
Crystal Structure Refinements of the Lead(II) Oxoarsenates(V) Pb2As2O7, Pb(H2AsO4)2, Pb5(AsO4)3OH and NaPb4(AsO4)3 from Single-Crystal X-ray Data


Abstract
:1. Introduction
2. Materials and Methods
2.1. Syntheses and Single-Crystal Growth Procedures
2.2. Single-Crystal X-ray Diffraction and Structure Analysis
2.3. Powder X-ray Powder Diffraction (PXRD)
3. Results and Discussion
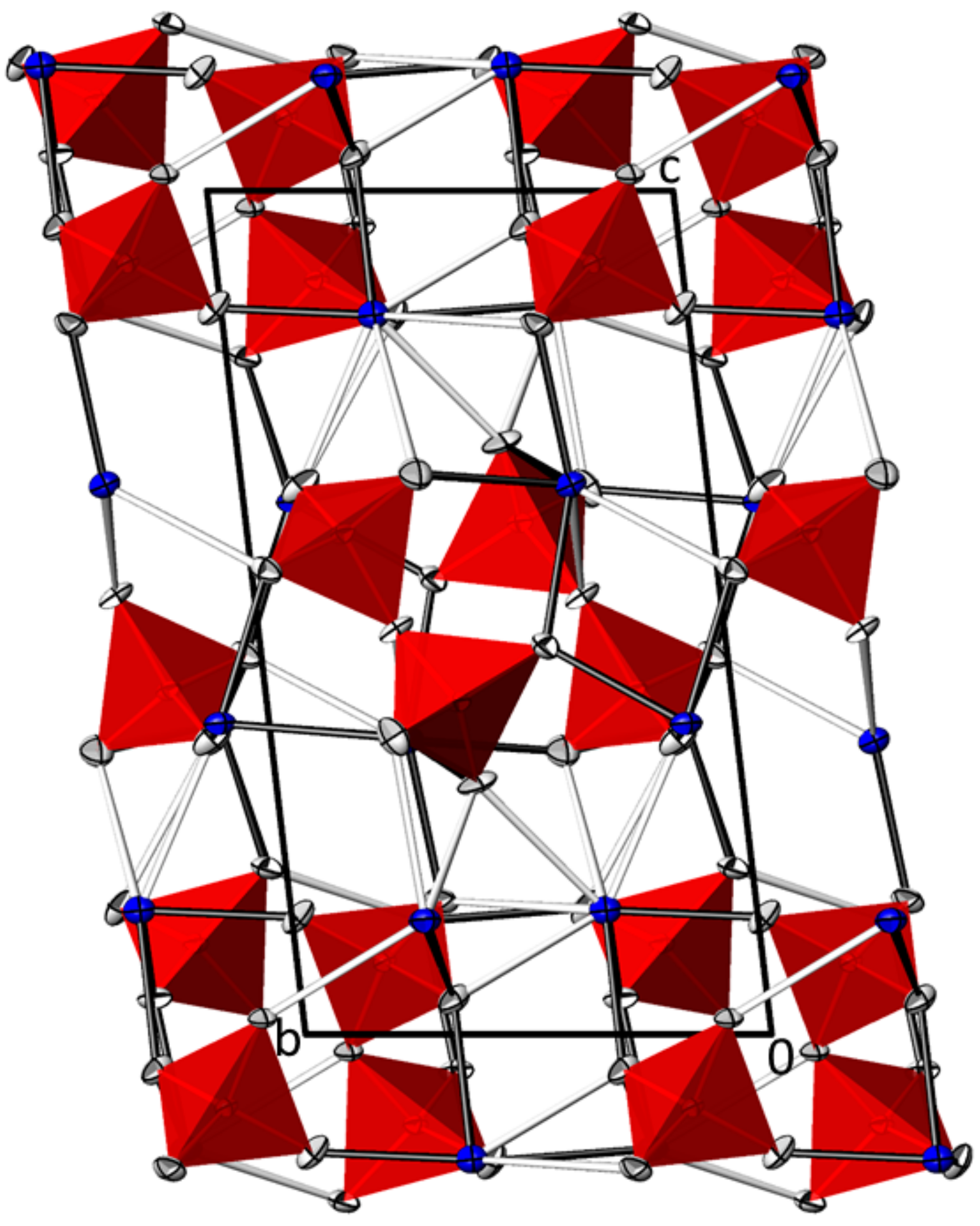
3.1. PbII2AsV2O7
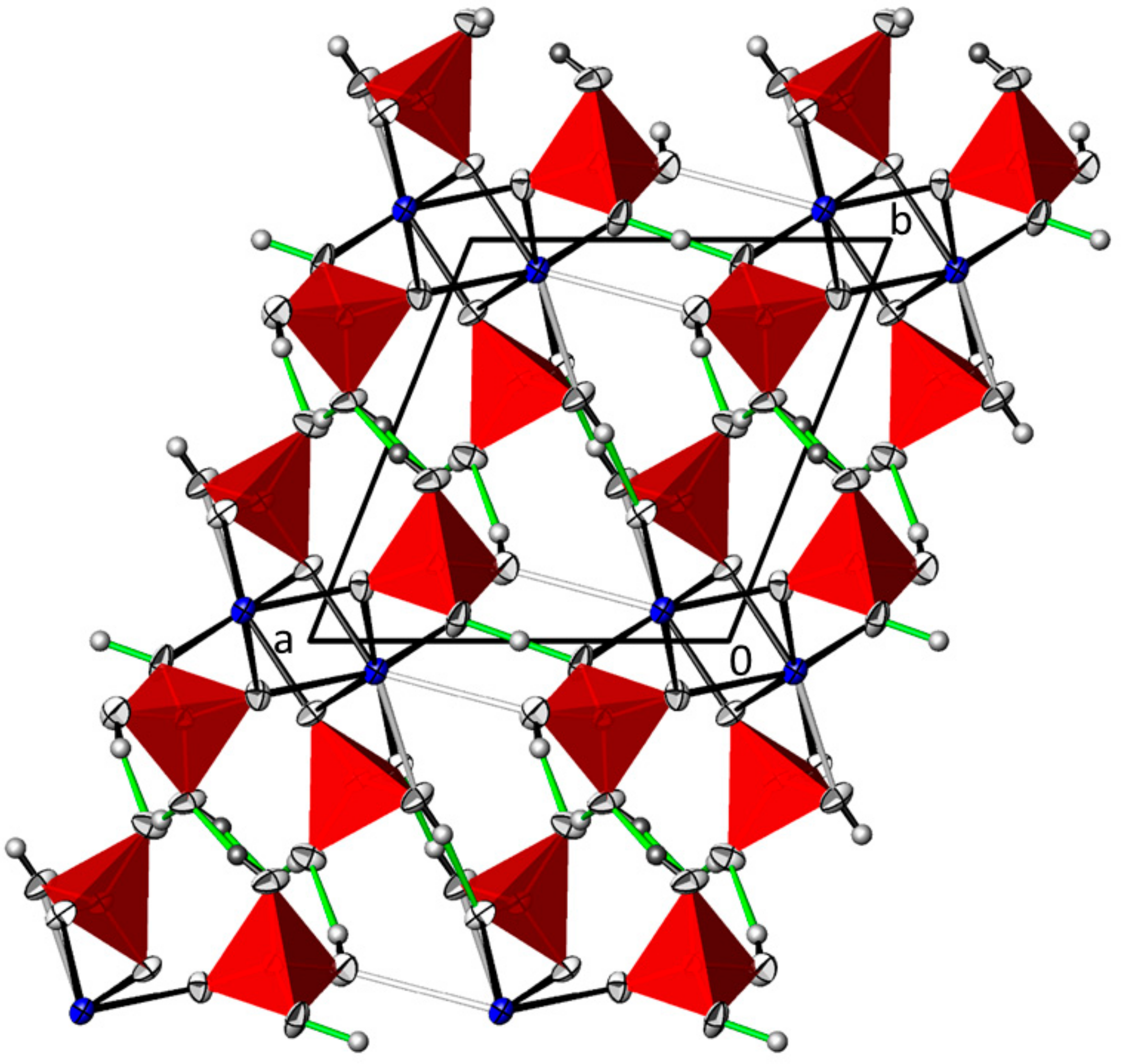
3.2. Pb(H2AsO4)2
3.3. Structural Comparison of Pb2As2O7 and Pb(H2AsO4)2 with Their Isotypic Ba and as Analogues
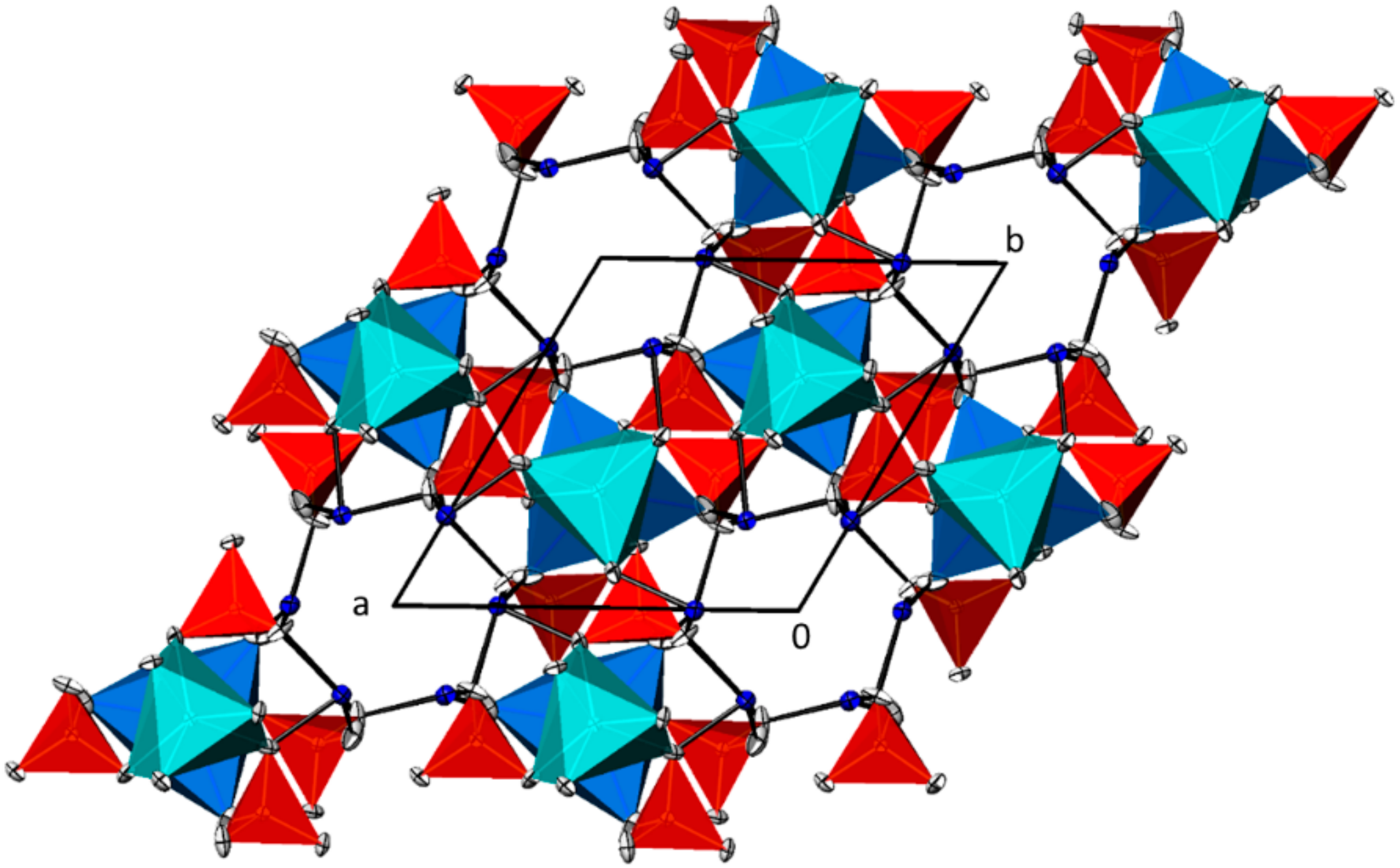
3.4. Pb5(AsO4)3OH
3.5. NaPb4(AsO4)3
3.6. Pb4As2O9
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Araki, T.; Moore, P.B.; Brunton, G.D. The crystal structure of paulmooreite, Pb2(As2O5): Dimeric arsenite groups. Am. Mineral. 1980, 65, 340–345. [Google Scholar]
- Effenberger, H.; Pertlik, F. Schultenit, PbHAsO4 und PbHPO4: Synthesen und Kristallstrukturen nebst einer Diskussion zur Symmetrie. Tschermaks Mineral. Petrogr. Mitt. 1986, 35, 157–166. [Google Scholar] [CrossRef]
