Spectroscopic Studies of Synthetic and Natural Saponites: A Review


Abstract
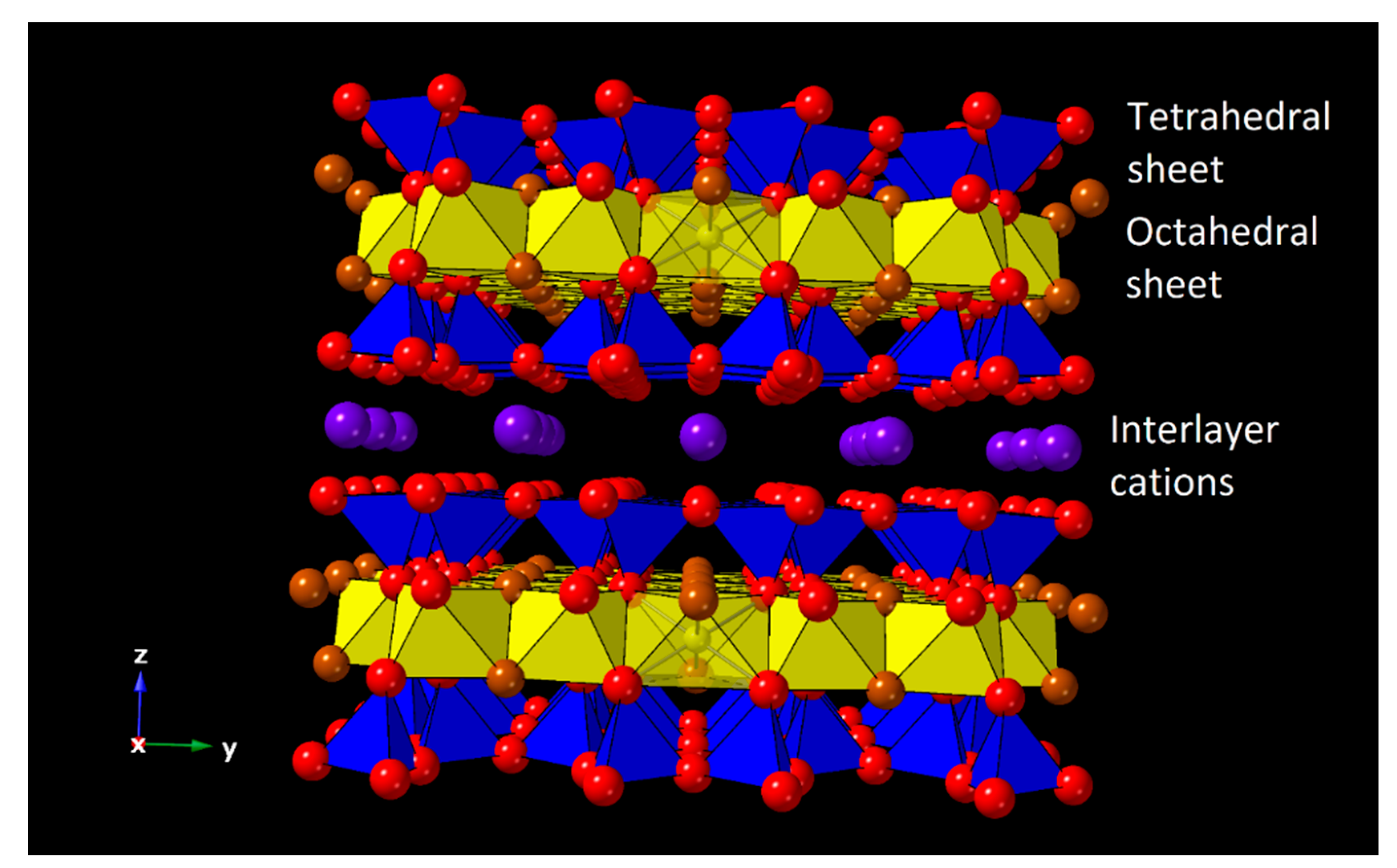
:1. Introduction
2. Materials and Methods
2.1. Hydrothermal Synthesis
2.2. Urea-Assisted Synthesis
2.3. Natural Saponites
2.4. Spectroscopic Methods
2.4.1. Mid-Infrared Spectroscopy
2.4.2. Raman Spectroscopy
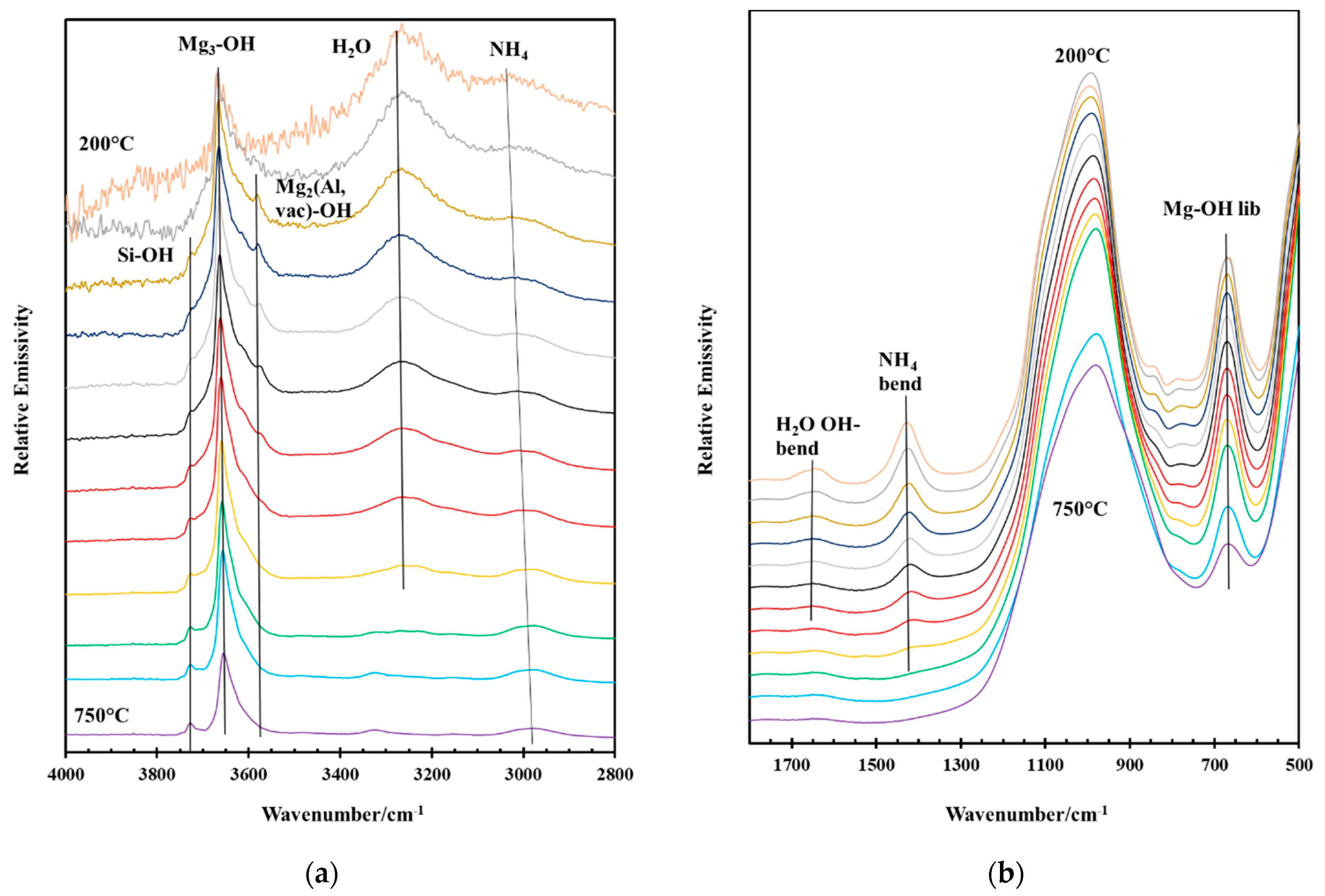
2.4.3. Infrared Emission Spectroscopy (IES)
2.4.4. Spectral Manipulation
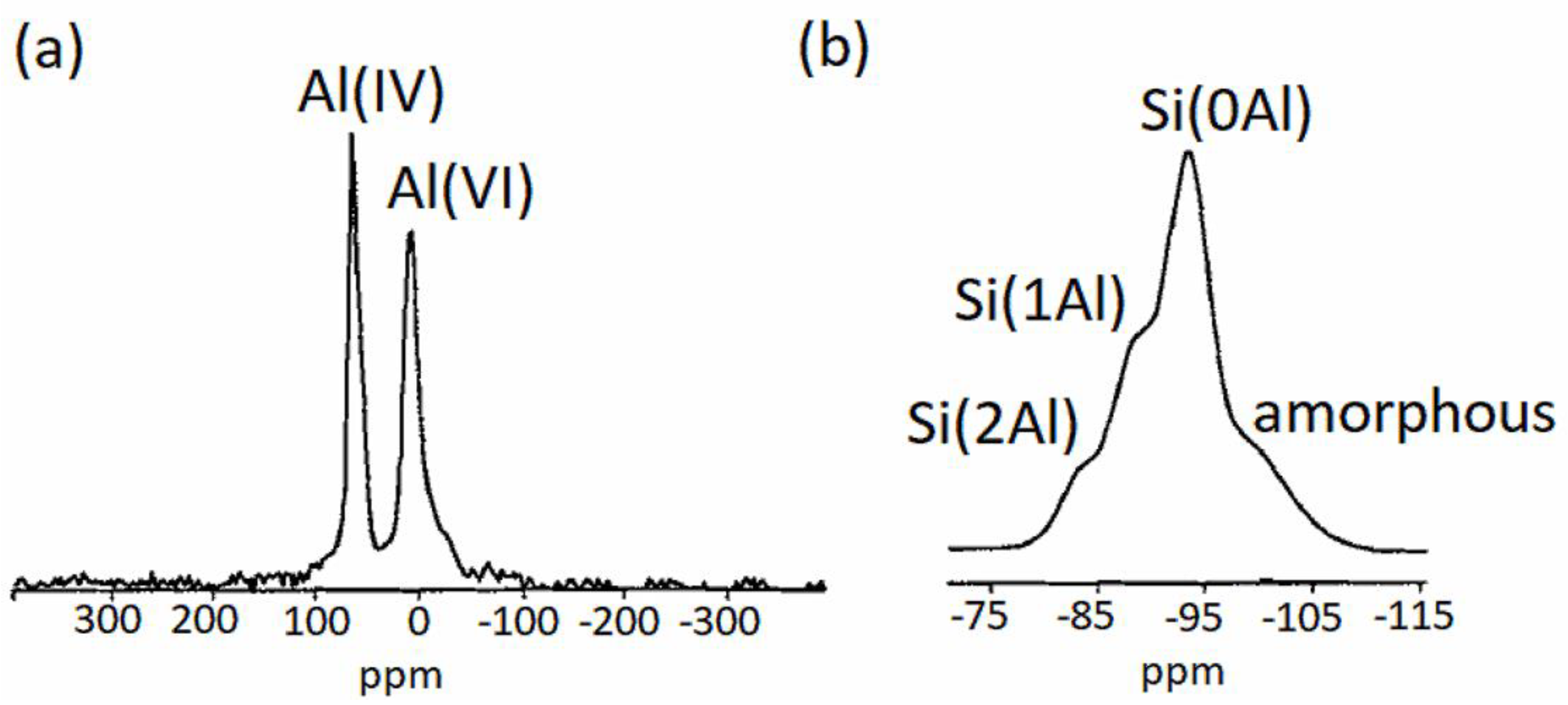
2.4.5. Magic-Angle-Spinning Nuclear Magnetic Resonance Spectroscopy (MAS-NMR)
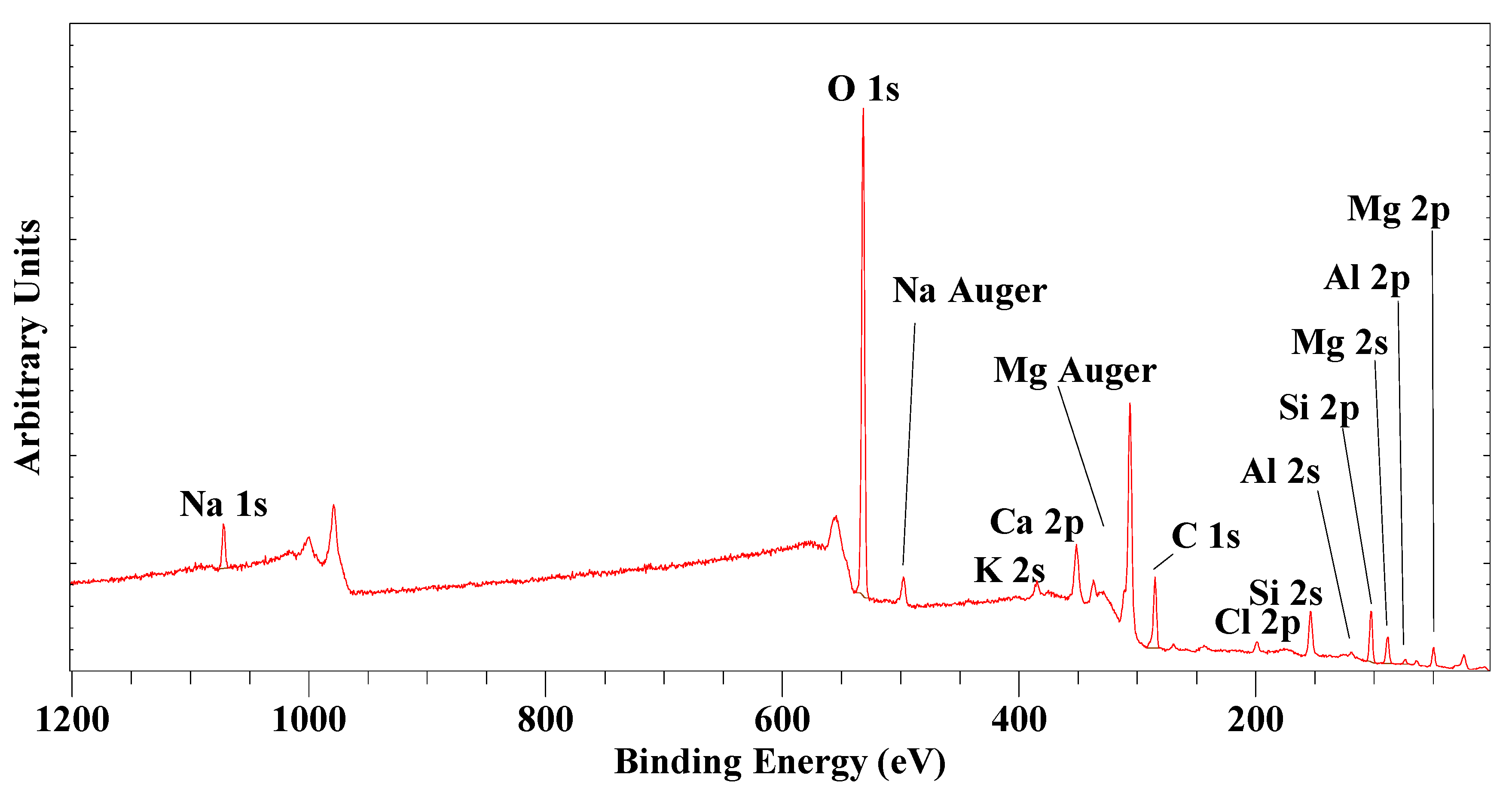
2.4.6. X-ray Photoelectron Spectroscopy (XPS)
3. Mid-Infrared and Raman Spectroscopy
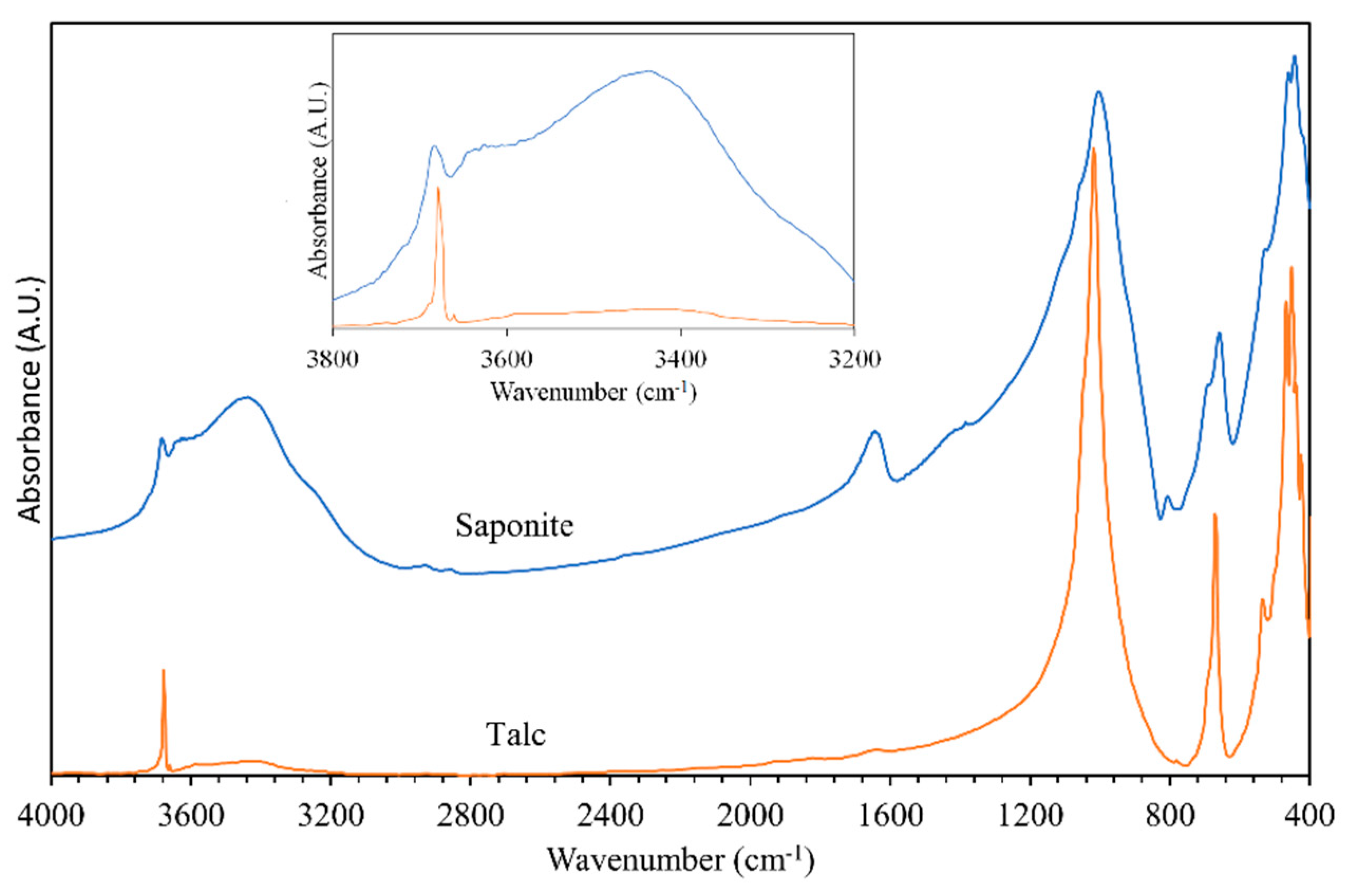
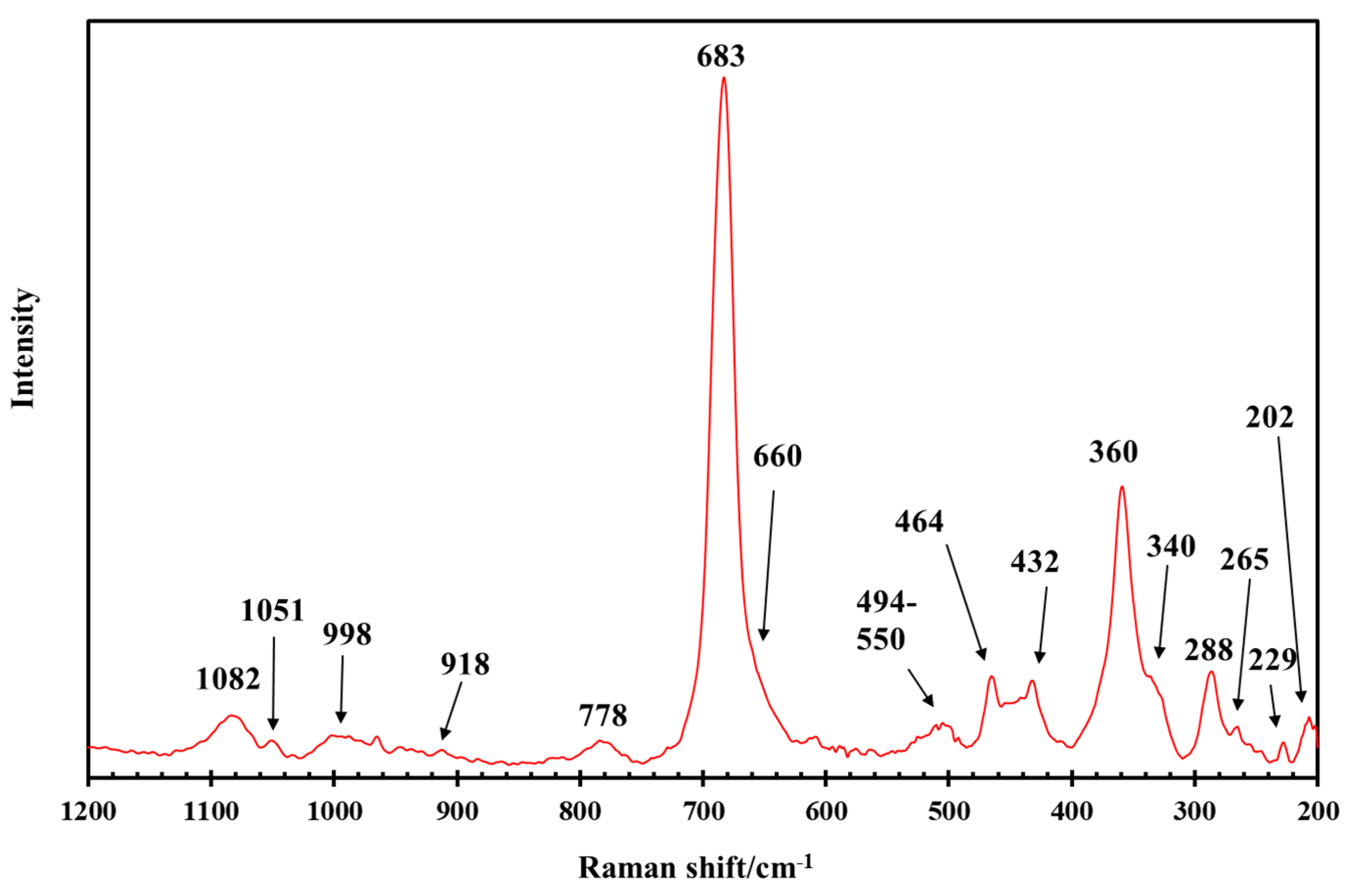
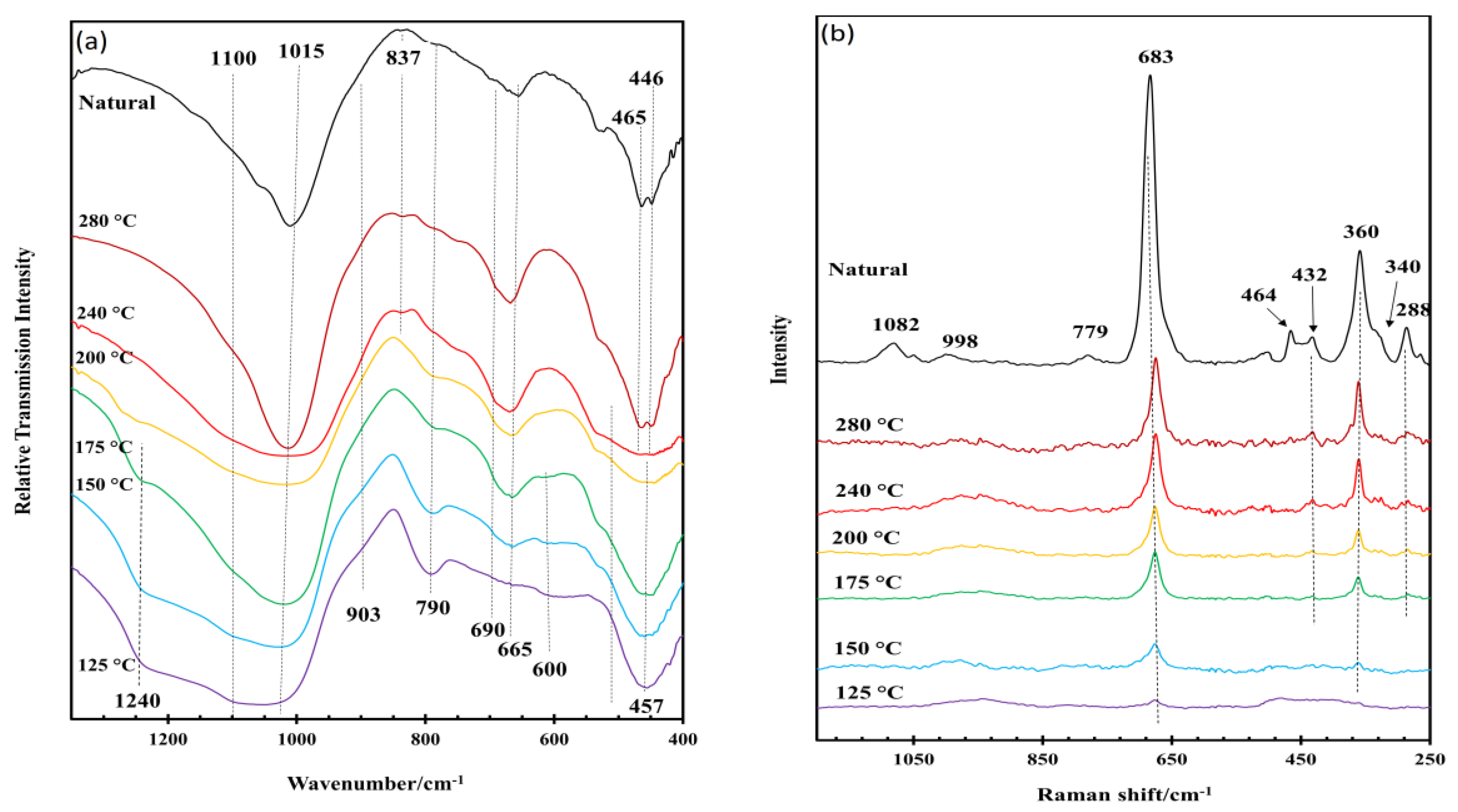
3.1. General Characterization of the IR and Raman Spectra of Saponite
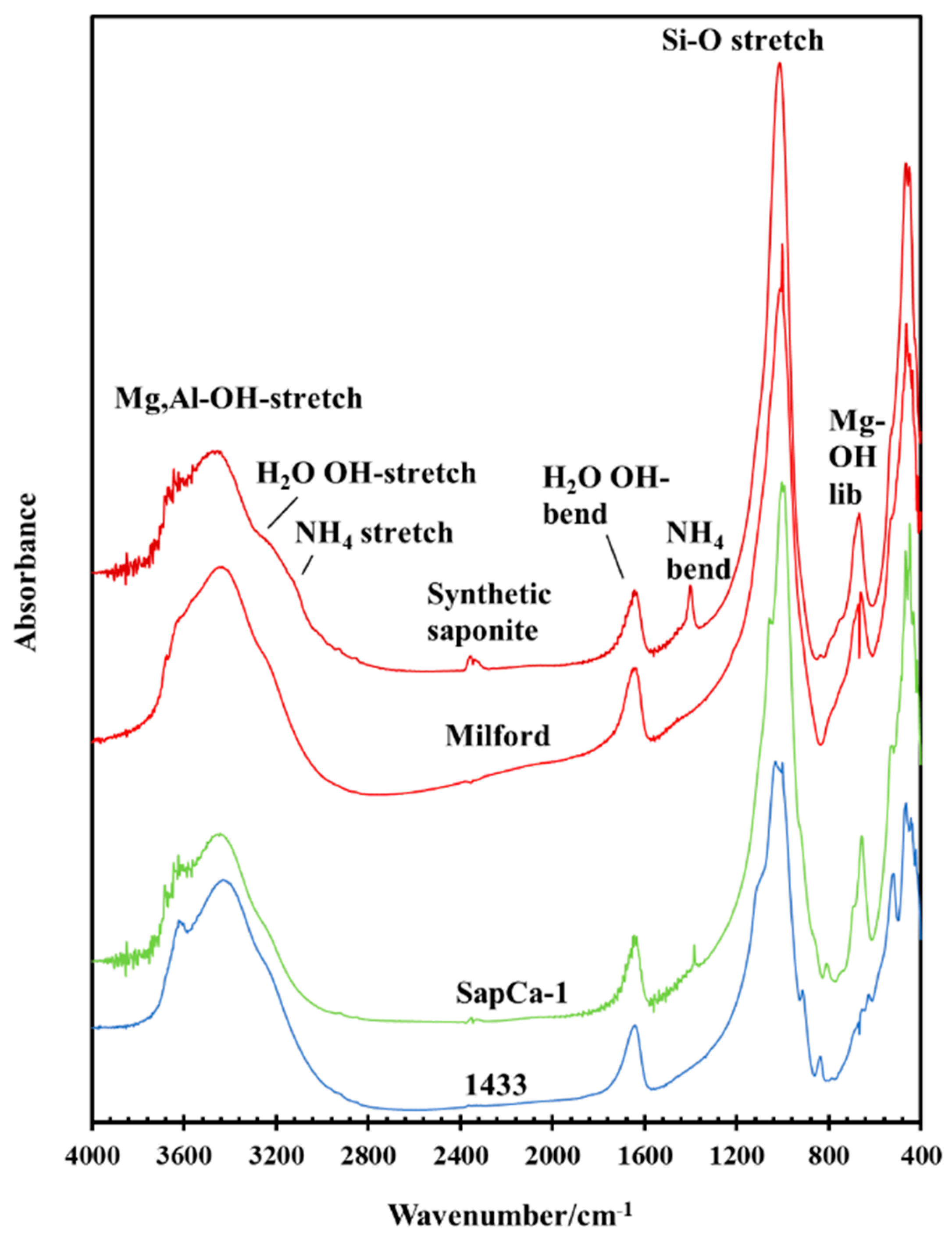
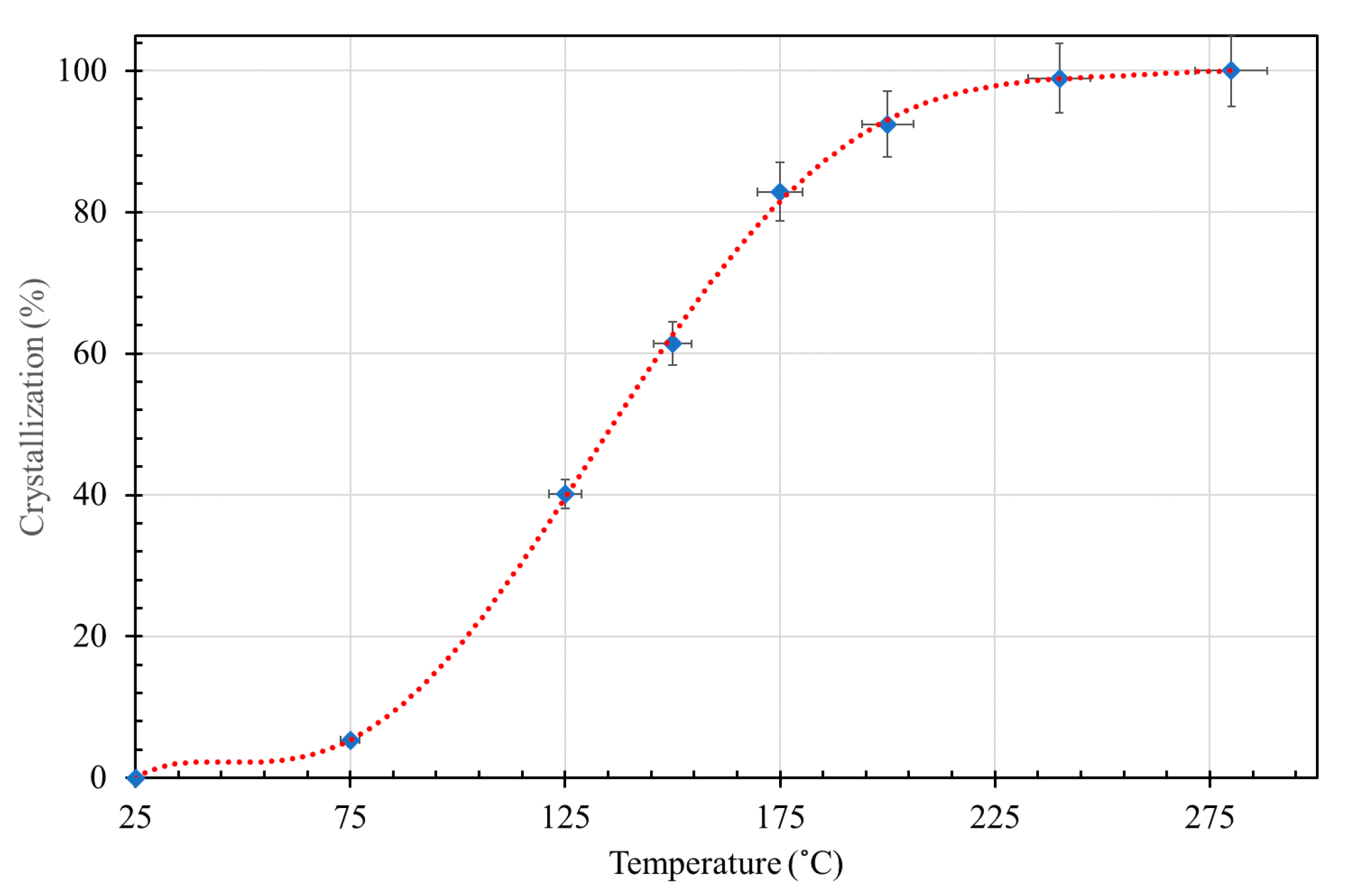
3.2. Effect of Hydrothermal Synthesis Temperature
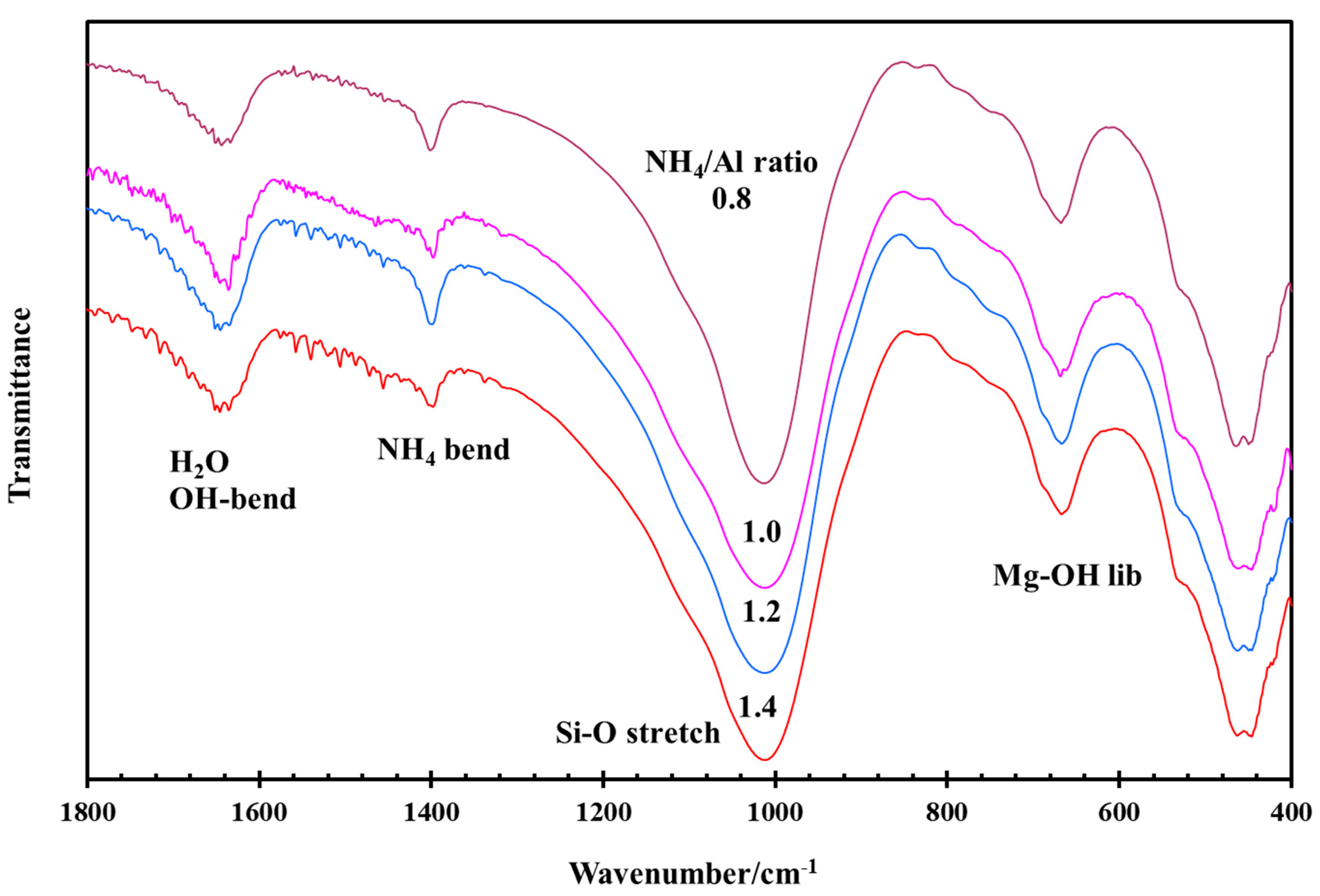
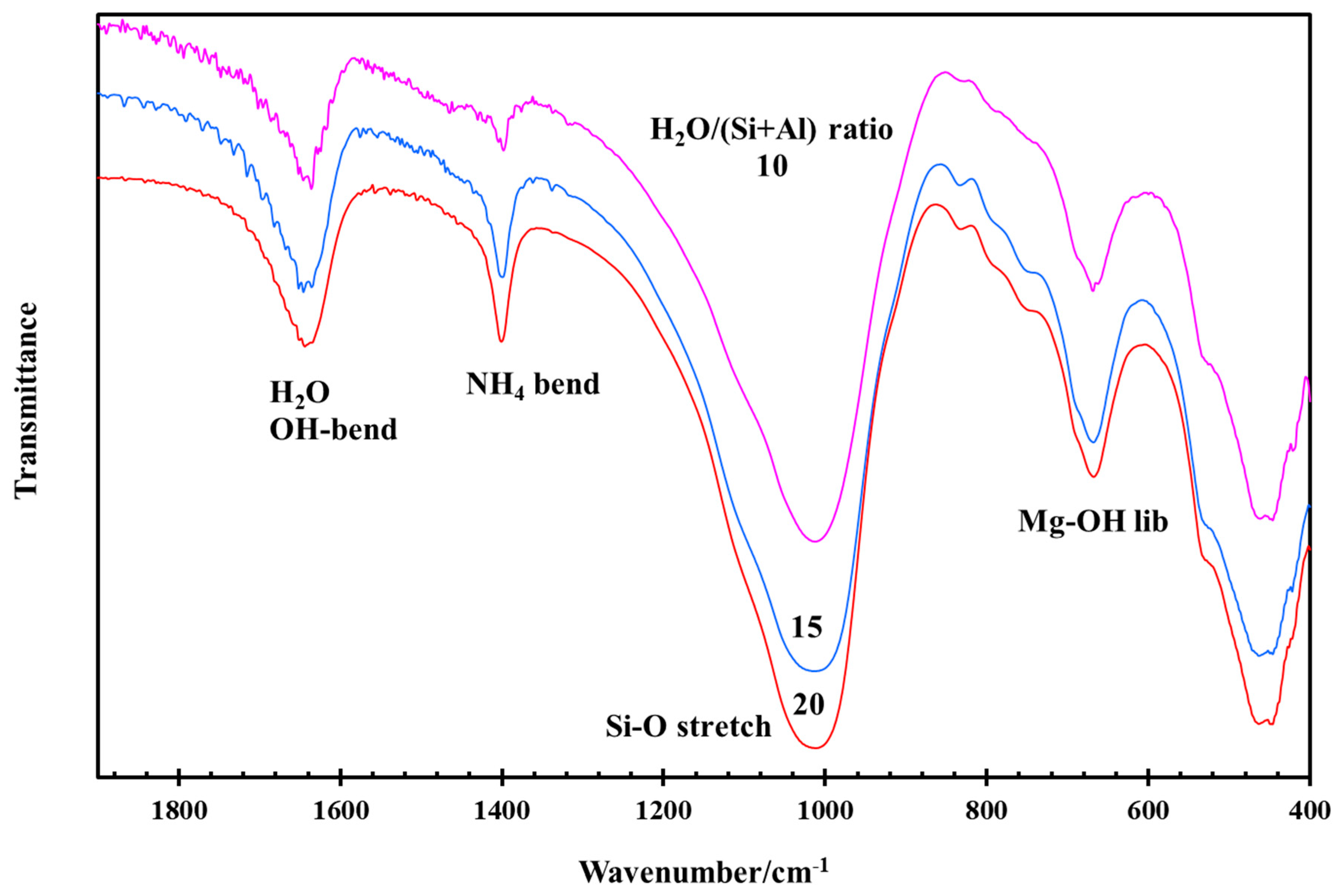
3.3. Effect of NH4/Al and H2O/(Si + Al) Ratios during Hydrothermal Synthesis
3.4. Thermal Decomposition of Natural and Synthetic Saponites
4. MAS-NMR of Synthetic Saponites
5. XPS Characterization of Natural Saponite
6. Final Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, F.; Sun, S.; Chen, X.; Chang, Y.; Zha, F.; Lei, Z. Mg–Al layered double hydroxides modified clay adsorbents for efficient removal of Pb2+, Cu2+ and Ni2+ from water. Appl. Clay Sci. 2016, 123, 134–140. [Google Scholar] [CrossRef]
- Franco, F.; Benítez-Guerrero, M.; Gonzalez-Triviño, I.; Pérez-Recuerda, R.; Assiego, C.; Cifuentes-Melchor, J.; Pascual-Cosp, J. Low-cost aluminum and iron oxides supported on dioctahedral and trioctahedral smectites: A comparative study of the effectiveness on the heavy metal adsorption from water. Appl. Clay Sci. 2016, 119, 321–332. [Google Scholar] [CrossRef]
- Seki, Y.; Ide, Y.; Okada, T.; Ogawa, M. Concentration of 2-phenylphenol by organoclays from aqueous sucrose solution. Appl. Clay Sci. 2015, 109–110, 64–67. [Google Scholar] [CrossRef] [Green Version]
- Epelde-Elezcano, N.; Martínez-Martínez, V.; Duque-Redondo, E.; Temiño, I.; Manzano, H.; López-Arbeloa, I. Strategies for modulating the luminescence properties of pyronin Y dye–clay films: An experimental and theoretical study. Phys. Chem. Chem. Phys. 2016, 18, 8730–8738. [Google Scholar] [CrossRef] [PubMed]
- Matejdes, M.; Himeno, D.; Suzuki, Y.; Kawamata, J. Controlled formation of pseudoisocyanine J-aggregates in the interlayer space of synthetic saponite. Appl. Clay Sci. 2017, 140, 119–123. [Google Scholar] [CrossRef]
- Kumaresan, S.; Rama Pawar, R.; Kevadiya, B.D.; Bajaj, H.C. Synthesis of Saponite Based Nanocomposites to Improve the Controlled Oral Drug Release of Model Drug Quinine Hydrochloride Dihydrate. Pharmaceuticals 2019, 12, 105. [Google Scholar]
- Vogels, R.J.M.J.; Kloprogge, J.T.; Geus, J.W. Catalytic activity of synthetic saponite clays: Effects of tetrahedral and octahedral composition. J. Catal. 2005, 231, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Kawi, S.; Yao, Y.Z. Saponite catalysts with systematically varied Mg/Ni ratio: Synthesis, characterization, and catalysis. Micropor. Mesopor. Mater. 1999, 33, 49–59. [Google Scholar] [CrossRef]
