
Relative Contributions of Mg Hydration and Molecular Structural Restraints to the Barrier of Dolomite Crystallization: A Comparison of Aqueous and Non-Aqueous Crystallization in (BaMg)CO3 and (CaMg)CO3 Systems

Abstract
:1. Introduction
2. Methods
2.1. Crystallization Experiments
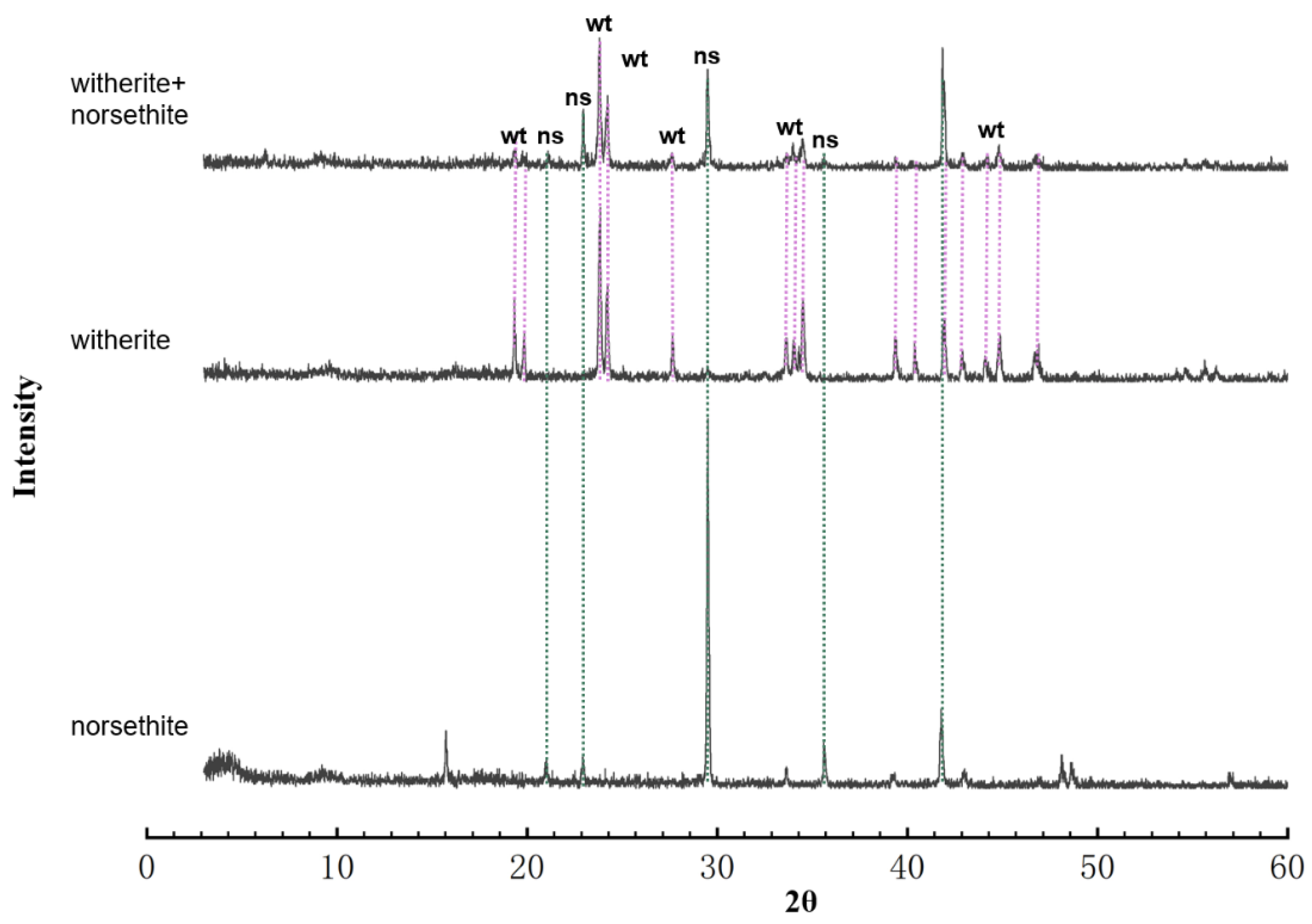
2.2. Precipitate Identification
3. Results
4. Discussion
4.1. Hydration Hindrance
4.2. Structural Restraints
4.3. Relative Effect of Mg Hydration and Structural Restraints
5. Summary
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sorby, H.C. XII.—On the cause of the production of different secondary forms of crystals. Mineral. Mag. J. Mineral. Soc. 1879, 3, 111–113. [Google Scholar] [CrossRef]
- Bragg, W. The analysis of crystals by the X-ray spectrometer. Proc. R. Soc. Lond. 1914, 89, 468–489. [Google Scholar]
- Wyckoff, R.W.G. The crystal structures of some carbonates of the calcite group. Am. J. Sci. 1920, 4, 317–360. [Google Scholar] [CrossRef]
- Lippmann, F. Crystal chemistry of sedimentary carbonate minerals. In Sedimentary Carbonate Minerals; Springer: Berlin/Heidelberg, Germany, 1973; pp. 5–96. [Google Scholar]
- Reeder, R.J.; Barber, D.J. Carbonates: Mineralogy and chemistry; Mineralogical Society of America: Washington, DC, USA, 1983; Volume 11. [Google Scholar]
- Plummer, L.; Wigley, T.; Parkhurst, D. The kinetics of calcite dissolution in CO2-water systems at 5° to 60 °C and 0.0 to 1.0 atm CO2. Am. J. Sci. 1978, 278, 179–216. [Google Scholar] [CrossRef]
- Busenberg, E.; Plummer, L.; Mumpton, F. A comparative study of the dissolution and crystal growth kinetics of calcite and aragonite. Stud. Diagenesis USGS Bull. 1986, 1578, 139–168. [Google Scholar]
- Teng, H.H.; Dove, P.M.; Orme, C.A.; De Yoreo, J. Thermodynamics of calcite growth: Baseline for understanding biomineral formation. Science 1998, 282, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Morse, J.W.; Arvidson, R.S.; Lüttge, A. Calcium carbonate formation and dissolution. Chem. Rev. 2007, 107, 342–381. [Google Scholar] [CrossRef]
- Urosevic, M.; Rodriguez-Navarro, C.; Putnis, C.V.; Cardell, C.; Putnis, A.; Ruiz-Agudo, E. In situ nanoscale observations of the dissolution of {1014} dolomite cleavage surfaces. Geochim. Cosmochim. Acta 2012, 80, 1–13. [Google Scholar] [CrossRef]
- Chang, B.; Li, C.; Liu, D.; Foster, I.; Tripati, A.; Lloyd, M.K.; Maradiaga, I.; Luo, G.; An, Z.; She, Z. Massive formation of early diagenetic dolomite in the Ediacaran ocean: Constraints on the “dolomite problem”. Proc. Natl. Acad. Sci. USA 2020, 117, 14005–14014. [Google Scholar] [CrossRef] [PubMed]