- Dinterer, F.; Effenberger, H.; Kugler, A.; Pertlik, F.; Spindler, P.; Wildner, M. Structure of lead(II) arsenate(III). Acta Crystallogr. C 1988, 44, 2043–2045. [Google Scholar] [CrossRef]
- Viswanathan, K.; Miehe, G. The crystal structure of low temperature Pb3(AsO4)2. Zeitschrift für Kristallographie 1978, 148, 275–280. [Google Scholar] [CrossRef]
- Krivovichev, S.V.; Armbruster, T.; Depmeier, W. Crystal structures of Pb8O5(AsO4)2 and Pb5O4(CrO4) and review of (PbO)-related structural units in inorganic compounds. J. Solid State Chem. 2004, 177, 1321–1332. [Google Scholar] [CrossRef]
- Losilla, E.R.; Aranda, M.A.G.; Ramirez, F.J.; Bruque, S. Crystal structure and spectroscopic characterization of MAs2O6 (M = Pb, Ca). Two simple salts with AsO6 groups. J. Phys. Chem. 1995, 99, 12975–12979. [Google Scholar] [CrossRef]
- Losilla, E.R.; Salvado, M.A.; Aranda, M.A.G.; Cabeza, A.; Pertierra, P.; Garcia Granda, S.; Bruque, S. Layered acid arsenates α-M(HAsO4)2·(H2O) (M = Ti, Sn, Pb): Synthesis optimization and crystal structures. J. Mol. Struct. 1998, 470, 93–104. [Google Scholar] [CrossRef]
