
Effect of Point Defects on Electronic Properties and Structure of Talc (001) Surface by First Principles

Abstract
:1. Introduction
2. Computational Methods and Models
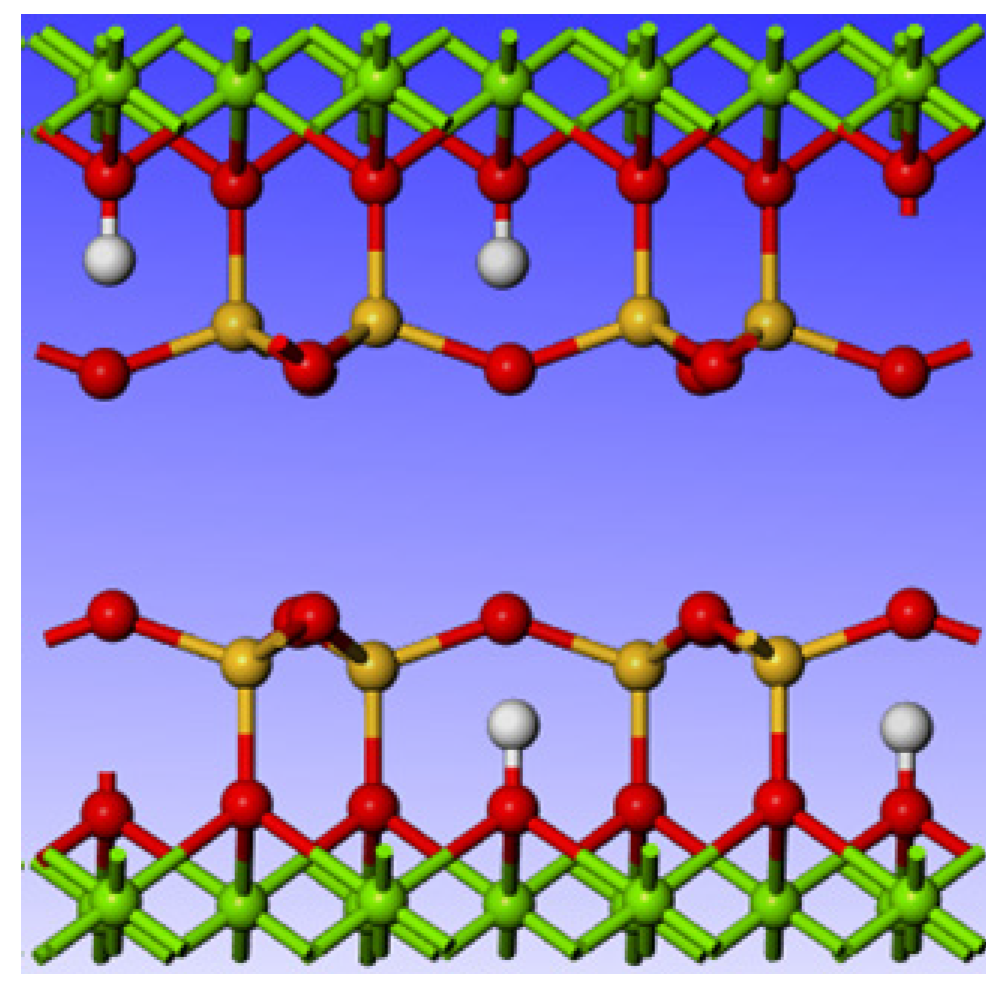
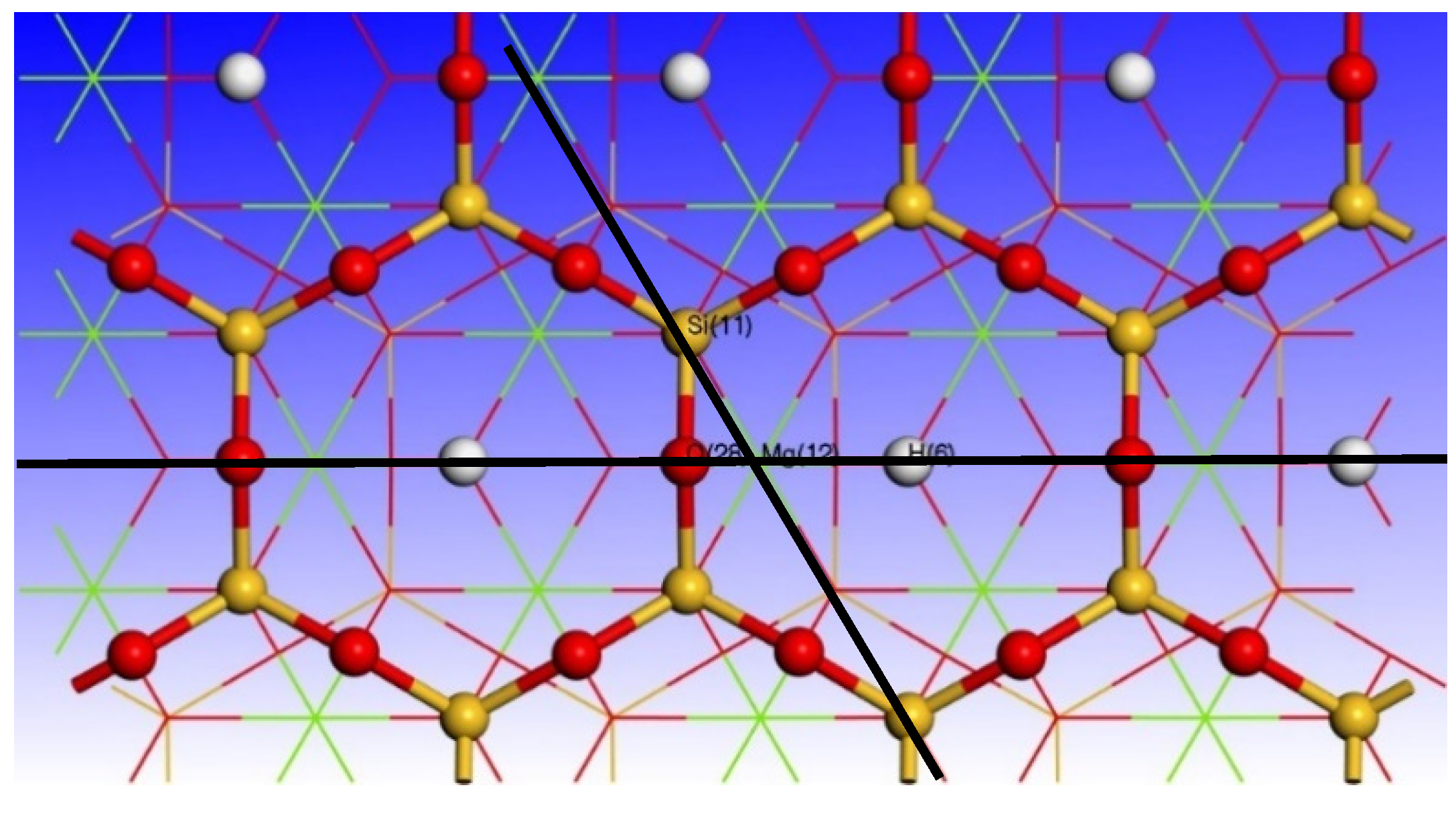
2.1. Calculation Parameters and Models
2.2. Energy Calculation Method
3. Results and Discussion
3.1. Formation Energy of Point Defects
3.2. Effect of Point Defects on the Surface Structure of Talc (001)
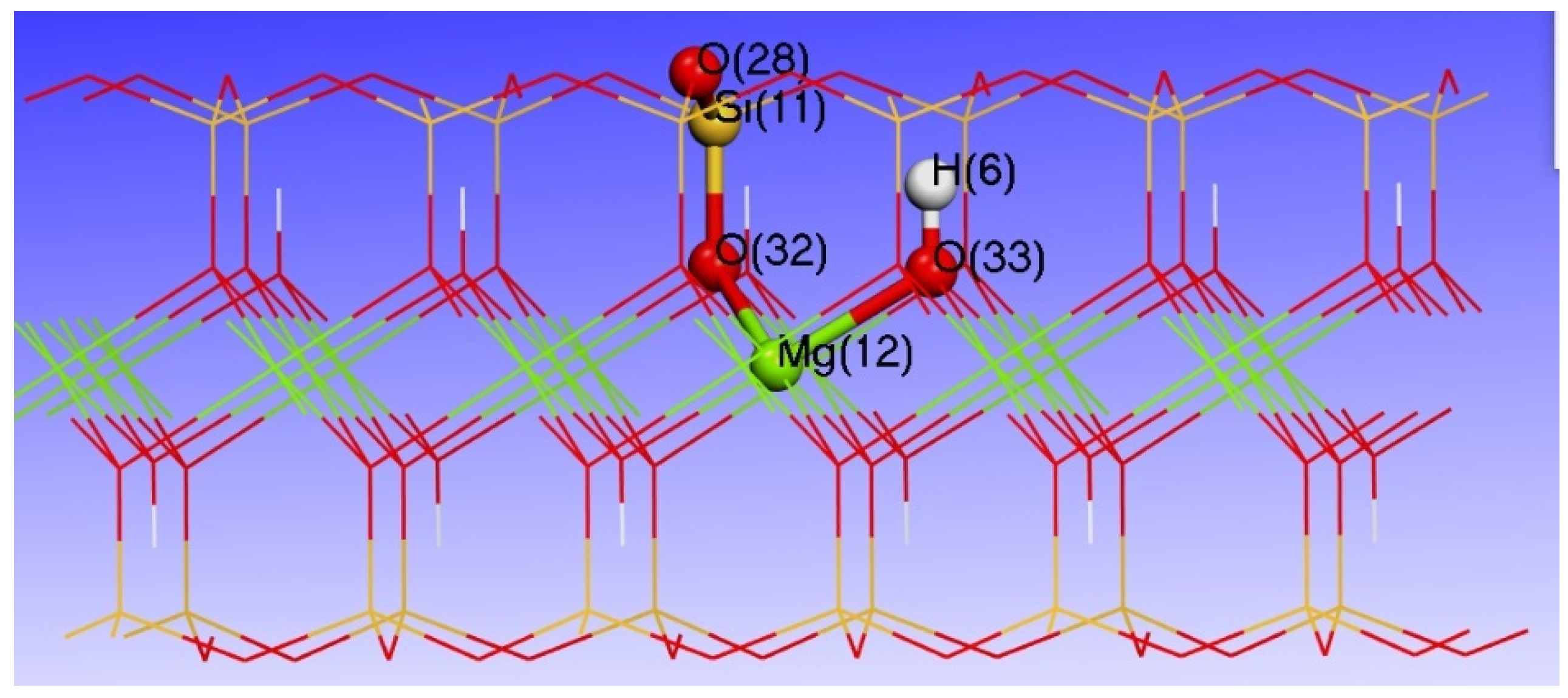
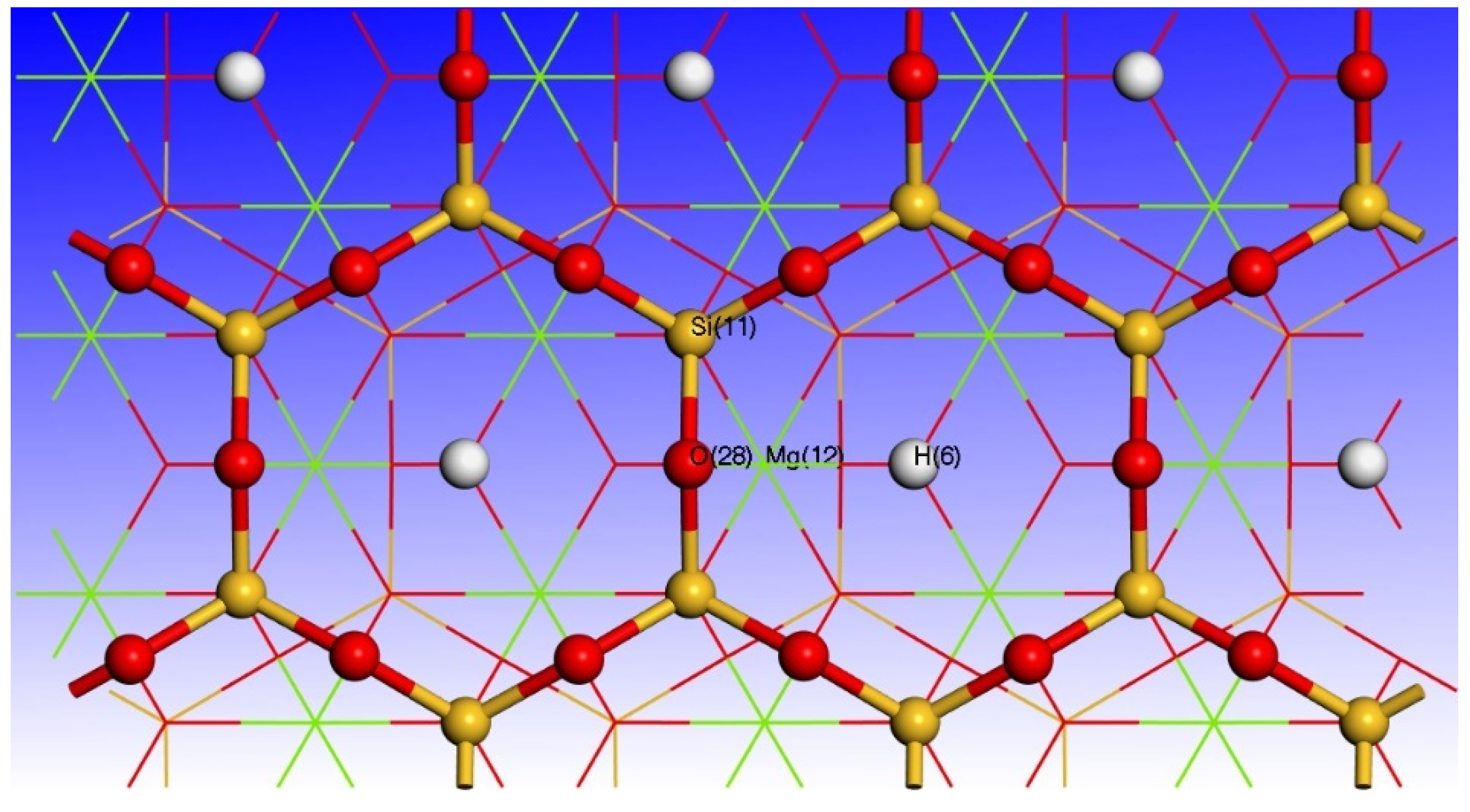
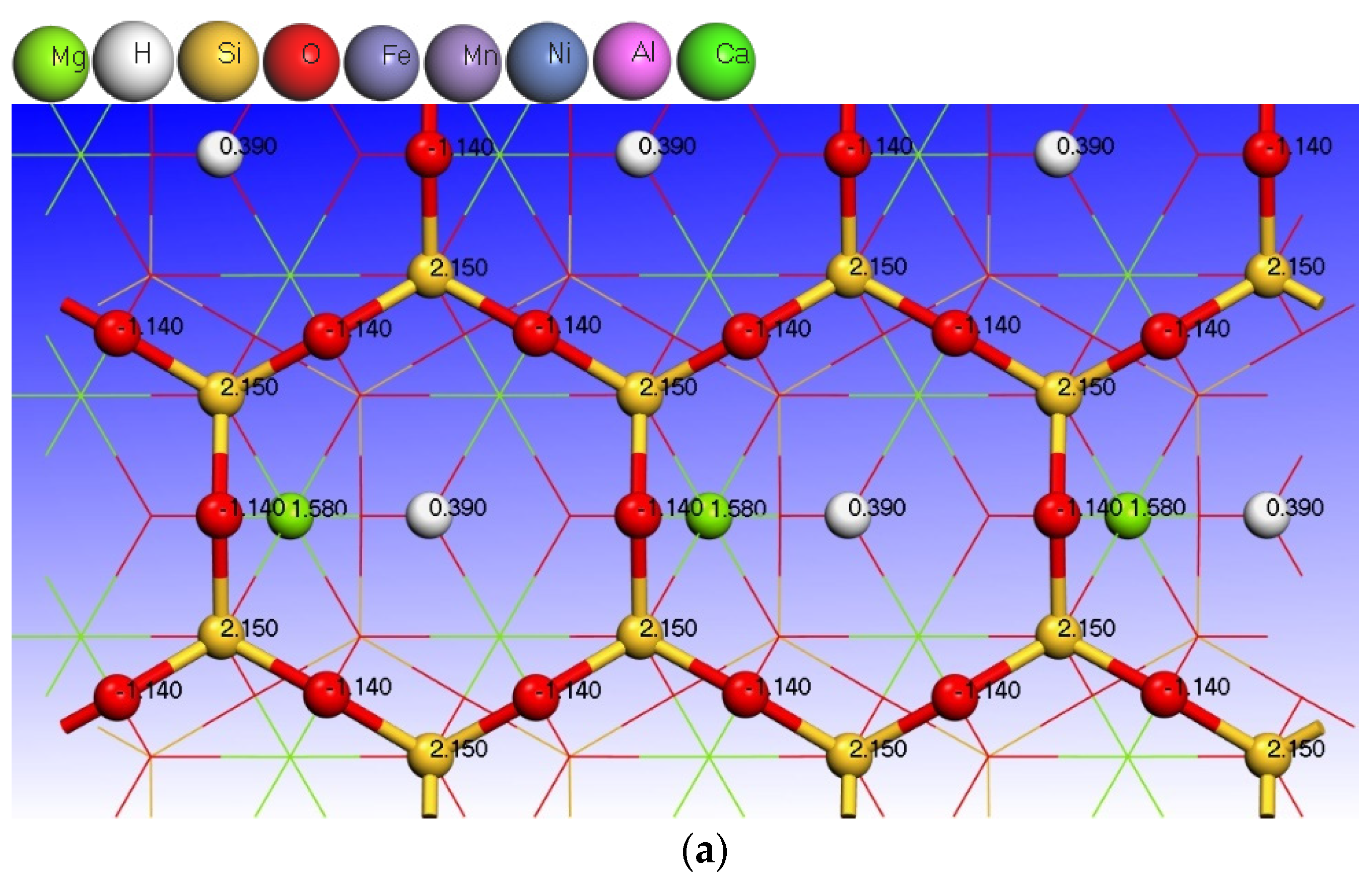
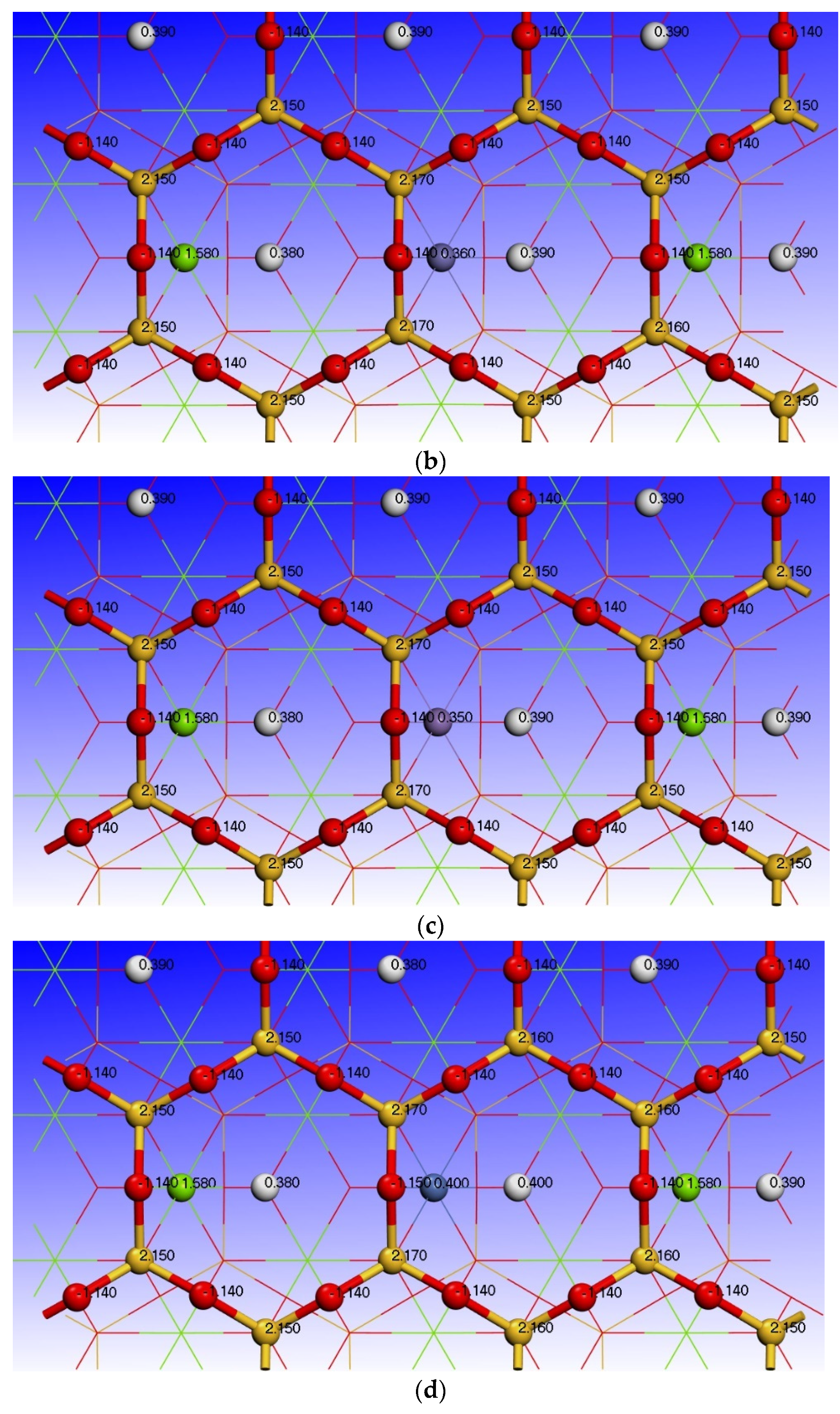
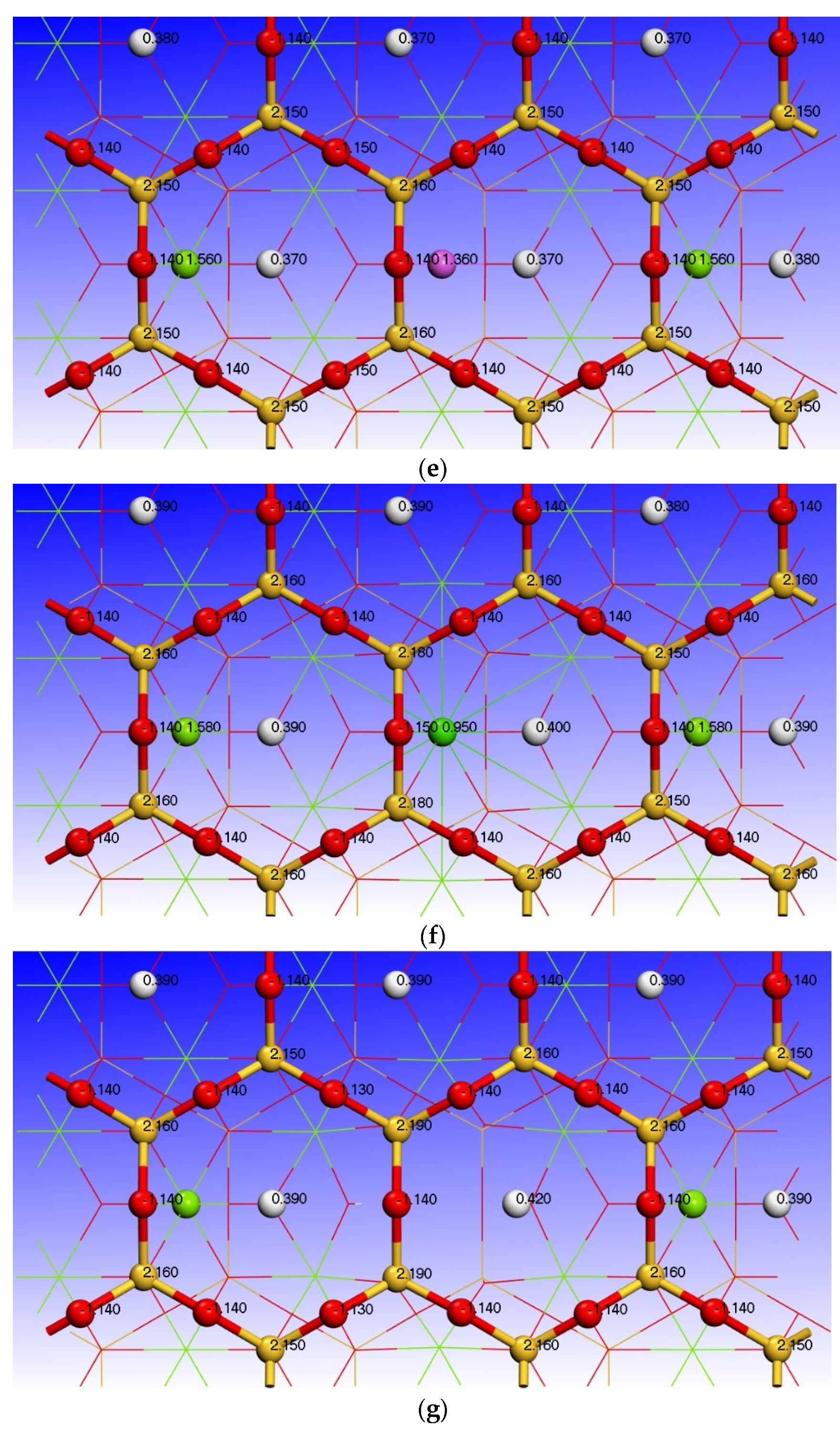
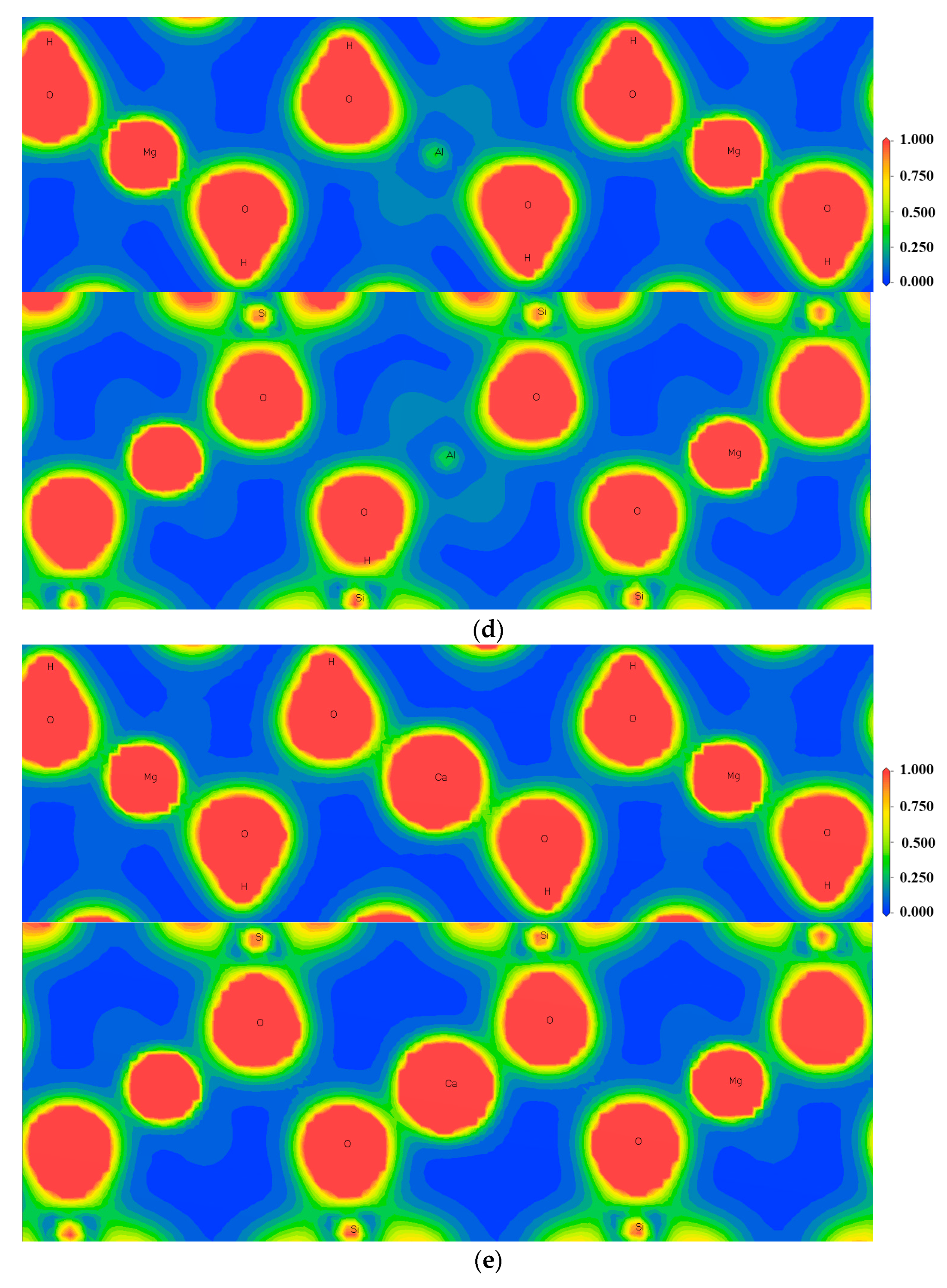
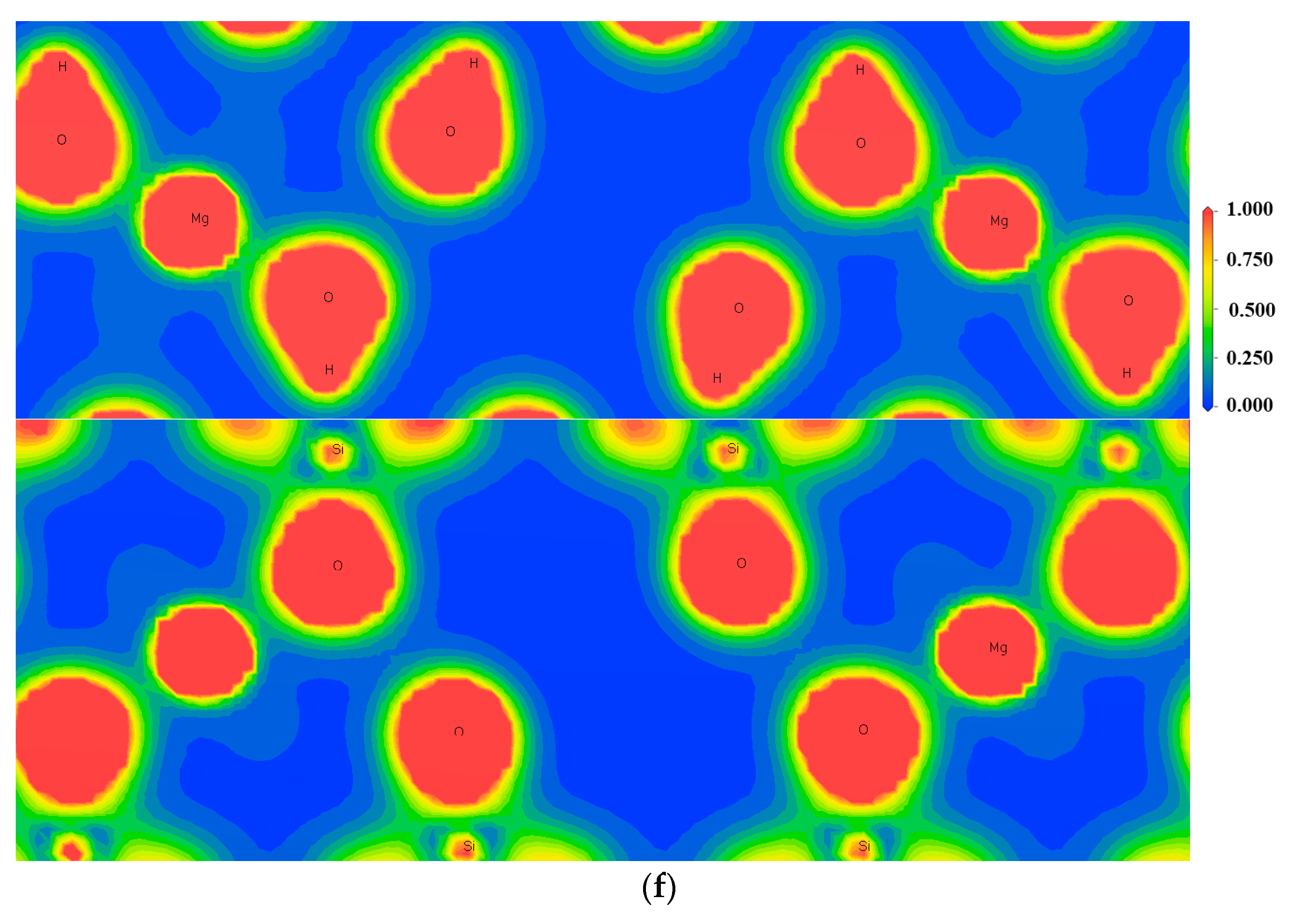
3.3. Effect of Point Defects on Surface Charge of Talc
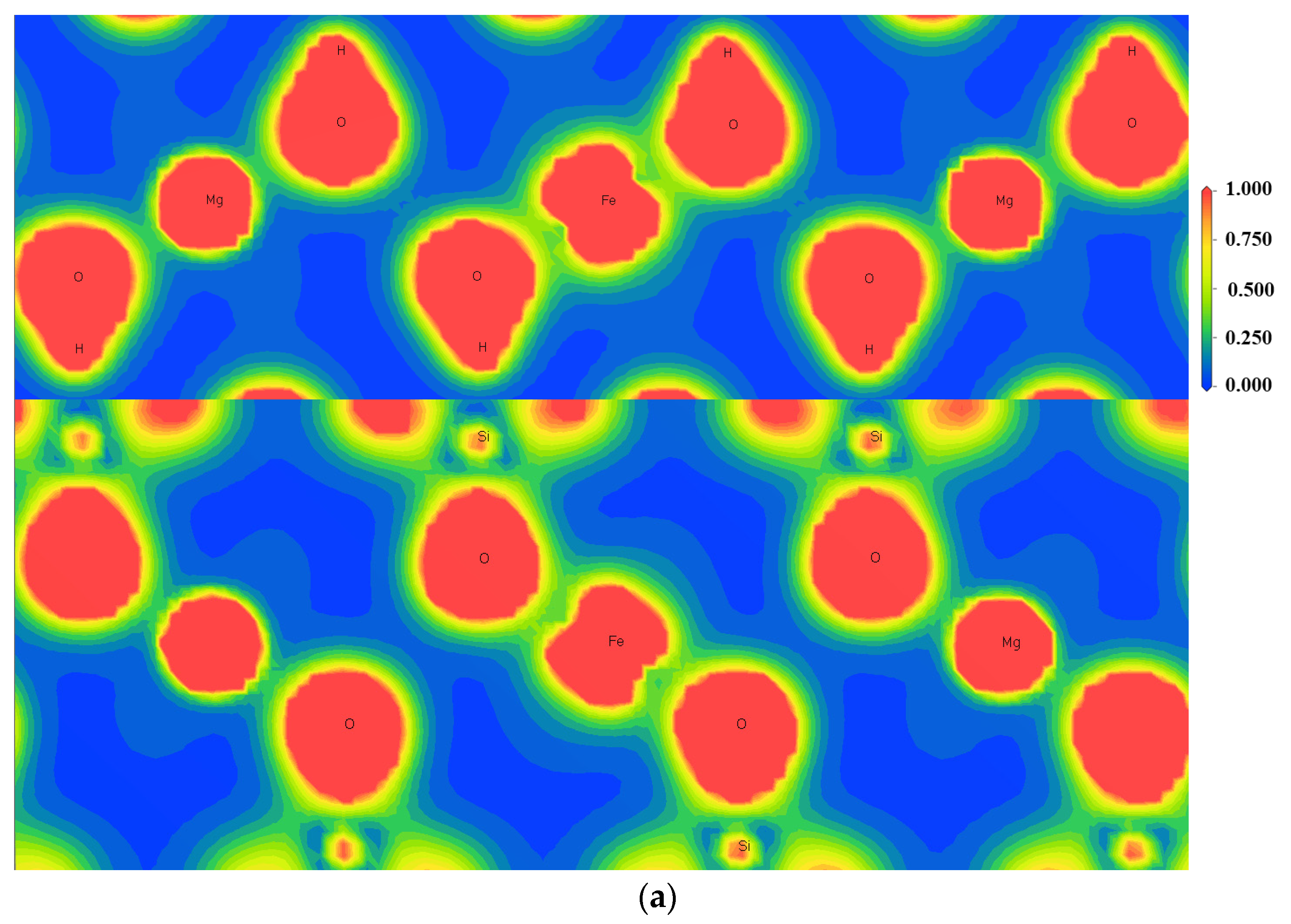
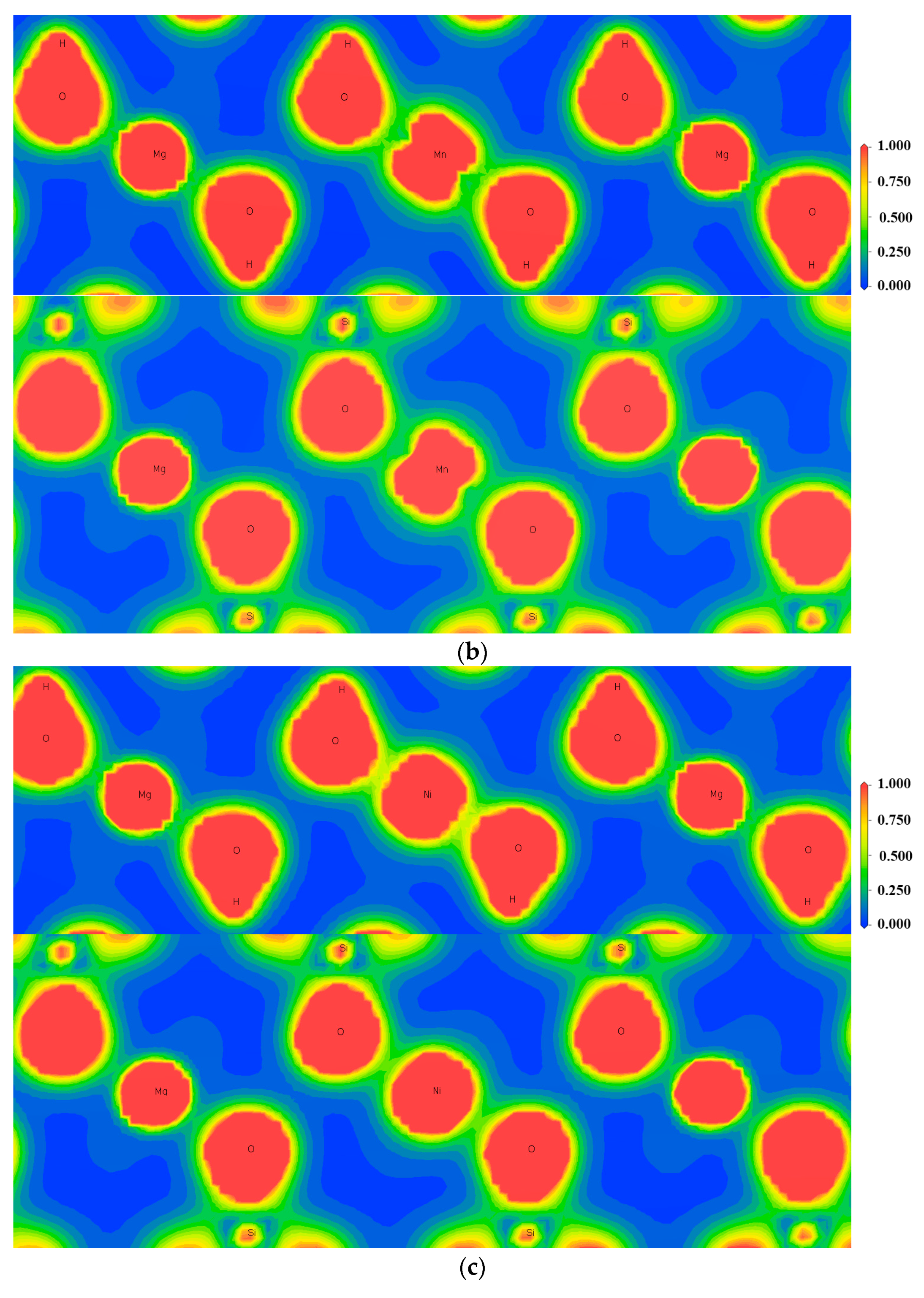
3.4. Charge Density Analysis
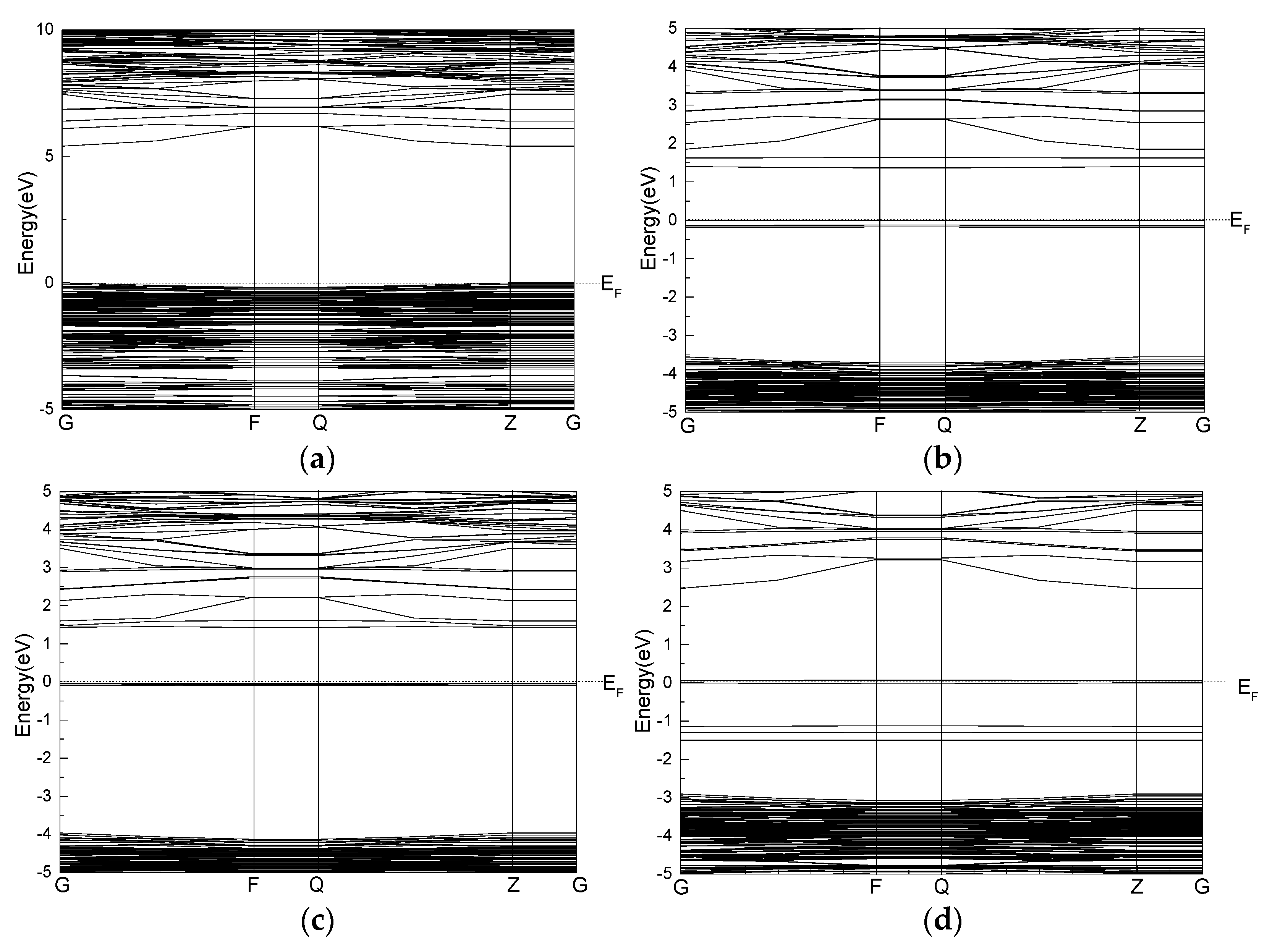
3.5. Effect of Point Defects on Surface Band Structure of Talc
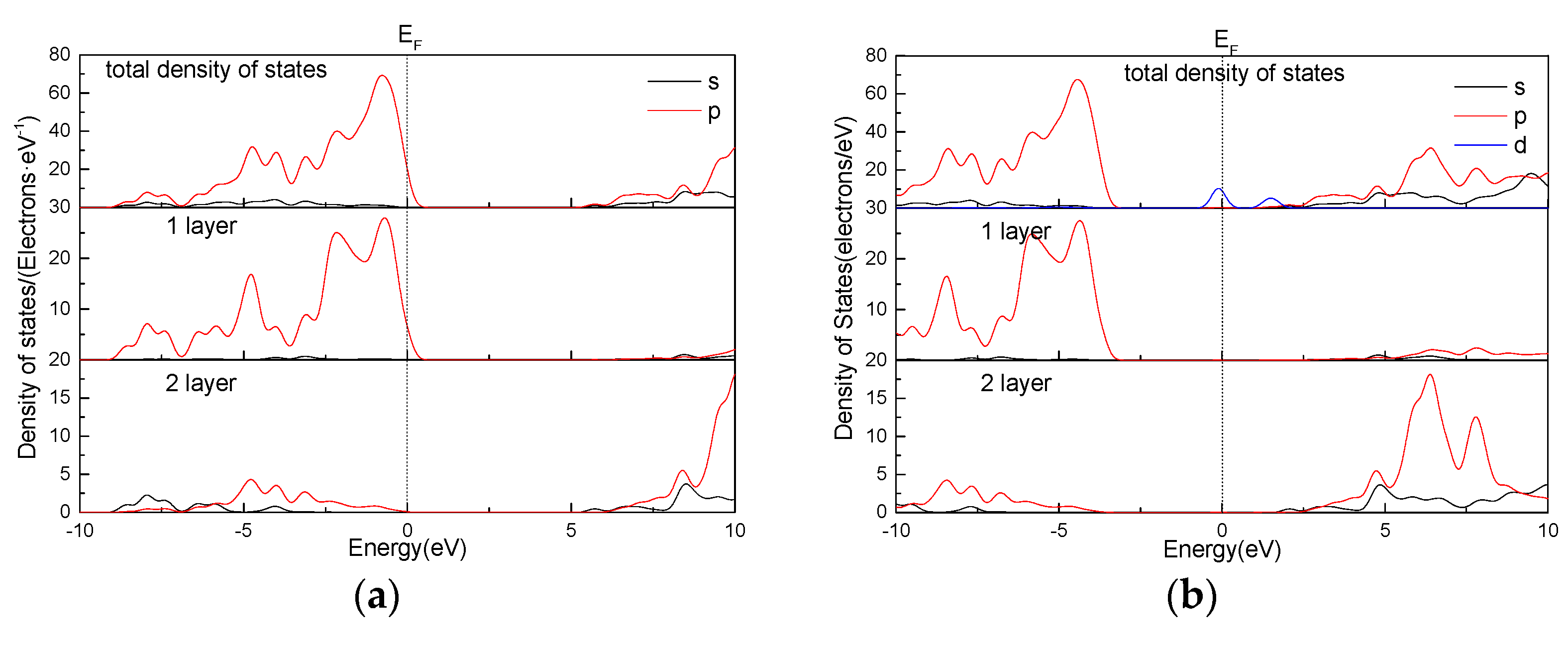
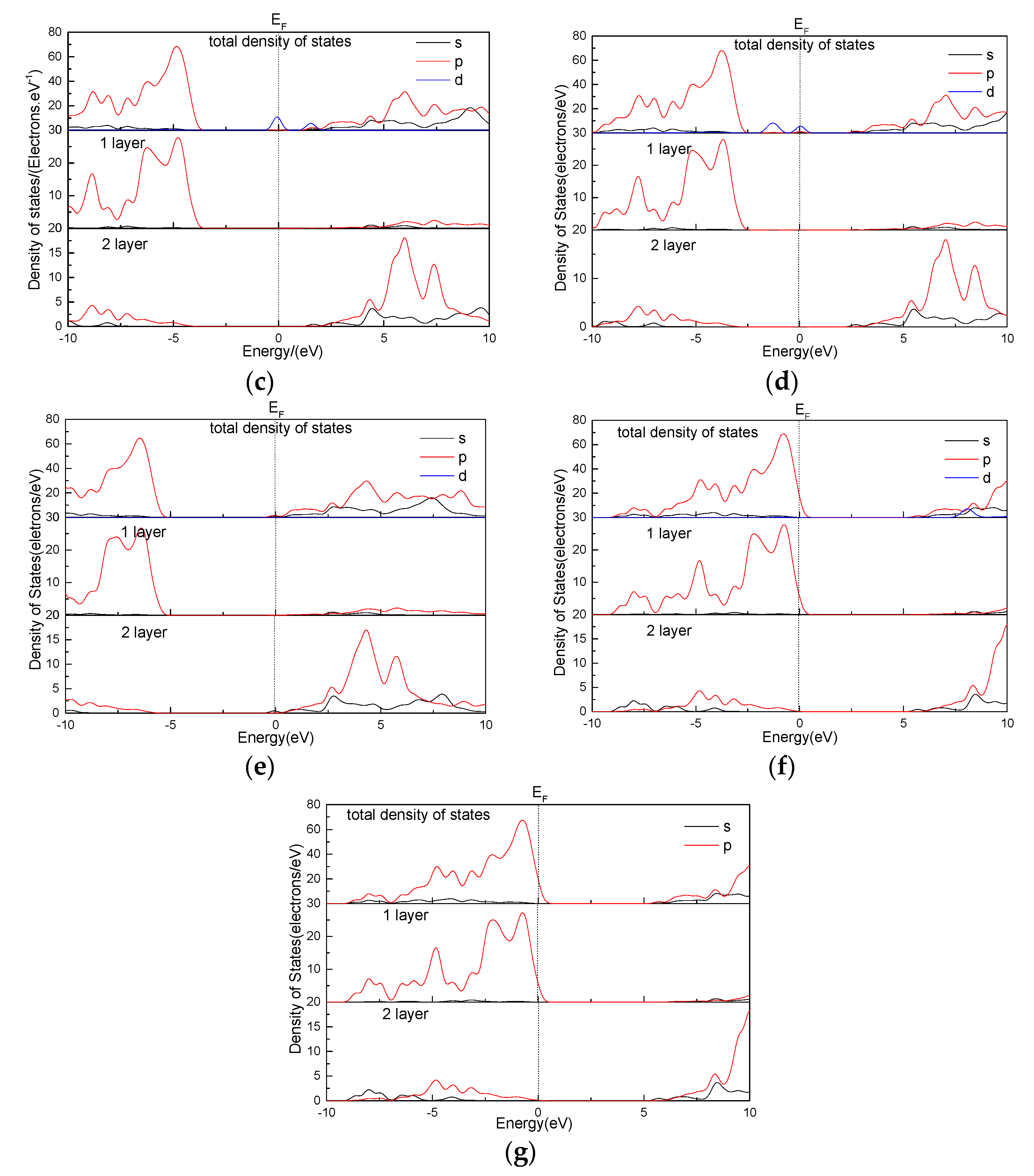
3.6. Effect of Point Defects on Talc State Density
4. Conclusions
- (1)