- Cusack, M.; Freer, A. Biomineralization: Elemental and organic influence in carbonate systems. Chem. Rev. 2008, 108, 4433–4454. [Google Scholar] [CrossRef] [PubMed]
- Raymo, M.E.; Ruddiman, W.F. Tectonic forcing of late Cenozoic climate. Nature 1992, 359, 117–122. [Google Scholar] [CrossRef]
- Royer, D.L.; Berner, R.A.; Park, J. Climate sensitivity constrained by CO2 concentrations over the past 420 million years. Nature 2007, 446, 530–532. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.W.; Kaufman, A.; Glusker, J.P. Coordination of water to magnesium cations. Inorg. Chem. 1994, 33, 419–427. [Google Scholar] [CrossRef]
- Zengler, D.; Dunham, J.; Ethington, R.L. Concepts and Models of Dolomitization; SEPM Society for Sedimentary Geology: Tulsa, OK, USA, 1980. [Google Scholar]
- Christ, C.; Hostetler, P. Studies in the system MgO-SiO2-CO2-H2O (II); the activity-product constant of magnesite. Am. J. Sci. 1970, 268, 439–453. [Google Scholar] [CrossRef]
- Sayles, F.; Fyfe, W. The crystallization of magnesite from aqueous solution. Geochim. Cosmochim. Acta 1973, 37, 87–99. [Google Scholar] [CrossRef]
- Deelman, J. Low-temperature synthesis of eitelite, Na2CO3·MgCO3. Neues Jahrbuch für Mineralogie. Monatshefte 1984, 10, 468–480. [Google Scholar]
- Deelman, J. Breaking Ostwald’s rule. Chem. Erde-Geochem. 2001, 61, 224–235. [Google Scholar]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A: Cryst. Phys. Diffr. Theor. Gen. Crystallogr. 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Pavlov, M.; Siegbahn, P.E.; Sandström, M. Hydration of beryllium, magnesium, calcium, and zinc ions using density functional theory. J. Phys. Chem. A 1998, 102, 219–228. [Google Scholar] [CrossRef]
- Markham, G.D.; Glusker, J.P.; Bock, C.W. The arrangement of first-and second-sphere water molecules in divalent magnesium complexes: Results from molecular orbital and density functional theory and from structural crystallography. J. Phys. Chem. B 2002, 106, 5118–5134. [Google Scholar] [CrossRef]
- Andersson, M.P.; Stipp, S.L. Predicting hydration energies for multivalent ions. J. Comput. Chem. 2014, 35, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Boero, M.; Terakura, K. Hydration properties of magnesium and calcium ions from constrained first principles molecular dynamics. J. Chem. Phys. 2007, 127, 074503. [Google Scholar] [CrossRef] [PubMed]
- Katz, A.K.; Glusker, J.P.; Beebe, S.A.; Bock, C.W. Calcium ion coordination: A comparison with that of beryllium, magnesium, and zinc. J. Am. Chem. Soc. 1996, 118, 5752–5763. [Google Scholar] [CrossRef]