- Gmelins Handbuch der Anorganischen Chemie, 8. Auflage, System Nr. 47: Blei. Teil C.; Verlag Chemie: Weinheim, Germany, 1970; pp. 890–900.
- Kasenov, B.K.; Shashchanova, R.B.; Beilina, A.Z.; Malyshev, V.B. Phase equilibria in the As2O5–PbO System. Inorg. Mater. 1991, 27, 1236–1240. [Google Scholar]
- McDonnell, C.C.; Smith, C.M. The Arsenates of Lead. J. Am. Chem. Soc. 1916, 38, 2027–2038. [Google Scholar] [CrossRef] [Green Version]
- Degen, T.; Sadki, M.; Bron, E.; König, U.; Nenert, G. The HighScore suite. Powder Diff. 2014, 29 (Suppl. S2), S13–S18. [Google Scholar] [CrossRef] [Green Version]
- Bette, S.; Eggert, G.; Fischer, A.; Dinnebier, R.E. Glass-induced lead corrosion of heritage objects: Structural characterization of K(OH)·2PbCO3. Inorg. Chem. 2017, 56, 5762–5770. [Google Scholar] [CrossRef]
- Martinetto, P.; Anne, M.; Dooryhée, E.; Walter, P.; Tsoucaris, G. Synthetic hydrocerussite, 2PbCO3·Pb(OH)2, by X-ray powder diffraction. Acta Crystallogr. C 2002, 58, i82–i84. [Google Scholar] [CrossRef] [PubMed]
- Apex2 and SAINT; Bruker AXS Inc.: Madison, WI, USA, 2014.
