem, O.; Caponi, J.J.; Chaillout, C.; Marezio, M.; Tholence, J.L.; Santoro, A. Neutron powder diffraction study of the crystal structure of HgBa2Ca4Cu5O12+d at room temperature and at 10K. Physica C 1994, 227, 1–9. [Google Scholar] [CrossRef]
- Hesse, K.F. Refinement of the crystal structure of wollastonite-2M (parawollastonite). Zeitschr. Kristallogr. 1984, 168, 93–98. [Google Scholar] [CrossRef]
- Merlini, M.; Gemmi, M.; Cruciani, G.; Artioli, G. High-temperature behaviour of melilite: In situ X-ray diffraction study of gehlenite-åkermanite-Na melilite solid solution. Phys. Chem. Miner. 2008, 35, 147–155. [Google Scholar] [CrossRef]
- Grice, J.-D. The structure of spurrite, tilleyite and scawtite, and relationships to other silicate-carbonate minerals. Can. Mineral. 2005, 43, 1489–1500. [Google Scholar] [CrossRef]
- Colville, A.A.; Geller, S. The crystal structure of brownmillerite, Ca2FeAlO5. Acta Cryst. 1971, 27, 2311–2315. [Google Scholar] [CrossRef]
- Cuesta, A.; de la Torre, A.G.; Losilla, E.R.; Santacruz, I.; Aranda, M.A.G. Pseudocubic crystal structure and phase transition in doped ye’elimite. Cry. Growth Des. 2014, 14, 5158–5163. [Google Scholar] [CrossRef]
- Desgranges, L.; Grebille, D.; Calvarin, G.; Chevrier, G.; Floquet, N.; Niepce, J.C. Hydrogen thermal motion in calcium hydroxide: Ca(OH)2. Acta Cryst. Sect. B 1993, 49, 812–817. [Google Scholar] [CrossRef]
- Moore, P.B.; Araki, T. Atomic arrangement of merwinite, Ca3Mg(SiO4)2, an unusual dense-packed structure of geophysical interest. Am. Mineral. 1972, 57, 1355–1374. [Google Scholar]
- Kusachi, I.; Henmi, C.; Kawahara, A.; Henmi, K. The structure of rankinite. Mineral. J. 1975, 8, 38–47. [Google Scholar] [CrossRef]
- Moore, P.B.; Araki, T. The crystal structure of Bredigite and the genealogy of some alkaline earth orthosilicates. Am. Mineral. 1976, 61, 74–87. [Google Scholar]
- Lafuente, B.; Downs, R.T.; Yang, H.; Stone, N. The power of databases: The RRUFF project. In Highlights in Mineralogical Crystallography; Armbruster, T., Danisi, R.M., Eds.; W. De Gruyter: Berlin, Germany, 2015; pp. 1–30. [Google Scholar]
- Schmit, H.; Pfeffer, W.; Rathgeber, C.; Hiebler, S. Experimental Investigation of the Concentration Dependent Maximum Storage Capacity of two Inorganic Phase Change Materials. Energy Procedia 2015, 73, 231–238. [Google Scholar] [CrossRef]
- Hill, R.J.; Flack, H.D. The Use of the Durbin-Watson d Statistic in Rietveld Analysis. J. Appl. Cryst. 1987, 20, 356–361. [Google Scholar] [CrossRef]
- Banno, Y.; Miyawaki, R.; Momma, K.; Bunno, M. A CO3-bearing member of the hydroxylapatite-hydroxylellestadite series from Tadano, Fukushima Prefecture, Japan: CO3-SO4 substitution in the apatite-ellestadite series. Mineral. Mag. 2016, 80, 363–370. [Google Scholar] [CrossRef]
- Pöhler, K. Fremdionenstabilisierte Dicalciumsilicate: Synthese und Hydraulische Reaktion. Ph.D. Thesis, Martin-Luther-Universität Halle-Wittenberg, Halle, Germany, 2016. [Google Scholar]
- Morsli, K.; De la Torre, A.G.; Zahir, M.; Aranda, M.A.G. Mineralogical phase analysis of alkali and sulfate bearing belite rich laboratory clinkers. Cem. Concr. Res. 2007, 37, 639–646. [Google Scholar] [CrossRef]
- Zhao, Y.; Lu, L.; Wang, S.; Gong, C.; Huang, Y. Modification of Dicalcium Silicates Phase Composition by BaO, SO3 and MgO. J. Inorg. Organomet. Polym. 2013, 23, 930–936. [Google Scholar] [CrossRef]
- Galuskin, E.V.; Galuskina, I.O.; Gfeller, F.; Krüger, B.; Kusz, J.; Vapnik, Y.; Dulski, M.; Dzierżanowski, P. Silicocarnotite, Ca5 [(SiO4)(PO4)](PO4), a new “old” mineral from the Negev Desert, Israel, and the ternesite–silicocarnotite solid solution: Indicators of high-temperature alteration of pyrometamorphic rocks of the Hatrurim Complex, Southern Levant. Eur. J. Mineral. 2016, 28, 105–123. [Google Scholar] [CrossRef]