- Bruni, F.; Imberti, S.; Mancinelli, R.; Ricci, M. Aqueous solutions of divalent chlorides: Ions hydration shell and water structure. J. Chem. Phys. 2012, 136, 064520. [Google Scholar] [CrossRef]
- Di Tommaso, D.; de Leeuw, N.H. First principles simulations of the structural and dynamical properties of hydrated metal ions Me2+ and solvated metal carbonates (Me=Ca, Mg, and Sr). Cryst. Growth Des. 2010, 10, 4292–4302. [Google Scholar] [CrossRef]
- Ingham, B.; Ko, M.; Laycock, N.; Kirby, N.M.; Williams, D.E. First stages of siderite crystallisation during CO2 corrosion of steel evaluated using in situ synchrotron small-and wide-angle X-ray scattering. Faraday Discuss. 2015, 180, 171–190. [Google Scholar] [CrossRef]
- Neuberg, C.; Rewald, B. Ueber kolloide und gelatinöse Verbindungen der Erdalkalien. Z. Für Chem. Und Ind. Der Kolloide 1908, 2, 354–357. [Google Scholar] [CrossRef]
- Forsgren, J.; Frykstrand, S.; Grandfield, K.; Mihranyan, A.; Strømme, M. A template-free, ultra-adsorbing, high surface area carbonate nanostructure. PLoS ONE 2013, 8, e68486. [Google Scholar] [CrossRef] [Green Version]
- Ende, M.; Effenberger, H.; Miletich, R. Evolution of the α-BaMg(CO3)2 low-temperature superstructure and the tricritical nature of its α–β phase transition. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2017, 73, 827–835. [Google Scholar] [CrossRef]
- Effenberger, H.; Pippinger, T.; Libowitzky, E.; Lengauer, C.; Miletich, R. Synthetic norsethite, BaMg (CO3)2: Revised crystal structure, thermal behaviour and displacive phase transition. Mineral. Mag. 2014, 78, 1589–1611. [Google Scholar] [CrossRef]
- Lippmann, F. Die Kristallstruktur des Norsethit, BaMg (CO3)2, im Vergleich zum Dolomit, CaMg (CO3)2. Naturwissenschaften 1967, 54, 514. [Google Scholar] [CrossRef]
- Lippmann, F. Syntheses of BaMg (CO3)2 (Norsethite) at 20 °C and the Formation of Dolomite in Sediments. In Recent Developments in Carbonate Sedimentology in Central Europe; Springer: Berlin/Heidelberg, Germany, 1968; pp. 33–37. [Google Scholar]
- Hood, W.C.; Steidl, P.F.; Tschopp, D.G. Precipitation of norsethite at room temperature. Am. Mineral. J. Earth Planet. Mater. 1974, 59, 471–474. [Google Scholar]
- Bötcher, M.E. Stable isotope fractionation during experimental formation of norsethite (BaMg[CO3]2): A mineral analogue of dolomite. Aquat. Geochem. 2000, 6, 201–212. [Google Scholar] [CrossRef]
- Pimentel, C.; Pina, C.M. The formation of the dolomite-analogue norsethite: Reaction pathway and cation ordering. Geochim. Cosmochim. Acta 2014, 142, 217–223. [Google Scholar] [CrossRef]