- Mata, G.; Trujillano, R.; Vicente, M.A.; Belver, C.; Fernandez-Garcia, M.; Korili, S.A.; Gil, A. Chromium-saponite clay catalysts: Preparation, characterization and catalytic performance in propene oxidation. Appl. Catal. A 2007, 327, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ménez, B.; Pisapia, C.; Andreani, M.; Jamme, F.; Vanbellingen, Q.P.; Brunelle, A.; Richard, L.; Dumas, P.; Réfrégiers, M. Abiotic synthesis of amino acids in the recesses of the oceanic lithosphere. Nature 2018, 564, 59–63. [Google Scholar] [CrossRef]
- Ponce, C.P.; Kloprogge, J.T. Urea-Assisted Synthesis and Characterization of Saponite with Different Octahedral (Mg, Zn, Ni, Co) and Tetrahedral Metals (Al, Ga, B), a Review. Life 2020, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Nityashree, N.; Price, C.A.H.; Pastor-Perez, L.; Manohara, G.V.; Garcia, S.; Maroto-Valer, M.M.; Reina, T.R. Carbon stabilised saponite supported transition metal-alloy catalysts for chemical CO2 utilisation via reverse water-gas shift reaction. Appl. Catal. B Environ. 2020, 261, 118241. [Google Scholar] [CrossRef]
- Gebretsadik, F.B.; Cesteros, Y.; Salagre, P.; Gimenez-Manogil, J.; Garcia-Garcia, A.; Bueno-Lopez, A. Potential of Cu-saponite catalysts for soot combustion. Catal. Sci. Technol. 2016, 6, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Minz, S.; Gupta, R.; Garg, S. Al–Fe and Al–Ti Pillared Saponite Clay Catalysts: Preparation and Characterization. In Proceedings of the Sustainable Engineering, Singapore, 5 April 2019; pp. 37–48. [Google Scholar]
- Vicente, I.; Salagre, P.; Cesteros, Y. Ni nanoparticles supported on microwave-synthesised saponite for the hydrogenation of styrene oxide. Appl. Clay Sci. 2011, 53, 212–219. [Google Scholar] [CrossRef]
- Besselink, R.; Stawski, T.M.; Freeman, H.M.; Hövelmann, J.; Tobler, D.J.; Benning, L.G. Mechanism of Saponite Crystallization from a Rapidly Formed Amorphous Intermediate. Cryst. Growth Design 2020, 20, 3365–3373. [Google Scholar] [CrossRef] [Green Version]
- Schumann, D.; Hartman, H.; Eberl, D.D.; Sears, S.K.; Hesse, R.; Vali, H. Formation of replicating saponite from a gel in the presence of oxalate: Implications for the formation of clay minerals in carbonaceous chondrites and the origin of life. Astrobiology 2012, 12, 549–561. [Google Scholar] [CrossRef]
- Vogels, R.J.M.J.; Kloprogge, J.T.; Geus, J.W. Synthesis and characterization of saponite clays. Am. Mineral. 2005, 90, 931–944. [Google Scholar] [CrossRef]
- Xue, S.; Pinnavaia, T.J. Porous synthetic smectic clay for the reinforcement of epoxy polymers. Micropor. Mesopor. Mater. 2008, 107, 134–140. [Google Scholar] [CrossRef]
- Sychev, M.; Prihod’ko, R. Characterization and catalytic activity of non-hydrothermally synthesized saponite-like materials. Stud. Surf. Sci. Catal. 1998, 118, 967–974. [Google Scholar]
- Bisio, C.; Gatti, G.; Boccaleri, E.; Marchese, L.; Superti, G.B.; Pastore, H.O.; Thommes, M. Understanding physico–chemical properties of saponite synthetic clays. Micropor. Mesopor. Mater. 2008, 107, 90–101. [Google Scholar] [CrossRef]
- Carniato, F.; Bisio, C.; Gatti, G.; Roncoroni, S.; Recchia, S.; Marchese, L. On the Properties of a Novel V-Containing Saponite Catalyst for Propene Oxidative Dehydrogenation. Catal. Lett. 2009, 131, 42–48. [Google Scholar] [CrossRef]