- After the parameter optimization test, the optimal parameters of cell optimization were obtained. The exchange correlation function was the GGA-PBE (DFT-D modification), and the lattice parameters of talc were obtained as a = 5.33 Å, b = 9.21 Å, and c = 9.46 Å.
- (2)
- According to the substitution energy data, the order of Mg atoms replaced by impurity atoms is Fe > Ca > Al > Ni > Mn. Through the Mulliken population analysis of atoms, it is found that point defects increase the electron loss of Si atoms on the talc surface. The Mulliken overlapping population analysis of atom bonding on the talc surface shows that the covalency of Si-O bonds above the defects of Fe, Mn, Ni, and Al is weakened and the ionic property is enhanced. The results show that point defects can change the hydrophilic and hydrophobic properties of the talc surface.
- (3)
- The band gap and state density of the talc surface are analyzed. The results show that the defects of Fe, Mn, and Ni decrease the band gap of the talc surface to below 2 eV, and the gold property of the talc surface is enhanced. This indicates that the point defects have a certain effect on the electrical conductivity of the talc surface.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- William, F.B.; Roald, H. Orbital Interactions in Phyllosilicates: Perturbations of an Idealized Two-Dimensional, Infinite Silicate Frame. Phys. Chem. Miner. 1988, 15, 398–408. [Google Scholar]