- Pimentel, C.; Pina, C.M. Reaction pathways towards the formation of dolomite-analogues at ambient conditions. Geochim. Cosmochim. Acta 2016, 178, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Lindner, M.; Saldi, G.D.; Carrocci, S.; Bénézeth, P.; Schott, J.; Jordan, G. On the growth of anhydrous Mg-bearing carbonates–Implications from norsethite growth kinetics. Geochim. Cosmochim. Acta 2018, 238, 424–437. [Google Scholar] [CrossRef]
- Liu, C.; Li, W. Transformation of amorphous precursor to crystalline carbonate: Insights from Mg isotopes in the dolomite-analogue mineral norsethite [BaMg (CO3)2]. Geochim. Cosmochim. Acta 2020, 272, 1–20. [Google Scholar] [CrossRef]
- Zhang, Y.-F.; Yao, Q.-Z.; Qian, F.-J.; Li, H.; Zhou, G.-T.; Fu, S.-Q. Formation pathway of norsethite dominated by solution chemistry under ambient conditions. Am. Mineral. J. Earth Planet. Mater. 2021, 106, 1306–1318. [Google Scholar] [CrossRef]
- Rock, P.A.; Mandell, G.K.; Casey, W.H.; Walling, E.M. Gibbs energy of formation of dolomite from electrochemical cell measurements and theoretical calculations. Am. J. Sci. 2001, 301, 103–111. [Google Scholar] [CrossRef]
- Bada, J.L.; Chalmers, J.H.; Cleaves, H.J. Is formamide a geochemically plausible prebiotic solvent? Phys. Chem. Chem. Phys. 2016, 18, 20085. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.; Ricketts, B. Chemical controls on the precipitation of mineral analogues of dolomite: The sulfate enigma. Geology 1986, 14, 408–410. [Google Scholar] [CrossRef]
- Longo, J.M.; Voight, K.C. Synthesis of mixed-metal carbonates by grinding. Solid State Ion. 1989, 32, 409–412. [Google Scholar] [CrossRef]
- Mercero, J.M.; Fowler, J.E.; Ugalde, J.M. Aluminum (III) interactions with the acid derivative amino acid chains. J. Phys. Chem. A 2000, 104, 7053–7060. [Google Scholar] [CrossRef]
- Peschke, M.; Blades, A.T.; Kebarle, P. Hydration energies and entropies for Mg2+, Ca2+, Sr2+, and Ba2+ from gas-phase ion-water molecule equilibria determinations. J. Phys. Chem. A 1998, 102, 9978–9985. [Google Scholar] [CrossRef]
- Grushka, E.; Grinberg, N. Advances in Chromatography; CRC Press: Boca Raton, FL, USA, 2006; Volume 45. [Google Scholar]
- Mergelsberg, S.T.; Yoreo, J.; Miller, Q.; Michel, F.M.; Dove, P.M. Metastable solubility and local structure of amorphous calcium carbonate (ACC). Geochim. Cosmochim. Acta 2020, 289. [Google Scholar] [CrossRef]