- Higashi, S.; Miki, H.; Komarneni, S. Mn-smectites: Hydrothermal synthesis and characterization. Appl. Clay Sci. 2007, 38, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Kloprogge, J.T.; Breukelaar, J.; Geus, J.W.; Jansen, J.B.H. Characterization of Mg-Saponites Synthesized from Gels Containing Amounts of Na+, K+, Rb+, Ca2+, Ba2+, or Ce4+ Equivalent to The CEC of the Saponite. Clays Clay Miner. 1994, 42, 18–22. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Breukelaar, J.; Jansen, J.B.H.; Geus, J.W. Development of Ammonium-Saponites from Gels with Variable Ammonium Concentration and Water Content at Low Temperatures. Clays Clay Miner. 1993, 41, 103–110. [Google Scholar] [CrossRef]
- Vogels, R.J.M.J.; Breukelaar, J.; Kloprogge, J.T.; Jansen, J.B.H.; Geus, J.W. Hydrothermal Crystallization of Ammonium-Saponite at 200 °C and Autogenous Water Pressure. Clays Clay Miner. 1997, 45, 1–7. [Google Scholar] [CrossRef]
- Gebretsadik, F.B.; Mance, D.; Baldus, M.; Salagre, P.; Cesteros, Y. Microwave synthesis of delaminated acid saponites using quaternary ammonium salt or polymer as template. Study of pH influence. Appl. Clay Sci. 2015, 114, 20–30. [Google Scholar] [CrossRef]
- Trujillano, R.; Rico, E.; Vicente, M.A.; Herrero, M.; Rives, V. Microwave radiation and mechanical grinding as new ways for preparation of saponite-like materials. Appl. Clay Sci. 2010, 48, 32–38. [Google Scholar] [CrossRef]
- Trujillano, R.; Rico, E.; Vicente, M.A.; Rives, V.; Ciuffi, K.J.; Cestari, A.; Gil, A.; Korili, S.A. Rapid microwave-assisted synthesis of saponites and their use as oxidation catalysts. Appl. Clay Sci. 2011, 53, 326–330. [Google Scholar] [CrossRef]
- Trujillano, R.; Rico, E.; Vicente, M.A.; Rives, V.; Sobrados, I.; Sanz, J. Saponites containing divalent transition metal cations in octahedral positions—Exploration of synthesis possibilities using microwave radiation and NMR characterization. Appl. Clay Sci. 2015, 115, 24–29. [Google Scholar] [CrossRef]
- Vicente, I.; Salagre, P.; Cesteros, Y.; Medina, F.; Sueiras, J.E. Microwave-assisted synthesis of saponite. Appl. Clay Sci. 2010, 48, 26–31. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Komarneni, S.; Amonette, J.E. Synthesis of Smectite Clay Minerals: A Critical Review. Clays Clay Miner. 1999, 47, 529–554. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Jansen, J.B.H.; Schuiling, R.D.; Geus, J.W. The interlayer collapse during dehydration of synthetic Na0.7-beidellite: A sodium-23 solid-state magic-angle spinning NMR study. Clays Clay Miner. 1992, 40, 561–566. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Frost, R.L. The effect of synthesis temperature on the FT-Raman and FT-IR spectra of saponites. Vibr. Spectrosc. 2000, 23, 119–127. [Google Scholar] [CrossRef]
- Vogels, R.J.M.J.; Kerkhoffs, M.J.H.V.; Geus, J.W. Non-hydrothermal synthesis, characterization and catalytic properties of saponite clays. Stud. Surf. Sci. Catal. 1995, 91, 1153–1161. [Google Scholar]
- Vogels, R.J.M.J.; Kloprogge, J.T.; Geus, J.W. Homogeneous forced hydrolysis of aluminum through the thermal decomposition of urea. J. Coll. Interf. Sci. 2005, 285, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Vogels, R.J.M.J.; Kloprogge, J.T.; Geus, J.W.; Beers, A.W.F. Synthesis and characterization of saponite clays: Part 2. Thermal stability. Am. Mineral. 2005, 90, 945–953. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Frost, R.L. Infrared emission spectroscopy of clay minerals. CMS Workshop Lect. 2005, 13, 99–124. [Google Scholar]