- Perdikatsis, B.; Burzlaff, H. Strukturverfeinerung Am Talk Mg3[(OH)2Si4O10]. Z. Krist.-Cryst. Mater. 1981, 156, 3–4. [Google Scholar] [CrossRef]
- Lee, J.H.; Guggenheim, S. Single-Crystal X-Ray Refinement of Pyrophyllite-1tc. Am. Mineral. 1981, 66, 350–357. [Google Scholar]
- Feng, B.; Lu, Y.P.; Feng, Q.M.; Zhang, M.Y.; Gu, Y.L. Talc–Serpentine Interactions and Implications for Talc Depression. Miner. Eng. 2012, 32, 68–73. [Google Scholar] [CrossRef]
- Deer, W.A.; Howie, R.A.; Zussman, J. An Introduction to the Rock-Forming Minerals, 2nd Edition. Longman Sci. Tech. 1992, 385, 617–619. [Google Scholar]
- Farrokhpay, S.; Ndlovu, B.; Bulelwa, D.N. Behavior of Talc and Mica in Copper Ore Flotation. Appl. Clay Sci. 2018, 160, 270–275. [Google Scholar] [CrossRef]
- Zhang, Q.D.; Yuan, Z.T.; Liu, J.T.; Li, X.L.; Lu, Y.W.; Lu, S.S. Effect of glucan on flotation separation of molybdenite and talc. Chin. J. Nonferrous Met. 2016, 4, 7. [Google Scholar]
- Long, T.; Feng, Q.M.; Lu, P.Y.; Zhang, G.F.; Ou, L.M.; Pan, G.C. Depression and dispersion effect of carboxy methyl cellulose on flotation of layered magnesium-silicates. Chin. J. Nonferrous Met. 2011, 21, 1145–1150. [Google Scholar]