- Xu, J.; Yan, C.; Zhang, F.F.; Konishi, H.; Xu, H.F.; Teng, H.H. Testing the cation-hydration effect on the crystallization of Ca-Mg-CO3 systems. Proc. Natl. Acad. Sci. USA 2013, 110, 17750–17755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, M.; Xu, J.; Teng, H.H. Evolution of calcite growth morphology in the presence of magnesium: Implications for the dolomite problem. Geochim. Cosmochim. Acta 2016, 172, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Speer, J.A. Crystal chemistry and phase relations of orthorhombic carbonates. Rev. Mineral. Geochem. 1983, 11, 145–190. [Google Scholar]
- Reeder, R.J. Crystal chemistry of the rhombohedral carbonates. Rev. Mineral. Geochem. 1983, 11, 1–47. [Google Scholar]
- Speer, J.A. Crystal chemistry and phase relations of orthorhombic carbonates. In Carbonates; De Gruyter: Berlin, Germany, 2018; pp. 145–190. [Google Scholar]
- Lindner, M.; Jordan, G. On the growth of witherite and its replacement by the Mg-bearing double carbonate norsethite: Implications for the dolomite problem. Am. Mineral. 2018, 103, 252–259. [Google Scholar] [CrossRef]
- Lindner, M.; Saldi, G.D.; Jordan, G.; Schott, J. On the effect of aqueous barium on magnesite growth—A new route for the precipitation of the ordered anhydrous Mg-bearing double carbonate norsethite. Chem. Geol. 2017, 460, 93–105. [Google Scholar] [CrossRef]
- Graf, D.L.; Goldsmith, J.R. Some hydrothermal syntheses of dolomite and protodolomite. J. Geol. 1956, 64, 173–186. [Google Scholar] [CrossRef]
- Tribble, J.S.; Arvidson, R.S.; Lane III, M.; Mackenzie, F.T. Crystal chemistry, and thermodynamic and kinetic properties of calcite, dolomite, apatite, and biogenic silica: Applications to petrologic problems. Sediment. Geol. 1995, 95, 11–37. [Google Scholar] [CrossRef]
- Land, L.S. The Isotopic and Trace Element Geochemistry of Dolomite: The State of the Art; SEPM Society for Sedimentary Geology: Tulsa, OK, USA, 1980. [Google Scholar]
- Rosenberg, P.; Holland, H. Calcite-dolomite-magnesite stability relations in solutions at elevated temperatures. Science 1964, 145, 700–701. [Google Scholar] [CrossRef] [PubMed]
- Nancollas, G.H.; Reddy, M.M. The crystallization of calcium carbonate. II. Calcite growth mechanism. J. Colloid Interface Sci. 1971, 37, 824–830. [Google Scholar] [CrossRef]
- Wiechers, H.; Sturrock, P.; Marais, G. Calcium carbonate crystallization kinetics. Water Res. 1975, 9, 835–845. [Google Scholar] [CrossRef]
- Kazmierczak, T.; Tomson, M.; Nancollas, G. Crystal growth of calcium carbonate. A controlled composition kinetic study. J. Phys. Chem. 1982, 86, 103–107. [Google Scholar] [CrossRef]
- Arvidson, R.S.; Mackenzie, F.T. The dolomite problem; control of precipitation kinetics by temperature and saturation state. Am. J. Sci. 1999, 299, 257–288. [Google Scholar] [CrossRef] [Green Version]