- Kloprogge, J.T.; Breukelaar, J.; Wilson, A.E.; Geus, J.W.; Jansen, J.B.H. Solid-state nuclear magnetic resonance spectroscopy on synthetic ammonium /aluminum-saponites. Clays Clay Miner. 1994, 42, 416–420. [Google Scholar] [CrossRef]
- Madejová, J.; Gates, W.P.; Petit, S. Chapter 5—IR Spectra of Clay Minerals. In Developments in Clay Science; Gates, W.P., Kloprogge, J.T., Madejová, J., Bergaya, F., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 8, pp. 107–149. [Google Scholar]
- Repacholi, M.H. Clay Mineralogy: Spectroscopic and Chemical Determinative Methods; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Russell, J.D.; Fraser, A. Infrared methods. In Clay Mineralogy: Spectroscopic and Chemical Determinative Methods; Wilson, M.J., Ed.; Chapman and Hall: London, UK, 1994; pp. 11–67. [Google Scholar]
- Pelletier, M.; Michot, L.J.; Barres, O.; Humbert, B.; Petit, S.; Robert, J.-L. Influence of KBr conditioning on the infrared hydroxyl-stretching region of saponites. Clay Miner. 1999, 34, 439–445. [Google Scholar] [CrossRef]
- Pelletier, M.; Michot, L.J.; Humbert, B.; Barres, O.; d’Espinose de la Caillerie, J.-B.; Robert, J.-L. Influence of layer charge on the hydroxyl stretching of trioctahedral clay minerals: A vibrational study of synthetic Na- and K-saponites. Am. Mineral. 2003, 88, 1801–1808. [Google Scholar] [CrossRef]
- Wang, A.; Jolliff, B.L.; Haskin, L.A. Raman spectroscopic characterization of a highly weathered basalt: Igneous mineralogy, alteration products, and a microorganism. J. Geophys. Res. Planets 1999, 104, 27067–27077. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Freeman, J.J.; Jolliff, B.L. Understanding the Raman spectral features of phyllosilicates. J. Raman Spectrosc. 2015, 46, 829–845. [Google Scholar] [CrossRef]
- Rinnert, E.; Carteret, C.; Humbert, B.; Fragneto-Cusani, G.; Ramsay, J.D.F.; Delville, A.; Robert, J.-L.; Bihannic, I.; Pelletier, M.; Michot, L.J. Hydration of a synthetic clay with tetrahedral charges: A multidisciplinary experimental and numerical study. J. Phys. Chem. B 2005, 109, 23745–23759. [Google Scholar] [CrossRef] [PubMed]
- Farmer, V.C.; Russell, J.D. Interlayer complexes in layer silicates. The structure of water in lamellar ionic solutions. Trans. Faraday Soc. 1971, 67, 2737–2749. [Google Scholar] [CrossRef]
- Farmer, V.C. The layer silicates. In The Infrared Spectra of Minerals; Farmer, V.C., Ed.; Mineralogical Society: London, UK, 1974; pp. 331–363. [Google Scholar]
- Loh, E. Optical vibrations in sheet silicates. J. Phys. C 1973, 6, 1091–1104. [Google Scholar] [CrossRef]
- Arab, M.; Bougeard, D.; Smirnov, K.S. Experimental and computer simulation study of the vibrational spectra of vermiculite. Phys. Chem. Chem. Phys. 2002, 4, 1957–1963. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Frost, R.L. Infrared emission spectroscopic study of the dehydroxylation of some natural and synthetic saponites. Neues Jahrb. Fuer Mineral. Mon. 2001, 446–463. [Google Scholar]
- Kloprogge, J.T. Raman spectroscopy of clay minerals. In Infrared and Raman Spectroscopies of Clay Minerals; Gates, W.P., Kloprogge, J.T., Madejova, J., Bergaya, F., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 8, pp. 150–199. [Google Scholar]
- Kloprogge, J.T.; Hammond, M.; Frost, R.L. Low temperature synthesis and characterisation of pecoraite, a nickel containing phyllosilicate. Neues Jahrb. Fuer Mineral. Mon. 2000, 5, 193–206. [Google Scholar]