- Ahmed, M.M.; Galal, A.I.; Mohamed, M.A. Improvement of Egyptian Talc Quality for Industrial Uses by Flotation Process and Leaching. Int. J. Miner. Processing 2007, 83, 132–145. [Google Scholar] [CrossRef]
- Burdukova, E.; Van Leerdam, G.C.; Prins, F.E.; Smeink, R.G.; Bradshaw, D.J.; Laskowski, J.S. Effect of Calcium Ions on the Adsorption of Cmc onto the Basal Planes of New York Talc—A Tof-Sims Study. Miner. Eng. 2008, 21, 1020–1025. [Google Scholar] [CrossRef]
- Vidal, C.G.; Pawlik, M. Molecular Weight Effects in Interactions of Guar Gum with Talc. Int. J. Miner. Process. 2015, 138, 38–43. [Google Scholar] [CrossRef]
- Rayner, J.H.; Brown, G. The Crystal Structure of Talc. Acta Crystallogr. 1969, 25, 163–165. [Google Scholar] [CrossRef]
- Yan, H.J.; Luo, X.P.; Zhu, X.W.; Weng, C.J.; Zhang, W.P.; Feng, B. Research Progress on Talc Inhibitors in Sulfide Ore Flotation. Conserv. Util. Miner. Resour. 2020, 40, 138–144. [Google Scholar]
- Chen, J.H.; Chen, Y.; Li, Y.Q. Effect of Vacancy Defects on Electronic Properties and Activation of Sphalerite (110) Surface by First-Principles. Trans. Nonferrous Met. Soc. China 2010, 20, 502–506. [Google Scholar] [CrossRef]