- Arvidson, R.S.; Mackenzie, F.T. Tentative kinetic model for dolomite precipitation rate and its application to dolomite distribution. Aquat. Geochem. 1997, 2, 273–298. [Google Scholar] [CrossRef]
- Gregg, J.M.; Bish, D.L.; Kaczmarek, S.E.; Machel, H.G. Mineralogy, nucleation and growth of dolomite in the laboratory and sedimentary environment: A review. Sedimentology 2015, 62, 1749–1769. [Google Scholar] [CrossRef]
- Mucci, A.; Morse, J.W. The incorporation of Mg2+ and Sr2+ into calcite overgrowths: Influences of growth rate and solution composition. Geochim. Cosmochim. Acta 1983, 47, 217–233. [Google Scholar] [CrossRef]
- Mucci, A. Growth kinetics and composition of magnesian calcite overgrowths precipitated from seawater: Quantitative influence of orthophosphate ions. Geochim. Cosmochim. Acta 1986, 50, 2255–2265. [Google Scholar] [CrossRef]
- Hartley, G.; Mucci, A. The influence of PCO2 on the partitioning of magnesium in calcite overgrowths precipitated from artificial seawater at 25° and 1 atm total pressure. Geochim. Cosmochim. Acta 1996, 60, 315–324. [Google Scholar] [CrossRef]
- Huang, Y.; Fairchild, I.J. Partitioning of Sr2+ and Mg2+ into calcite under karst-analogue experimental conditions. Geochim. Cosmochim. Acta 2001, 65, 47–62. [Google Scholar] [CrossRef]
- Katz, A. The interaction of magnesium with calcite during crystal growth at 25–90 °C and one atmosphere. Geochim. Cosmochim. Acta 1973, 37, 1563–1586. [Google Scholar] [CrossRef]
- Berner, R. The role of magnesium in the crystal growth of calcite and aragonite from sea water. Geochim. Cosmochim. Acta 1975, 39, 489–504. [Google Scholar] [CrossRef]
- Oomori, T.; Kaneshima, H.; Maezato, Y.; Kitano, Y. Distribution coefficient of Mg2+ ions between calcite and solution at 10–50 °C. Mar. Chem. 1987, 20, 327–336. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| (Mg,Ba)Cl2 [M] | NaHCO3 [M] | Mg/Ba | SInrs | SIwit | aMg2+ [mM] | aBa2+ [mM] | aCO32− [mM] | Mineral Phase |
|---|---|---|---|---|---|---|---|---|
| 0.001 | 0.25 | 9:1 | 2.03 | 0.24 | 0.103 | 0.012 | 0.405 | N |
| 8:2 | 2.28 | 0.54 | 0.092 | 0.023 | 0.406 | N | ||
| 7:3 | 2.4 | 0.71 | 0.080 | 0.035 | 0.406 | N | ||
| 6:4 | 2.45 | 0.84 | 0.069 | 0.047 | 0.406 | W + N | ||
| 5:5 | 2.47 | 0.94 | 0.058 | 0.059 | 0.406 | W | ||
| 0.1 | 9:1 | 1.88 | 0.16 | 0.158 | 0.018 | 0.225 | N | |
| 8:2 | 2.13 | 0.47 | 0.140 | 0.036 | 0.225 | N | ||
| 7:3 | 2.25 | 0.64 | 0.123 | 0.054 | 0.225 | N + W | ||
| 6:4 | 2.31 | 0.77 | 0.105 | 0.072 | 0.226 | W | ||
| 5:5 | 2.33 | 0.87 | 0.088 | 0.089 | 0.226 | W | ||
| 0.05 | 9:1 | 1.66 | 0.05 | 0.205 | 0.023 | 0.135 | N | |
| 8:2 | 1.92 | 0.36 | 0.183 | 0.046 | 0.135 | N | ||