- Kloprogge, J.T.; Hammond, M.; Hickey, L.; Frost, R.L. A new low temperature synthesis route of fraipontite (Zn,Al)3(Si,Al)2O5(OH)4. Mater. Res. Bull. 2001, 36, 1091–1098. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Hammond, M.; Hickey, L.; Frost, R.L. Low temperature synthesis of cobalt clays. J. Mater. Sci. Lett. 2002, 21, 931–933. [Google Scholar] [CrossRef] [Green Version]
- Kloprogge, J.T.; Frost, R.L.; Fry, R. Infrared emission study of the thermal transformation mechanism of Al13-pillared clay. Analyst 1999, 124, 381–384. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Jansen, J.B.H.; Geus, J.W. Characterization of synthetic sodium-beidellite. Clays Clay Miner. 1990, 38, 409–414. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Komarneni, S.; Yanagisawa, K.; Frost, R.L.; Fry, R. Infrared study of some synthetic and natural beidellites. J. Mater. Sci. Lett. 1998, 17, 1853–1855. [Google Scholar] [CrossRef]
- Van der Marel, H.W.; Beutelspacher, H. Atlas of Infrared Spectroscopy of Clay Minerals and Their Admixtures; Elsevier: Amsterdam, The Netherlands, 1974; p. 396. [Google Scholar]
- Velde, B. Infrared OH-stretch bands in potassic micas, talcs and saponites; influence of electronic configuration and site of charge compensation. Am. Mineral. 1983, 68, 1169–1173. [Google Scholar]
- Borade, R.B.; Clearfield, A. Characterization of acid sites in Beta and ZSM-20 zeolites. J. Phys. Chem. C 1992, 96, 6729–6737. [Google Scholar] [CrossRef]
- Baldermann, A.; Dohrmann, R.; Kaufhold, S.; Nickel, C.; Letofsky-Papst, I.; Dietzel, M. The Fe-Mg-saponite solid solution series—A hydrothermal synthesis study. Clay Miner. 2014, 49, 391–415. [Google Scholar] [CrossRef]
- Dill, H.G.; Dohrmann, R.; Kaufhold, S. Disseminated and faultbound autohydrothermal ferroan saponite in Late Paleozoic andesites of the Saar-Nahe Basin, SW Germany: Implications for the economic geology of intermediate (sub)volcanic rocks. Appl. Clay Sci. 2011, 51, 226–240. [Google Scholar] [CrossRef]
- Madejová, J. FTIR techniques in clay mineral studies. Vibr. Spectrosc. 2003, 31, 1–10. [Google Scholar] [CrossRef]
- Voncken, J.H.L.; Wevers, J.M.A.R.; van der Eerden, A.M.J.; Bos, A.M.J.; Jansen, J.B.H. Hydrothermal synthesis of tobelite, NH4Al2Si3AlO10(OH)2, from various starting materials and implications for its occurrence in nature. Geol. Mijnb. 1987, 66, 259–269. [Google Scholar]
- Kloprogge, J.T.; Frost, R.L. Infrared emission spectroscopy of Al-pillared beidellite. Appl. Clay Sci. 1999, 15, 431–445. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Frost, R.L. Infrared emission spectroscopic study of some natural and synthetic paragonites. Appl. Spectrosc. 1999, 53, 1071–1077. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Frost, R.L. An infrared emission spectroscopic study of synthetic and natural pyrophyllite. Neues Jahrb. Fuer Mineral. Mon. 1999, 2, 62–74. [Google Scholar]
- Kloprogge, J.T.; Frost, R.L.; Hickey, L. Infrared absorption and emission study of synthetic mica-montmorillonite in comparison to rectorite, beidellite and paragonite. J. Mater. Sci. Lett. 1999, 18, 1921–1923. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Frost, R.L.; Kristof, J. Application of infrared emission spectroscopy for the study of the dehydroxylation of synthetic paragonite. Can. J. Anal. Sci. Spectrosc. 1999, 44, 33–36. [Google Scholar]