- Chen, J.H.; Long, X.H.; Zhao, C.H.; Kang, D.; Guo, J. DFT Calculation on Relaxation and Electronic Structure of Sulfide Minerals Surfaces in Presence of H2O Molecule. J. Cent. S. Univ. 2014, 21, 3945–3954. [Google Scholar] [CrossRef]
- Zhao, C.H.; Chen, J.H.; Wu, B.Z.; Long, X.H. Density Functional Theory Study on Natural Hydrophobicity of Sulfide Surfaces. Trans. Nonferrous Met. Soc. China 2014, 24, 491–498. [Google Scholar] [CrossRef]
- Chen, J.H.; Li, Y.Q.; Lan, L.H.; Guo, J. Interactions of Xanthate with Pyrite and Galena Surfaces in the Presence and Absence of Oxygen. J. Ind. Eng. Chem. 2014, 20, 268–273. [Google Scholar] [CrossRef]
- Clark, S.J.; Segallii, M.D.; Pickardii, C.J.; Hasnipiii, P.J.; Probertiv, M.I.J. First Principles Methods Using Castep. Z. Krist.-Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical Gga-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Terentjev, A.V.; Constantin, L.A.; Pitarke, J.M. Dispersion-corrected PBEsol exchange-correlation functional. Phys. Rev. B 2018, 98, 21. [Google Scholar] [CrossRef]
- Lan, L.H.; Chen, J.H.; Chen, J.H.; Li, Y.Q.; Lan, P.; Yang, Z.; Ai, G.Y. Microthermokinetic Study of Xanthate Adsorption onpurity-Doped Galena. Trans. Nonferrous Met. Soc. China 2016, 26, 272–281. [Google Scholar] [CrossRef]