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar]
- Mullica, D.F.; Perkins, H.O.; Grossie, D.A.; Boatner, L.A.; Sales, B.C. Structure of dichromate-type lead pyrophosphate Pb2P2O7. J. Solid State Chem. 1986, 62, 371–376. [Google Scholar] [CrossRef]
- Worzala, H.; Jost, K.H. On the mechanism of the conversion of lead dihydrogenphosphate, Pb(H2PO4)2, into lead monohydrogenphosphate, PbHPO4, and phosphoric acid. Z. Anorg. Allg. Chem. 1982, 486, 165–176. [Google Scholar] [CrossRef]
- Mighell, A.D. The reduced cell: Its use in the identification of crystalline materials. J. Appl. Crystallogr. 1976, 9, 491–498. [Google Scholar] [CrossRef]
- Coelho, A.A. TOPAS and TOPAS-Academic: An optimization program integrating computer algebra and crystallographic objects written in C++. J Appl. Crystallogr. 2018, 51, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Sordyl, J.; Puzio, B.; Manecki, M.; Borkiewicz, O.; Topolska, J.; Zelek-Pogudz, S. Structural assessment of fluorine, chlorine, bromine, iodine and hydroxide substitutions in lead arsenate apatites (Mimetites)–Pb5(AsO4)3X. Minerals 2020, 10, 494. [Google Scholar] [CrossRef]
- Nabar, M.A.; Dalvi, A.P. Crystal data for lead diarsenate [Pb2As2O7]. J. Appl. Crystallogr. 1978, 11, 162. [Google Scholar] [CrossRef]
- Brown, I.D.; Calvo, C. The crystal chemistry of large cation dichromates, pyrophosphates, and related compounds with stoichiometry X2Y2O7. J. Solid State Chem. 1970, 1, 173–179. [Google Scholar] [CrossRef]
- Clark, G.M.; Morley, R. Inorganic pyro-compounds Ma[(X2O7)b]. Chem. Soc. Rev. 1976, 5, 269–295. [Google Scholar] [CrossRef]
- Effenberger, H.; Pertlik, H. Crystal structures of Ag5Cu(AsO4)(As2O7) and Ag7Cu(As2O7)2Cl with a survey on pyroarsenate anions. Z. Kristallogr. 1993, 207, 223–236. [Google Scholar] [CrossRef]
- Weil, M.; Stöger, B. Crystal chemistry of transition metal diarsenates M2As2O7 (M = Mn, Co, Ni, Zn): Variants of the thortveitite structure. Acta Crystallogr. B 2010, 66, 603–614. [Google Scholar] [CrossRef]
- Weil, M. The isotypic family of the diarsenates MM’As2O7 (M = Sr, Ba; M’ = Cd, Hg). Z. Naturforsch. 2016, 71b, 403–409. [Google Scholar] [CrossRef]
- Boudin, S.; Grandin, A.; Borel, M.M.; Leclaire, A.; Raveau, B. Redetermination of the β-Ca2P2O7 structure. Acta Crystallogr. C 1993, 49, 2062–2064. [Google Scholar] [CrossRef]
- Weil, M. Insights into Formation conditions, crystal structures and thermal behavior of hydrous and anhydrous barium arsenates. Crystal Growth Des. 2016, 16, 908–921. [Google Scholar] [CrossRef]
- Dickens, B.; Prince, E.; Schroeder, L.W.; Brown, W.E. Ca(H2PO4)2, a crystal structure containing unusual hydrogen bonding. Acta Crystallogr. B 1973, 29, 2057–2070. [Google Scholar] [CrossRef]
- Vasić, P.; Prelesnik, B.; Herak, R.; Čurić, M. Structure of lead bis(dihydrogenphosphate). Acta Crystallogr. B 1981, B37, 660–662. [Google Scholar] [CrossRef]
- Arbib, E.; Elouadi, B.; Chaminade, J.-P. New refinement of lead bis(dihydrogenphosphate) Pb(H2PO4)2 and crystal chemistry of M(H2PO4)2 (M = Ca, Sr, Ba, Pb, Sn). Mat. Res. Bull. 1998, 33, 1483–1493. [Google Scholar] [CrossRef]
- Pollitt, S.; Weil, M. Polymorphism of H2SeO3, NaHSeO4 and Na5H3(SeO4)4(H2O)2, and Re-refinement of the crystal structure of Te2O4(OH)2. Z. Anorg. Allg. Chem. 2014, 640, 1622–1631. [Google Scholar] [CrossRef]
- Baran, J.; Lis, T.; Starynowicz, P. Structure and vibrational spectra of Na5H3(SeO4)4·2H2O crystal. J. Mol. Struct. 1989, 213, 51–61. [Google Scholar] [CrossRef]
- Kozlova, N.P.; Iskhakova, L.D.; Marugin, V.V.; Zhadanov, B.V.; Polyakova, I.A. Structure and thermal stability of the hydrogenselenate Na5H3(SeO4)4·2H2O. Zh. Neorg. Khim. 1990, 35, 1363–1368. [Google Scholar]