| 7:3 | 2.04 | 0.53 | 0.160 | 0.070 | 0.135 | W | ||
| 6:4 | 2.09 | 0.66 | 0.137 | 0.093 | 0.135 | W | ||
| 5:5 | 2.11 | 0.76 | 0.114 | 0.016 | 0.136 | W | ||
| 0.01 | 9:1 | 0.8 | −0.38 | 0.305 | 0.034 | 0.034 | N | |
| 8:2 | 1.05 | −0.08 | 0.271 | 0.068 | 0.034 | N | ||
| 7:3 | 1.17 | 0.1 | 0.237 | 0.102 | 0.034 | W | ||
| 6:4 | 1.24 | 0.23 | 0.203 | 0.136 | 0.034 | W | ||
| 5:5 | 1.26 | 0.33 | 0.169 | 0.170 | 0.034 | W | ||
| 0.005 | 9:1 | 0.3 | −0.63 | 0.334 | 0.037 | 0.017 | np | |
| 8:2 | 0.55 | −0.33 | 0.387 | 0.074 | 0.017 | np | ||
| 7:3 | 0.68 | −0.15 | 0.260 | 0.115 | 0.018 | np | ||
| 6:4 | 0.74 | −0.02 | 0.223 | 0.148 | 0.018 | np | ||
| 5:5 | 0.76 | 0.08 | 0.185 | 0.186 | 0.018 | np | ||
| 0.01 | 0.25 | 9:1 | 3.93 | 1.18 | 1.035 | 0.116 | 0.363 | N |
| 8:2 | 4.18 | 1.49 | 0.919 | 0.232 | 0.365 | N | ||
| 7:3 | 4.30 | 1.67 | 0.804 | 0.348 | 0.366 | N | ||
| 6:4 | 4.36 | 1.79 | 0.689 | 0.463 | 0.368 | N + W | ||
| 5:5 | 4.39 | 1.89 | 0.574 | 0.579 | 0.369 | W | ||
| 4:6 | 4.37 | 1.97 | 0.459 | 0.694 | 0.371 | W | ||
| 0.1 | 9:1 | 3.66 | 1.05 | 1.554 | 0.173 | 0.179 | N | |
| 8:2 | 3.92 | 1.35 | 1.380 | 0.345 | 0.180 | N + W | ||
| 7:3 | 4.04 | 1.53 | 1.207 | 0.517 | 0.182 | W + N | ||
| 6:4 | 4.11 | 1.66 | 1.033 | 0.671 | 0.183 | W | ||
| 5:5 | 4.13 | 1.76 | 0.861 | 0.862 | 0.185 | W | ||
| 4:6 | 4.12 | 1.84 | 0.688 | 1.032 | 0.186 | W | ||
| 0.05 | 9:1 | 3.31 | 0.87 | 1.954 | 0.215 | 0.096 | N + W | |
| 8:2 | 3.57 | 1.18 | 1.736 | 0.431 | 0.097 | W | ||
| 7:3 | 3.70 | 1.36 | 1.513 | 0.655 | 0.098 | W | ||
| 6:4 | 3.77 | 1.49 | 1.300 | 0.860 | 0.099 | W | ||
| 5:5 | 3.80 | 1.59 | 1.082 | 1.074 | 0.101 | W | ||
| 4:6 | 2.79 | 1.68 | 0.865 | 1.288 | 0.102 | W | ||
| 0.01 | 9:1 | 2.18 | 0.3 | 2.575 | 0.280 | 0.019 | W | |
| 8:2 | 2.44 | 0.61 | 2.288 | 0.560 | 0.020 | W | ||
| 7:3 | 2.57 | 0.79 | 2.001 | 0.840 | 0.20 | W | ||
| 6:4 | 2.65 | 0.93 | 1.715 | 1.120 | 0.21 | W | ||
| 5:5 | 2.68 | 1.03 | 1.429 | 1.400 | 0.021 | W | ||
| 4:6 | 2.66 | 1.12 | 1.143 | 1.680 | 0.022 | W | ||
| 0.005 | 9:1 | 1.61 | 0.02 | 2.694 | 0.293 | 0.010 | np | |
| 8:2 | 1.87 | 0.33 | 2.395 | 0.585 | 0.010 | np | ||
| 7:3 | 2.00 | 0.51 | 2.095 | 0.877 | 0.010 | np | ||
| 6:4 | 2.08 | 0.64 | 1.795 | 1.170 | 0.010 | W | ||
| 5:5 | 2.11 | 0.75 | 1.496 | 1.462 | 0.011 | W | ||
| 4:6 | 2.11 | 0.83 | 1.197 | 1.754 | 0.011 | W | ||
| 0.04 | 0.25 | 9:1 | 4.87 | 1.65 | 4.125 | 0.448 | 0.275 | N |
| 8:2 | 5.13 | 1.96 | 3.662 | 0.895 | 0.279 | N + W | ||
| 7:3 | 5.26 | 2.14 | 3.200 | 1.341 | 0.282 | W | ||
| 0.1 | 9:1 | 4.41 | 1.41 | 5.824 | 0.617 | 0.115 | N | |
| 8:2 | 4.67 | 1.72 | 5.171 | 1.233 | 0.117 | W + N | ||