- Kloprogge, J.T.; Fry, R.; Frost, R.L. An Infrared Emission Spectroscopic Study of the Thermal Transformation Mechanisms in Al13-Pillared Clay Catalysts with and without Tetrahedral Substitutions. J. Catal. 1999, 184, 157–171. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Komarneni, S.; Yanagisawa, K.; Fry, R.; Frost, R.L. Infrared Emission Spectroscopic Study of the Dehydroxylation via Surface Silanol Groups of Synthetic and Natural Beidellite. J. Coll. Interf. Sci. 1999, 212, 562–569. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Frost, R.L. Infrared emission spectroscopy study of the dehydroxylation of 10 Å halloysite from a Neogene cryptokarst of southern Belgium. Geol. Belg. 2000, 2, 213–220. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Hickey, L.; Frost, R.L. Heating stage Raman and infrared emission spectroscopic study of the dehydroxylation of synthetic Mg-hydrotalcite. Appl. Clay Sci. 2001, 18, 37–49. [Google Scholar] [CrossRef]
- Newman, A.C.D. Chemistry of Clay and Clay Minerals; Longman Scientific & Technical: Harlow, UK, 1987; p. 480. [Google Scholar]
- Frost, R.L.; Collins, B.M.; Finnie, K.; Vassallo, A.J. Infrared emission spectroscopy of clay minerals and their thermal transformations. In Proceedings of the 10th International Clay Conference, Adelaide, Australia, 18–23 July 1993; pp. 219–224. [Google Scholar]
- Kloprogge, J.T.; Frost, R.L.; Hickey, L. Infrared emission spectroscopic study of the dehydroxylation of some hectorites. Thermochim. Acta 2000, 345, 145–156. [Google Scholar] [CrossRef]
- Lipsicas, M.; Raythatha, R.H.; Pinnavaia, T.J.; Johnson, I.D.; Giese, R.F., Jr.; Costanzo, P.M.; Roberts, J.L. Silicon and aluminium site distributions in 2:1 layered silicate clays. Nature 1984, 309, 604–607. [Google Scholar] [CrossRef]
- Loewenstein, W. The distribution of aluminum in the tetrahedra of silicates and aluminates. Am. Mineral. 1954, 39, 92–96. [Google Scholar]
- Sanz, J.; Serratosa, J.M. 29Si and 27Al high resolution MAS-NMR spectra of phyllosilicates. J. Amer. Chem. Soc 1984, 106, 4790–4793. [Google Scholar] [CrossRef]
- Alma, N.C.M.; Hays, G.R.; Samoson, A.V.; Lippmaa, E.T. Characterization of synthetic dioctahedral clays by solid-state silicon-29 and aluminum-27 nuclear magnetic resonance spectrometry. Anal. Chem. 1984, 56, 729–733. [Google Scholar] [CrossRef]
- Weiss, C.A.; Altaner, S.P.; Kirkpatrick, R.J. High resolution 295i NMR spectroscopy of 2:1 layer silicates: Correlation among chemical shift, structural distortions, and chemical variations. Am. Mineral. 1987, 72, 935–942. [Google Scholar]
- Guggenheim, S. The brittle micas. In Micas; Bailey, S.W., Ed.; Mineralogical Society of America: Chantilly, VA, USA, 1984; Volume 13, pp. 61–104. [Google Scholar]
- Suquet, H.; Malard, C.; Copin, E.; Pezerat, H. Variation du parametre bet de la distance basale d001 dans une serie de saponites a charge croissante. I. Etats hydrates. Clay Miner. 1981, 16, 53–67. [Google Scholar] [CrossRef]
- Vogels, R.J.M.J.; Kloprogge, J.T.; Geus, J.W. Synthesis and characterization of boron and gallium substituted saponite clays below 100˚C at one atmosphere. Micropor. Mesopor. Mater. 2005, 77, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Bradley, S.M.; Howe, R.F.; Kydd, R.A. Correlation between 27Al and 71Ga NMR chemical shifts. Magn. Reson. Chem. 1993, 31, 883–886. [Google Scholar] [CrossRef]