- Kazume, N.; Yoshizawa, M.; Hasegawa, M. Energetics of Mg and B Adsorption on Polar Zinc Oxide Surfaces from First Principles. Phys. Rev. 2008, 77, 35330. [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Point Defect Type | Point Defect Formation Energy (KJ·mol−1) |
|---|---|
| Fe | −162.72 |
| Mn | 265.77 |
| Ni | 139.78 |
| Al | 13.04 |
| Ca | −32.84 |
| Mg vacancy | 1111.68 |
| Type | (001) Surf | Fe Impurity | Mn Impurity | Ni Impurity | Al Impurity | Ca Impurity | Mg Vacancy | Experimental Value |
|---|---|---|---|---|---|---|---|---|
| Unitlayer thickness | 6.62 | 6.65 (0.03) | 6.64 (0.02) | 6.61 (0.01) | 6.68 (0.06) | 6.49 (0.13) | 6.50 (0.12) | 6.50 |
| Si-Ob/Å | 1.63 | 1.63 (0.00) | 1.63 (0.00) | 1.63 (0.00) | 1.63 (0.00) | 1.63 (0.00) | 1.63 (0.00) | 1.61 |
| Si-Oa/Å | 1.63 | 1.63 (0.00) | 1.6 (0.00) | 1.63 (0.00) | 1.66 (0.03) | 1.62 (0.01) | 1.61 (0.02) | 1.63 |
| X-Oa/Å | 2.11 | 2.06 | 2.10 | 2.14 | 1.97 | 2.25 | —— | 2.08 |
| X-O(O-H)/Å | 2.06 | 2.01 | 2.05 | 1.99 | 1.90 | 2.25 | —— | 2.05 |
| O-H/Å | 0.97 | 0.98 (0.01) | 0.98 (0.01) | 0.98 (0.01) | 0.98 (0.01) | 0.97 (0.00) | 0.97 (0.00) | —— |
| Oaup-Oadown/Å | 2.27 | 2.23 (0.04) | 2.26 (0.01) | 2.29 (0.02) | 2.19 (0.08) | 2.33 (0.06) | 2.34 (0.07) | 2.20 |
| Type | Atoms | s | p | d | Charge/e |
|---|---|---|---|---|---|
| Talc (001) surface | H (6) | 0.61 | 0.00 | 0.00 | 0.39 |