| 7:3 | 4.8 | 1.90 | 4.518 | 1.847 | 0.119 | W | ||
| 0.05 | 9:1 | 3.93 | 1.17 | 6.843 | 0.715 | 0.057 | W | |
| 8:2 | 4.20 | 1.48 | 6.077 | 1.430 | 0.058 | W | ||
| 7:3 | 4.33 | 1.67 | 5.312 | 2.143 | 0.059 | W | ||
| 0.01 | 9:1 | 2.63 | 0.52 | 7.993 | 0.824 | 0.011 | W | |
| 8:2 | 2.90 | 0.83 | 7.104 | 1.648 | 0.013 | W | ||
| 7:3 | 3.04 | 1.02 | 6.215 | 2.471 | 0.016 | W | ||
| 0.005 | 9:1 | 2.03 | 0.22 | 8.166 | 0.840 | 0.005 | W | |
| 8:2 | 2.31 | 0.53 | 7.259 | 1.680 | 0.006 | W | ||
| 7:3 | 2.45 | 0.72 | 6.351 | 2.520 | 0.006 | W | ||
| 0.05 | 0.25 | 9:1 | 5.00 | 1.71 | 5.145 | 0.554 | 0.256 | N |
| 8:2 | 5.26 | 2.02 | 4.566 | 1.105 | 0.260 | N + W | ||
| 7:3 | 5.39 | 2.2 | 3.989 | 1.656 | 0.263 | W | ||
| 6:4 | 5.46 | 2.33 | 3.414 | 2.205 | 0.267 | W | ||
| 0.1 | 9:1 | 4.49 | 1.45 | 7.142 | 0.747 | 0.104 | W + N | |
| 8:2 | 4.76 | 1.76 | 6.340 | 1.493 | 0.107 | W | ||
| 7:3 | 4.89 | 1.95 | 5.540 | 2.236 | 0.109 | W | ||
| 6:4 | 4.97 | 2.08 | 4.742 | 2.978 | 0.111 | W | ||
| 0.05 | 9:1 | 4.00 | 1.2 | 8.277 | 0.854 | 0.051 | W | |
| 8:2 | 4.27 | 1.51 | 7.351 | 1.706 | 0.053 | W | ||
| 7:3 | 4.41 | 1.7 | 6.427 | 2.558 | 0.054 | W | ||
| 6:4 | 4.48 | 1.83 | 5.504 | 3.408 | 0.055 | W |
| Mineral | T(K) | ΔH≠ (kJmol−1) | ΔS≠ (JK−1mol−1) | ΔG≠ (kJmol−1) |
|---|---|---|---|---|
| calcite | 298 | 44.2 | −120.3 | 81.3 |
| norsethite | 298 | 77.5 | −18.1 | 82.9 |
| dolomite | 298 | 132.0 | 29.7 | 125.4 |
| Mg/Ba (Approximate Number) | T(K) | ΔH≠ (kJmol−1) | ΔS≠ (JK−1mol−1) | ΔG≠ (kJmol−1) |
|---|---|---|---|---|
| 10 | 298 | 95.8 | 39.0 | 84.1 |
| 20 | 298 | 77.6 | −9.0 | 80.3 |
| 40 | 298 | 70.1 | −29.6 | 78.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, S.; Wang, Y.; Teng, H. Relative Contributions of Mg Hydration and Molecular Structural Restraints to the Barrier of Dolomite Crystallization: A Comparison of Aqueous and Non-Aqueous Crystallization in (BaMg)CO3 and (CaMg)CO3 Systems. Minerals 2021, 11, 1214. https://doi.org/10.3390/min11111214
Zhou S, Wang Y, Teng H. Relative Contributions of Mg Hydration and Molecular Structural Restraints to the Barrier of Dolomite Crystallization: A Comparison of Aqueous and Non-Aqueous Crystallization in (BaMg)CO3 and (CaMg)CO3 Systems. Minerals. 2021; 11(11):1214. https://doi.org/10.3390/min11111214
Chicago/Turabian StyleZhou, Shi, Yuebo Wang, and Henry Teng. 2021. "Relative Contributions of Mg Hydration and Molecular Structural Restraints to the Barrier of Dolomite Crystallization: A Comparison of Aqueous and Non-Aqueous Crystallization in (BaMg)CO3 and (CaMg)CO3 Systems" Minerals 11, no. 11: 1214. https://doi.org/10.3390/min11111214