- Van Olphen, H.; Fripiat, J.J. Data handbook for Clay Minerals and other Non-Metallic Materials; Pergamon Press: Oxford, UK, 1979; p. 346. [Google Scholar]
- Kogel, J.E.; Lewis, S.A. Baseline Studies of the Clay Minerals Society Source Clays: Chemical Analysis by Inductively Coupled Plasma-Mass Spectroscopy (ICP-MS). Clays Clay Miner. 2001, 49, 387–392. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Wood, B.J. Baseline studies of the Clay Minerals Society Source Clays by X-Ray Photoelectron Spectroscopy. Clay Sci. 2018, 22, 85–94. [Google Scholar]
- Kloprogge, J.T.; Wood, B.J. Handbook of Mineral Spectroscopy Volume 1. X-Ray Photoelectron Spectra; Elsevier: Amsterdam, The Netherlands, 2020; p. 505. [Google Scholar]
- Vantelon, D.; Belkhou, R.; Bihannic, I.; Michot, L.J.; Montarges-Pelletier, E.; Robert, J.-L. An XPEEM study of structural cation distribution in swelling clays. I. Synthetic trioctahedral smectites. Phys. Chem. Miner. 2009, 36, 593–602. [Google Scholar] [CrossRef]
- Barr, T.L.; Seal, S.; He, H.; Klinowski, J. X-ray photoelectron spectroscopic studies of kaolinite and montmorillonite. Vacuum 1995, 46, 1391–1395. [Google Scholar] [CrossRef]
- He, H.; Zhou, Q.; Frost, R.L.; Wood, B.J.; Duong, L.V.; Kloprogge, J.T. A X-ray photoelectron spectroscopy study of HDTMAB distribution within organoclays. Spectrochim. Acta A 2007, 66, 1180–1188. [Google Scholar] [CrossRef] [Green Version]
- Dove, M.T.; Thayaparam, S.; Heine, V.; Hammonds, K.D. The phenomenon of low Al-Si ordering temperatures in aluminosilicate framework structures. Am. Mineral. 1996, 81, 349–362. [Google Scholar] [CrossRef]
- Palin, E.J.; Dove, M.T.; Redfern, S.A.T.; Bosenick, A.; Sainz-Diaz, C.I.; Warren, M.C. Computational study of tetrahedral Al-Si ordering in muscovite. Phys. Chem. Miner. 2001, 28, 534–544. [Google Scholar] [CrossRef]
- Gonzales-Elipe, A.R.; Espinos, J.P.; Munuera, G.; Sanz, J.; Serratosa, J.M. Bonding-state characterization of constituent elements in phyllosilicate minerals by XPS and NMR. J. Phys. Chem. B 1988, 92, 3471–3476. [Google Scholar] [CrossRef]
- Ebina, T.; Iwasaki, T.; Chatterjee, A.; Katagiri, M.; Stucky, G.D. Comparative study of XPS and DFT with reference to the distribution of Al in tetrahedral and octahedral sheets of phyllosilicates. J. Phys. Chem. B 1997, 101, 1125–1129. [Google Scholar] [CrossRef]
- Kloprogge, J.T. Spectroscopic studies of synthetic and natural beidellites: A review. Appl. Clay Sci. 2006, 31, 165–179. [Google Scholar]
- Kloprogge, J.T.; Duong, L.V.; Wood, B.J.; Frost, R.L. XPS study of the major minerals in bauxite: Gibbsite, bayerite and (pseudo-)boehmite. J. Coll. Interf. Sci. 2006, 296, 572–576. [Google Scholar]
- Mosser, C.; Mosser, A.; Romeo, M.; Petit, S.; Decarreau, A. Natural and synthetic copper phyllosilicates studied by XPS. Clays Clay Miner. 1992, 40, 593–599. [Google Scholar] [CrossRef]
- He, H.; Barr, T.L.; Klinowski, J. ESCA and Solid-State NMR studies of allophane. Clay Miner. 1995, 30, 201–209. [Google Scholar] [CrossRef]
- Johns, W.D.; Gier, S. X-ray photoelectron spectroscopic study of layer-charge magnitude in micas and illite-smectite clays. Clay Miner. 2001, 36, 355–367. [Google Scholar] [CrossRef]
- Bhattacharyya, K.G. XPS study of mica surfaces. J. Electron Spectrosc. Rel. Phenom. 1993, 63, 289–306. [Google Scholar] [CrossRef]
- Liu, Z.H.; Brown, N.M.D. XPS characterization of mica surfaces processed using a radio-frequency (rf) argon plasma. J. Phys. D Appl. Phys. 1998, 31, 1771–1781. [Google Scholar] [CrossRef]