| O (33) | 1.90 | 5.24 | 0.00 | −1.14 | |
| Mg(12) | 0.43 | 5.99 | 0.00 | 1.58 | |
| O (32) | 1.91 | 5.34 | 0.00 | −1.25 | |
| Si (11) | 0.65 | 1.20 | 0.00 | 2.15 | |
| O (28) | 1.83 | 5.31 | 0.00 | −1.14 | |
| Fe impurity defect surface | H (6) | 0.61 | 0.00 | 0.00 | 0.39 |
| O (33) | 1.88 | 5.13 | 0.00 | −1.01 | |
| Fe | 0.38 | 0.54 | 6.72 | 0.36 | |
| O (32) | 1.88 | 5.25 | 0.00 | −1.14 | |
| Si (11) | 0.64 | 1.19 | 0.00 | 2.17 | |
| O (28) | 1.83 | 5.31 | 0.00 | −1.14 | |
| Mn impurity defect surface | H (6) | 0.61 | 0.00 | 0.00 | 0.39 |
| O (33) | 1.88 | 5.13 | 0.00 | −1.01 | |
| Mn | 0.38 | 0.45 | 5.82 | 0.35 | |
| O (32) | 1.88 | 5.25 | 0.00 | −1.13 | |
| Si (11) | 0.64 | 1.19 | 0.00 | 2.17 | |
| O (28) | 1.83 | 5.31 | 0.00 | −1.14 | |
| Ni impurity defect surface | H (6) | 0.60 | 0.00 | 0.00 | 0.40 |
| O (33) | 1.88 | 5.12 | 0.00 | −1.00 | |
| Ni | 0.45 | 0.59 | 8.55 | 0.40 | |
| O (32) | 1.89 | 5.27 | 0.00 | −1.16 | |
| S i(11) | 0.64 | 1.19 | 0.00 | 2.17 | |
| O (28) | 1.83 | 5.32 | 0.00 | −1.15 | |
| Al impurity defect surface | H (6) | 0.63 | 0.00 | 0.00 | 0.37 |
| O (33) | 1.87 | 5.20 | 0.00 | −1.08 | |
| Al | 0.60 | 1.04 | 0.00 | 1.36 | |
| O (32) | 1.88 | 5.30 | 0.00 | −1.18 | |
| Si (11) | 0.65 | 1.18 | 0.00 | 2.16 | |