- Remy, C.; Reynard, B.; Madon, M. Raman Spectroscopic Investigations of Dicalcium Silicate: Polymorphs and High-Temperature Phase Transformations. J. Am. Ceram. Soc. 2005, 80, 413–423. [Google Scholar] [CrossRef]
- Pilz, W. Raman spectra of silicates. Acta Physica Hungarica 1987, 61, 27–30. [Google Scholar] [CrossRef]
- Dowty, E. Vibrational interactions of tetrahedra in silicate glasses and crystals. Phys. Chem. Miner. 1987, 14, 542–552. [Google Scholar] [CrossRef]
- Burshtein, Z.; Shimony, Y.; Morganau, S.; Henderson, D.O.; Mu, R.; Silberman, E. Symmetry Lowering due to Site-Occupation Disorder in Vibrational Spectra of Gehlenite, Ca2(AlSi)AlO7. Phys. Chem. Solids 1993, 54, 1043–1049. [Google Scholar] [CrossRef]
- Sharma, S.K.; Yoder, H.S., Jr.; Matson, D.W. Raman study of some melilites in crystalline and glassy states. Geochim. Cosmochim. Acta 1988, 52, 1961–1967. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D2 | P | D1 | |
|---|---|---|---|
| Waste AAC | 46.8 | 47.7 | 40.8 |
| calcite | 53.2 | 52.3 | 59.2 |
| LoI | 27.57 | 25.74 | 31.40 |
| SiO2 | 22.55 | 23.91 | 22.83 |
| CaO | 41.8 | 44.28 | 42.42 |
| SO3 | 5.04 | 2.11 | 0.58 |
| Al2O3 | 1.24 | 1.64 | 1.04 |
| Fe2O3 | 0.50 | 0.71 | 0.45 |
| MgO | 0.38 | 0.6 | 0.33 |
| Cl | 1.08 | 1.14 | 1.12 |
| Na2O | <0.1 | n.d. | n.d. |
| K2O | <0.3 | <0.1 | <0.1 |
| Amorphous | 33.7(9) | 39.2(6) | 30.1(6) |
| Quartz | 9.6(1) | 6.98(7) | 10.78(8) |
| Calcite | 47.5(4) | 44.8(3) | 52.3(3) |
| Vaterite | 1.5(1) | 1.15(9) | 1.65(8) |
| Aragonite | 1.4(2) | 0.4(1) | 1.8(1) |
| Tobermorite | 1.6(2) | 4.3(2) | 2.3(1) |
| Anhydrite | 4.3(1) | 1.94(9) | 0.51(7) |
| Bassanite | 0.3(1) | 0.42(9) | 0.18(6) |
| Sinjarite | 0.3(1) | 0.7(1) | 0.40(8) |
| WL 30–950 °C | 28.2 | 26.4 | 32.2 |
| H2O | 3.7 | 3.8 | 4.7 |
| CO2 | 24.4 | 22.4 | 27.3 |
| SO3 | 5.4 | 2.3 | 0.7 |
| AmCaCO3 | 5.0 | 4.5 | 6.2 |
| Am CaSO4 | 4.6 | 1.6 | 0.5 |
| Am CaCl2(+xH2O) | 1.4 | 1.2 | 1.4 |
| Am Feldspar | 3.3 | 4.4 | 2.8 |
| Am C-S-H | 10.2 | 24.7 | 10.4 |
| Am SiO2 | 5.8 | 1.5 | 4.7 |
| HT[D2-]_1000 | HT[D2]_1000 | HT[P-]_1000 | HT[P]_1000 | HT[D1-]_1000 | HT[D1]_1000 | |
|---|---|---|---|---|---|---|
| Amorphous | 11.8(6) | 14.2(9) | 16.6(6) | 15.0(7) | 17.1(7) | 18.2(9) |
| β-C2S | 31.4(3) | 48.0(3) | 45.6(4) | 63.8(4) | 40.1(5) | 67.7(5) |
| Ellestadite | 0 | 19.4(2) | 0 | 9.9(1) | 0 | 1.5(9) |
| Ternesite | 32.4(3) | 4.7(1) | 10.7(2) | 0.12(7) | 4.3(2) | 0.16(7) |
| Quartz | 7.05(7) | 3.81(5) | 4.82(6) | 2.11(4) | 9.72(8) | 1.06(5) |
| Lime | 9.51(7) | 2.44(4) | 9.83(7) | 0.17(5) | 18.4(1) | 0.07(2) |
| Wollastonite | 1.1(1) | 0.5(1) | 6.2(2) | 0.6(1) | 5.0(1) | 0.4(1) |
| Portlandite | 0.51(5) | 0 | 1.14(8) | 0 | 2.62(8) | 0 |
| α’H-C2S | 2.5(2) | 0 | 0 | 0 | 1.2(2) | 0 |
| Melilite | 0.9(1) | 0.08(5) | 2.8(1) | 0.06(5) | 0 | 0 |
| Chlormayenite | 0 | 2.60(7) | 0 | 3.90(9) | 0 | 2.56(9) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullrich, A.; Garbev, K.; Schweike, U.; Köhler, M.; Bergfeldt, B.; Stemmermann, P. CaCl2 as a Mineralizing Agent in Low-Temperature Recycling of Autoclaved Aerated Concrete: Cl-Immobilization by Formation of Chlorellestadite. Minerals 2022, 12, 1142. https://doi.org/10.3390/min12091142
Ullrich A, Garbev K, Schweike U, Köhler M, Bergfeldt B, Stemmermann P. CaCl2 as a Mineralizing Agent in Low-Temperature Recycling of Autoclaved Aerated Concrete: Cl-Immobilization by Formation of Chlorellestadite. Minerals. 2022; 12(9):1142. https://doi.org/10.3390/min12091142
Chicago/Turabian StyleUllrich, Angela, Krassimir Garbev, Uwe Schweike, Michael Köhler, Britta Bergfeldt, and Peter Stemmermann. 2022. "CaCl2 as a Mineralizing Agent in Low-Temperature Recycling of Autoclaved Aerated Concrete: Cl-Immobilization by Formation of Chlorellestadite" Minerals 12, no. 9: 1142. https://doi.org/10.3390/min12091142