- Nessbitt, H.W.; Bancroft, G.M. High Resolution Core- and Valence-Level XPS Studies of the Properties (Structural, Chemical and Bonding) of Silicate Minerals and Glasses. In Spectroscopic Methods in Mineralogy and Materials Sciences; Henderson, G.S., Neuville, D.R., Downs, R.T., Eds.; Mineralogical Society of America: Chantilly, VA, USA, 2014; Volume 78, pp. 271–329. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raman | IR | IR | Assignment [34,52,53] |
|---|---|---|---|
| Milford Saponite | NH4-saponite | ||
| 202 | ν1(A1g) (Mg,Al)O6 | ||
| 229 | |||
| 265 | A1(ν1) OH-O triangle | ||
| 288 | SiO4 | ||
| 340 | ν2(e) SiO4 or SiO4 | ||
| 360 | ν5(e) SiO4, MgO6 | ||
| 424 | 422 | ? SiO4 | |
| 432 | ν3(a1) SiO4 or SiO4 | ||
| 447 | 449 | SiO4 | |
| 464 | 465 | 461 | ν6(e) SiO4 (R), Mg2Al-OH translation (IR) |
| 494–540 | 528 | 529 | SiO4 (R), Mg3-OH perpendicular (IR) |
| 659 | 654 | 665 | Mg3-OH libration |
| 683 | 696 | 691 | ν2(a1) SiO4 (R), SiO4 (IR) |
| 740 | Mg2Al-OH deformation | ||
| 776 | 787 | 779 | |
| 838 | 836 | δ(AlMgOH) (R), Al-O apical AIO4 (IR) | |
| 918 | 912 | δ(Al2OH), | |
| 998 | 1010 | 1020 | ν1(a1) SiO4 (R), SiO4 (IR) |
| 1050 | ν4(e) SiO4 | ||
| 1082 | 1110 | 1111 | ν3(f2) SiO4 (R), Si-O stretch (IR) |
| 1165 | Si-O stretch | ||
| 1430 | NH4 bend | ||
| 1635 | water OH-bend | ||
| 3118 | NH4 stretch | ||
| 3222 | 3218 | water OH stretch | |
| 3420 | 3431 | water OH stretch | |
| 3625 | 3589 | Mg2(Al,vac)-OH stretch | |
| 3674 | 3662 | Mg3-OH stretch | |
| 3778 | Si-OH stretch | ||
| NH4-Saponite | Natural Saponite | Tobellite | Assignment [60,66] |
|---|---|---|---|
| 2850 | NH4 stretch | ||
| 3118 | 3070 | NH4 stretch | |
| 3219 | 3222 | H-O-H stretch | |
| 3300 | NH4 stretch | ||
| 3432 | 3420 | H-O-H stretch | |
| 3590 | 3625 | 3630 | Al-OH |
| 3662 | 3680 | Mg-OH | |
| 3780 | Si-OH |
| Synthesis Temperature (°C) | Si/Al(IV) | Al(1Al) Fraction |
|---|---|---|
| 125 | 5.6 | 0.05 |
| 175 | 5.9 | 0.04 |
| 200 | 5.1 | 0.06 |
| 240 | 5.6 | 0.05 |
| 280 | 5.3 | 0.05 |
| Synthesis Temperature (°C) | θobserved | αobserved | bideal (Å) | bNMR (Å) | bXRD (Å) | ||
|---|---|---|---|---|---|---|---|
| Equation (4) | Equation (5) | Equation (4) | Equation (5) | ||||
| 125 | 120.68° | 11.21° | 9.261 | 9.212 | 9.084 | 9.036 | 9.175 |
| 175 | 120.03° | 10.56° | 9.254 | 9.208 | 9.097 | 9.052 | 9.175 |
| 200 | 121.16° | 10.69° | 9.270 | 9.220 | 9.078 | 9.029 | 9.184 |
| 240 | 119.71° | 10.24° | 9.261 | 9.217 | 9.113 | 9.070 | 9.174 |
| 280 | 119.39° | 9.92° | 9.267 | 9.219 | 9.128 | 9.081 | 9.188 |
| Element | XPS | Data Handbook/CCM |
|---|---|---|
| Si | 22.4 | 22.39 |
| Al | 2.4 | 2.21 |
| Mg | 16.4 | 15.74 |
| Fe | 0.46 | |
| Na | 3.6 | 2.02 |
| K | 0.5 | 0.32 |
| Ca | 0.5 | 0.64 |
| Cl | 2.2 | |
| Ti | 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kloprogge, J.T.; Ponce, C.P. Spectroscopic Studies of Synthetic and Natural Saponites: A Review. Minerals 2021, 11, 112. https://doi.org/10.3390/min11020112
Kloprogge JT, Ponce CP. Spectroscopic Studies of Synthetic and Natural Saponites: A Review. Minerals. 2021; 11(2):112. https://doi.org/10.3390/min11020112
Chicago/Turabian StyleKloprogge, J. Theo, and Concepcion P. Ponce. 2021. "Spectroscopic Studies of Synthetic and Natural Saponites: A Review" Minerals 11, no. 2: 112. https://doi.org/10.3390/min11020112