- Fukami, T.; Miyazaki, J.; Tomimura, T.; Chen, R.H. Crystal structures and isotope effect on Na5H3(SeO4)4·2H2O and Na5D3(SeO4)4·2H2O. Cryst. Res. Technol. 2010, 45, 856–862. [Google Scholar] [CrossRef]
- De la Flor, G.; Orobengoa, D.; Tasci, E.; Perez-Mato, J.M.; Aroyo, M.I. Comparison of structures applying the tools available at the Bilbao Crystallographic Server. J. Appl. Cryst. 2016, 49, 653–664. [Google Scholar] [CrossRef]
- Aroyo, M.I.; Perez-Mato, J.M.; Capillas, C.; Kroumova, E.; Ivantchev, S.; Madariaga, G.; Kirov, A.; Wondratschek, H. Bilbao crystallographic Server I: Databases and crystallographic computing programs. Zeitschrift für Kristallographie 2006, 221, 15–27. [Google Scholar] [CrossRef]
- Lima-de-Faria, J.; Hellner, E.; Liebau, F.; Makovicky, E.; Parthé, E. Nomenclature of inorganic structure types. Report of the international union of crystallography commission on crystallographic nomenclature subcommittee on the nomenclature of inorganic structure types. Acta Crystallogr. A 1990, A46, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Pasero, M.; Kampf, A.R.; Ferraris, C.; Pekov, I.V.; Rakovan, J.; White, T.J. Nomenclature of the apatite supergroup minerals. Eur. J. Mineral. 2010, 22, 163–179. [Google Scholar] [CrossRef]
- White, T.J.; ZhiLi, D. Structural derivation and crystal chemistry of apatites. Acta Crystallogr. B 2003, 59, 1–16. [Google Scholar] [CrossRef]
- Calos, N.J.; Kennard, C.H.L. Crystal structure of mimetite, Pb5(AsO4)3Cl. Zeitschrift für Kristallographie 1990, 191, 125–129. [Google Scholar] [CrossRef]
- Dai, Y.; Hughes, J.M.; Moore, P.B. The crystal structures of mimetite and clinomimetite, Pb5(AsO4)3Cl. Can. Mineral. 1991, 29, 369–376. [Google Scholar]
- Schachinger, T.; Kolitsch, U.; Bernhard, F.; Bojar, H.-P. Erzmineralisationen und ihre Verwitterungsprodukte aus dem weiteren Bereich der Steirischen und Lungauer Kalkspitze. Steir. Mineralog. 2014, 28, 8–21. [Google Scholar]
- Markl, G.; Marks, M.A.W.; Holzäpfel, J.; Wenzel, T. Major, minor and trace element composition of pyromorphite-group minerals as recorder of supergene weathering processes from the Schwarzwald mining district, SW Germany. Am. Mineral. 2014, 99, 1133–1146. [Google Scholar] [CrossRef]
- McDonnell, C.C.; Smith, C.M. The arsenates of lead III. Basic arsenates. J. Am. Chem. Soc. 1917, 39, 937–943. [Google Scholar] [CrossRef] [Green Version]
- Streeter, L.R.; Thatcher, R.W. Preparation of a basic arsenate of lead of definite composition. Ind. Eng. Chem. 1924, 16, 941–942. [Google Scholar] [CrossRef]
- Engel, G. Hydrothermalsynthese von Bleihydroxylapatiten Pb5(XO4)3OH mit X = P, As, V. Naturwissenschaften 1970, 57, 355. [Google Scholar] [CrossRef]
- Kwaśniak-Kominek, M.; Matusik, J.; Bajda, T.; Manecki, M.; Rakovan, J.; Marchlewski, T.; Szala, B. Fourier transform infrared spectroscopic study of hydroxylpyromorphite Pb10(PO4)6(OH)2–hydroxylmimetite Pb10(AsO4)6(OH)2 solid solution series. Polyhedron 2015, 99, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Wi, P.; Zeng, Y.Z.; Wang, C.M. Prediction of apatite lattice constants from their constituent elemental radii and artificial intelligence methods. Biomaterials 2004, 25, 1123–1130. [Google Scholar]
- Lim, S.C.; Baikie, T.; Pramana, S.S.; Smith, R.; White, T.J. Apatite metaprism twist angle (φ) as a tool for crystallochemical diagnosis. J. Solid State Chem. 2011, 184, 2978–2986. [Google Scholar] [CrossRef]
- Olds, T.A.; Kampf, A.R.; Rakovan, J.F.; Burns, P.C.; Mills, O.P.; Laughlin-Yurs, C. Hydroxylpyromorphite, a mineral important to lead remediation: Modern description and characterization. Am. Mineral. 2021, 106, 922–929. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Schwendtner, K.; Kolitsch, U. Three new acid M+ arsenates and phosphates with multiply protonated As/PO4 groups. Acta Crystallogr. C 2018, 75, 1134–1141. [Google Scholar] [CrossRef] [Green Version]