| O (28) | 1.84 | 5.30 | 0.00 | −1.14 | |
| Ca impurity defect surface | H (6) | 0.60 | 0.00 | 0.00 | 0.40 |
| O (33) | 1.88 | 5.21 | 0.00 | −1.09 | |
| Ca | 2.36 | 6.01 | 0.69 | 0.95 | |
| O (32) | 1.89 | 5.32 | 0.00 | −1.21 | |
| Si (11) | 0.64 | 1.19 | 0.00 | 2.18 | |
| O (28) | 1.83 | 5.33 | 0.00 | −1.15 | |
| Mg vacancy defect surface | H (6) | 0.58 | 0.00 | 0.00 | 0.42 |
| O (33) | 1.91 | 5.08 | 0.00 | −0.99 | |
| Mg (12) | —— | —— | —— | —— | |
| O (32) | 1.91 | 5.24 | 0.00 | −1.16 | |
| Si (11) | 0.63 | 1.18 | 0.00 | 2.19 | |
| O (28) | 1.83 | 5.32 | 0.00 | −1.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, X.; Du, H.; Lan, P.; Chen, J.; Lan, L. Effect of Point Defects on Electronic Properties and Structure of Talc (001) Surface by First Principles. Minerals 2022, 12, 69. https://doi.org/10.3390/min12010069
Ma X, Du H, Lan P, Chen J, Lan L. Effect of Point Defects on Electronic Properties and Structure of Talc (001) Surface by First Principles. Minerals. 2022; 12(1):69. https://doi.org/10.3390/min12010069
Chicago/Turabian StyleMa, Xindi, Huicong Du, Ping Lan, Jianhua Chen, and Lihong Lan. 2022. "Effect of Point Defects on Electronic Properties and Structure of Talc (001) Surface by First Principles" Minerals 12, no. 1: 69. https://doi.org/10.3390/min12010069