- Gagné, O.C.; Hawthorne, F.C. Bond-length distributions for ions bonded to oxygen: Metalloids and post-transition metals. Acta Crystallogr. B 2018, 74, 63–78. [Google Scholar] [CrossRef] [Green Version]
- Barinova, A.V.; Bonin, M.; Pushcharovskii, D.Y.; Rastsvetaeva, R.K.; Schenk, K.; Dimitrova, O.V. Crystal structure of synthetic hydroxylpyromorphite Pb5(PO4)3(OH). Crystallogr. Rep. 1998, 43, 189–192. [Google Scholar]
- Kim, J.Y.; Hunter, B.A.; Fenton, R.R.; Kennedy, B.J. Neutron powder diffraction study of lead hydroxyapatite. Austr. J. Chem. 1997, 50, 1061–1065. [Google Scholar] [CrossRef]
- Sudarsanan, K.; Young, R.A. Significant precision in crystal structural details: Holly springs hydroxyapatite. Acta Crystallogr. B 1969, 25, 1534–1543. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Stephens, P.W.; Tang, Y.; Li, W.; Phillips, B.L.; Parise, J.P.; Reeder, R.J. Arsenate substitution in hydroxylapatite: Structural characterization of the Ca5(PxAs1-xO3)4OH solid solution. Am. Mineral. 2009, 94, 666–675. [Google Scholar] [CrossRef]
- Sudarsanan, K.; Young, R.A. Structure of strontium hydroxide phosphate, Sr5(PO4)3OH. Acta Crystallogr. B 1972, 28, 3668–3670. [Google Scholar] [CrossRef]
- Weil, M.; Đorđević, T.; Lengauer, C.L.; Kolitsch, U. Investigations in the systems Sr–As–O–X (X = H, Cl): Preparation and crystal structure refinements of the anhydrous arsenates(V) Sr3(AsO4)2, Sr2As2O7, α- and β-SrAs2O6, and of the apatite-type phases Sr5(AsO4)3OH and Sr5(AsO4)3Cl. Solid State Sci. 2009, 11, 2111–2117. [Google Scholar] [CrossRef]
- Bondareva, O.S.; Malinovskii, Y.A. Crystal structure of synthetic Ba hydroxylapatite. Kristallografiya 1986, 31, 233–236. [Google Scholar]
- Hata, M.; Okada, K.; Iwai, S.I.; Akao, M.; Aoki, H. Cadmium hydroxyapatite. Acta Crystallogr. B 1978, 34, 3062–3064. [Google Scholar] [CrossRef]
- Mathew, M.; Brown, W.E.; Austin, M.; Negas, T. Lead alkali apatites without hexad anion: The crystal structure of Pb8K2(PO4)6. J. Solid State Chem. 1980, 35, 69–76. [Google Scholar] [CrossRef]
- Manoun, B.; Azdouz, M.; Azrour, M.; Essehli, R.; Benmokhtar, S.; El Ammari, L.; Ezzahi, A.; Ider, A.; Lazor, P. Synthesis, Rietveld refinements and Raman spectroscopic studies of tricationic lacunar apatites Na1−xKxPb4(AsO4)3 (0 ≤ x ≤ 1). J. Mol. Struct. 2011, 986, 1–9. [Google Scholar] [CrossRef]
- El Koumiri, M.; Oishi, S.; Sato, S.; El Ammari, L.; Elouadi, B. The crystal structure of the lacunar apatite NaPb4(PO4)3. Mat. Res. Bull. 2000, 35, 503–513. [Google Scholar] [CrossRef]
- Toumi, M.; Mhiri, T. Crystal structure and spectroscopic studies of Na2Pb8(PO4)6. J. Ceram. Soc. Jpn. 2008, 116, 904–906. [Google Scholar] [CrossRef] [Green Version]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Pb2(As2O7) | Pb(H2AsO4)2 | Pb5(AsO4)3OH | NaPb4(AsO4)3 | |
|---|---|---|---|---|---|
| Temperature/°C | — 23(1) — | ||||
| Diffractometer | — Bruker-AXS APEX-II CCD detector — | ||||
| Radiation; λ/Å | — Mo K; 0.71073 — | ||||
| Space group (# no.) | P (# 2) | P (# 2) | P63/m (# 176) | P (# 147) | |
| Formula units Z | 4 | 2 | 2 | 2 | |
| Formula weight | 676.22 | 489.06 | 1469.72 | 1268.51 | |
| Crystal dimensions/mm | 0.10 × 0.03 × 0.03 | 0.10 × 0.06 × 0.01 | 0.10 × 0.01 × 0.01 | 0.04 × 0.04 × 0.02 | |
| Crystal description | light-yellow fragment | colorless plate | light-yellow needle | colorless block | |
| a/Å | 7.13790(10) | 7.9497(10) | 10.1266(3) | 10.0230(14) | |
| b/Å | 7.14000(10) | 8.6137(10) | 10.1266(3) | 10.0230(14) | |
| c/Å | 13.0681(3) | 5.9984(8) | 7.5010(2) | 7.3117(15) | |
| α/° | 83.3602(11) | 108.888(5) | 90 | 90 | |
| β/° | 86.6710(10) | 96.128(5) | 90 | 90 | |
| γ/° | 89.9469(10) | 108.449(6) | 120 | 120 | |
| V/Å3 | 660.42(2) | 358.51(8) | 666.16(4) | 636.1(2) | |
| μ/mm − 1 | 60.851 | 32.693 | 70.429 | 60.606 | |
| X-ray density/g·cm–3 | 6.801 | 4.531 | 7.327 | 6.623 | |
| Range θmin–θmax | 1.57–40.18 | 2.69–36.73 | 2.32–30.00 | 2.35–30.28 | |
| Range | h | − 12 → 12 | − 13 → 13 | − 14 → 14 | − 14 → 14 |
| k | − 12 → 12 | − 14 → 14 | − 14 → 14 | − 14 → 14 | |
| l | −23 → 23 | − 10 → 10 | − 10 → 9 | − 10 → 10 | |
| Measured reflections | 70,283 | 24,488 | 9576 | 32,342 | |
| Independent reflections | 8240 | 3579 | 699 | 1279 | |
| Obs. reflections [I >2σ(I)] | 6951 | 3017 | 482 | 1207 | |
| Ri | 0.074 | 0.060 | 0.077 | 0.063 | |
| Transmis. coeff. Tmin; Tmax | 0.1644; 0.5697 | 0.3792; 0.7472 | 0.1107; 0.5411 | 0.2245; 0.6244 | |
| Structure solution and refinement | — Shelxs and Shelxl — | ||||
| Absorption correction | — Sadabs — | ||||
| Number of parameters | 200 | 114 | 40 | 64 | |
| Ext. coef. (Shelxl) | 0.00118(8) | 0.00095(16) | - | - | |
| Diff. elec. dens. max; min/e– Å–3 (dist./Å, atom) | 6.23 (0.61, Pb2); −5.53 (0.59, Pb1) | 1.47 (0.68, Pb1); − 1.24 (0.71, Pb1) | 5.60 (0.67, Pb2); −3.05 (0.76, Pb2) | 2.61 (0.43, (Pb/Na)3); − 1.69 (0.57, Pb1) | |
| R[F2 > 2σ(F2)] | 0.0346 | 0.0237 | 0.0380 | 0.0211 | |
| wR2(F2 all) | 0.0848 | 0.0403 | 0.0871 | 0.0452 | |
| Goof | 1.098 | 1.014 | 1.080 | 1.096 | |
| CSD number | 2099610 | 2099611 | 2099612 | 2099609 | |
| Pb2As2O7 | |||||
| Pb1 | O6 | 2.416(5) | Pb4 | O7 v | 2.463(4) |
| Pb1 | O2 i | 2.485(5) | Pb4 | O3 i | 2.479(5) |
| Pb1 | O10 | 2.521(5) | Pb4 | O7 | 2.514(5) |
| Pb1 | O3 | 2.563(5) | Pb4 | O5 ix | 2.523(5) |
| Pb1 | O1 ii | 2.673(5) | Pb4 | O14 | 2.673(5) |
| Pb1 | O7 iii | 2.878(5) | Pb4 | O2v | 2.695(5) |
| Pb1 | O4 ii | 2.965(5) | Pb4 | O6 | 3.140(5) |
| Pb1 | O14 iii | 3.319(5) | Pb4 | O12 | 3.226(5) |
| Pb1 | O5 iii | 3.464(5) | As1 | O1 | 1.657(4) |
| Pb2 | O13 iv | 2.417(4) | As1 | O3 | 1.669(4) |
| Pb2 | O12 i | 2.484(5) | As1 | O2 | 1.679(5) |
| Pb2 | O8 ii | 2.624(5) | As1 | O4 | 1.777(4) |
| Pb2 | O10 i | 2.631(5) | As2 | O5 | 1.649(5) |
| Pb2 | O14 v | 2.817(5) | As2 | O6 | 1.671(5) |
| Pb2 | O2 | 2.883(6) | As2 | O7 | 1.679(4) |
| Pb2 | O11 v | 2.903(4) | As2 | O4 | 1.760(4) |
| Pb2 | O1 | 2.987(5) | As3 | O9 | 1.649(4) |
| Pb2 | O9 ii | 3.369(5) | As3 | O10 | 1.674(4) |
| Pb3 | O9 vi | 2.441(5) | As3 | O8 | 1.683(4) |
| Pb3 | O13 | 2.546(5) | As3 | O11x | 1.760(4) |
| Pb3 | O8 vii | 2.555(5) | As4 | O14 | 1.651(4) |
| Pb3 | O12 viii | 2.642(5) | As4 | O13 | 1.669(4) |
| Pb3 | O8 | 2.753(5) | As4 | O12 | 1.672(4) |
| Pb3 | O5 | 2.871(6) | As4 | O11 | 1.778(4) |
| Pb3 | O1ii | 2.912(4) | |||
| Pb3 | O10 | 3.021(5) | |||
| Pb3 | O6 | 3.149(6) | |||
| Symmetry codes: (i) −x + 1, −y + 1, −z + 1; (ii) −x, −y + 1, −z + 1; (iii) x, y + 1, z; (iv) x, y, z + 1; (v) −x + 1, −y, −z + 1; (vi) x, y − 1, z; (vii) −x, −y + 1, −z; (viii) x − 1, y, z; (ix) x + 1, y, z; (x) −x + 1, −y + 1, −z. | |||||
| Pb(H2AsO4)2 | |||||
| Pb1 | O5 i | 2.442(2) | As1 | O2 | 1.652(2) |
| Pb1 | O1 ii | 2.529(2) | As1 | O1 | 1.657(2) |
| Pb1 | O2 | 2.559(2) | As1 | O4 vi | 1.719(2) |
| Pb1 | O5 iii | 2.575(2) | As1 | O3 vii | 1.727(2) |
| Pb1 | O1 iv | 2.584(2) | As2 | O5 | 1.665(2) |
| Pb1 | O7 v | 2.701(2) | As2 | O7 | 1.673(2) |
| Pb1 | O4 | 2.944(2) | As2 | O8 | 1.676(2) |
| Pb1 | O6 | 3.117(3) | As2 | O6 | 1.722(2) |
| Symmetry codes: (i) −x + 1, −y + 1, −z + 1; (ii) −x, −y + 1, −z + 1; (iii) x, y + 1, z; (iv) x, y, z + 1; (v) −x + 1, −y, −z + 1; (vi) x, y − 1, z; (vii) −x, −y + 1, −z; | |||||
| Pb5(AsO4)3(OH) | |||||
| Pb1 | O2 i | 2.534(8) 2.534 | Pb2 | O1 vi | 2.355(14) 2.398 |
| Pb1 | O2 ii | 2.534(8) | Pb2 | O3 vii | 2.616(9) 2.618 |
| Pb1 | O2 iii | 2.534(8) | Pb2 | O3 viii | 2.616(9) |
| Pb1 | O1 iv | 2.774(10) 2.754 | Pb2 | O3 iv | 2.664(11) 2.691 |
| Pb1 | O1 v | 2.774(10) | Pb2 | O3 v | 2.664(11) |
| Pb1 | O1 | 2.774(10) | Pb2 | O4 xi | 2.88(3) 2.693 |
| Pb1 | O3 v | 3.009(13) 2.944 | Pb2 | O4 xii | 2.88(3) |
| Pb1 | O3 iv | 3.009(13) | Pb2 | O2 vi | 2.937(12) 3.058 |
| Pb1 | O3 | 3.009(13) | |||
| As1 | O2 vi | 1.655(11) 1.674 | |||
| As1 | O3 xiii | 1.655(10) 1.691 | |||
| As1 | O3 | 1.655(10) | |||
| As1 | O1 | 1.681(13) 1.711 | |||
| Symmetry codes: (i) x − y, x, −z; (ii) −x + 1, −y + 1, −z; (iii) y, −x + y + 1, −z; (iv) −y + 1, x − y + 1, z; (v) −x + y, −x + 1, z; (vi) −y + 1, x − y, z; (vii) y, −x + y, z + 1/2; (viii) y, −x + y, −z; (xi) −x, −y, z − 1/2; (xii) −x, −y, −z + 1; (xiii) x, y, −z + 1/2. Values in italics are from the powder synchrotron study [22]. | |||||
| NaPb4(AsO4)3 | |||||
| (Na/Pb)1a | O2i | 2.428(7) | Pb2 | O1vi | 2.248(7) |
| (Na/Pb)1a | O2ii | 2.428(7) | Pb2 | O4ii | 2.433(7) |
| (Na/Pb)1a | O2iii | 2.428(7) | Pb2 | O4viii | 2.509(7) |
| (Na/Pb)1a | O1iv | 2.564(7) | Pb2 | O3xv | 2.568(7) |
| (Na/Pb)1a | O1v | 2.564(7) | Pb2 | O2ix | 2.805(6) |
| (Na/Pb)1a | O1vi | 2.564(7) | Pb2 | O3 | 2.912(8) |
| (Na/Pb)1a | O4vii | 3.202(8) | As1 | O2ix | 1.673(5) |
| (Na/Pb)1a | O4viii | 3.202(8) | As1 | O3xvii | 1.675(7) |
| (Na/Pb)1a | O4ix | 3.202(8) | As1 | O1xviii | 1.689(6) |
| (Na/Pb)1b | O2x | 2.435(7) | As1 | O4 | 1.699(7) |
| (Na/Pb)1b | O2xi | 2.435(7) | |||
| (Na/Pb)1b | O2xii | 2.435(7) | |||
| (Na/Pb)1b | O3xiii | 2.773(8) | |||
| (Na/Pb)1b | O3 | 2.773(8) | |||
| (Na/Pb)1b | O3xiv | 2.773(8) | |||
| (Na/Pb)1b | O1i | 3.070(8) | |||
| (Na/Pb)1b | O1v | 3.070(8) | |||
| (Na/Pb)1b | O1vi | 3.070(8) | |||
| Symmetry codes: (i) −x + 1, −y + 1, −z; (ii) x − y, x, −z; (iii) y, −x + y + 1, −z; (iv) y, −x + y, −z; (v) −x, −y + 1, −z; (vi) x − y + 1, x + 1, −z; (vii) x, y + 1, z; (viii) −y, x − y, z; (ix) −x + y + 1, −x + 1, z; (x) −x + 1, −y + 1, −z + 1; (xi) x − y, x, −z + 1; (xii) y, −x + y + 1, −z + 1; (xiii) −y + 1, x − y + 1, z; (xiv) −x + y, −x + 1, z; (xv) y, −x + y, −z + 1; (xvii) −x + y, −x, z; (xviii) −x, −y, −z. | |||||
| D–H···A | D–H | H···A | D···A | D–H···A |
|---|---|---|---|---|
| O3–H3···O8vi | 0.85 (1) | 1.81 (1) | 2.660 (3) | 176 (3) |
| O4–H4···O2ix | 0.85 (1) | 1.87 (2) | 2.685 (3) | 160 (4) |
| O6–H6···O3 | 0.85 (1) | 1.96 (1) | 2.805 (3) | 170 (5) |
| O7–H7···O7v | 1.23 (1) | 1.23 (1) | 2.450 (4) | 180 (1) |
| O8–H8···O8x | 0.85 (1) | 1.63 (2) | 2.458 (5) | 165 (7) |
| Symmetry codes: (v) −x + 1, −y, −z; (vi) x, y, z + 1; (ix) −x + 1, −y + 1, −z + 1; (x) −x + 2, −y + 1, −z + 1. | ||||
| Pb2As2O7 | Pb2P2O7 [18] | |u| | Ba2As2O7 [30] (1) | |u| |
| a = 7.13790, b = 7.14000, c = 13.06810 Å, α = 83.3602, β = 86.6710, γ = 89.9469° | a = 6.9140, b = 6.9660, c = 12.7510 Å, α = 83.180, β = 88.860, γ = 89.640° | a = 7.3996, b = 7.3812, c = 13.3261 Å, α = 83.116, β = 86.446, γ = 89.792° | ||
| Pb1 | Pb1 | 0.0609 | Ba2 | 0.2396 |
| O10 | O10 | 0.0699 | O7 | 0.1127 |
| Pb2 | Pb2 | 0.0709 | Ba3 | 0.0571 |
| As2 | P2 | 0.0751 | As2 | 0.0562 |
| O8 | O8 | 0.0792 | O5 | 0.0653 |
| As3 | P3 | 0.0845 | As4 | 0.0447 |
| O12 | O12 | 0.1220 | O14 | 0.2650 |
| O2 | O2 | 0.1266 | O3 | 0.0404 |
| O4 | O4 | 0.1332 | O4 | 0.0907 |
| Pb4 | Pb4 | 0.1367 | Ba1 | 0.1397 |
| As1 | P1 | 0.1390 | As1 | 0.0735 |
| O5 | O5 | 0.1459 | O2 | 0.0537 |
| O7 | O7 | 0.1561 | O11 | 0.1227 |
| As4 | P4 | 0.1721 | As3 | 0.1624 |
| O11 | O11 | 0.1756 | O13 | 0.1020 |
| Pb3 | Pb3 | 0.1780 | Ba4 | 0.1873 |
| O3 | O3 | 0.1803 | O9 | 0.1343 |
| O13 | O13 | 0.1873 | O6 | 0.2288 |
| O6 | O6 | 0.1977 | O8 | 0.1125 |
| O9 | O9 | 0.2333 | O12 | 0.1897 |
| O1 | O1 | 0.2390 | O10 | 0.0968 |
| O14 | O14 | 0.2918 | O1 | 0.1930 |
| S | 0.0189 | 0.0170 | ||
| dmax. (Å) | 0.2918 | 0.2650 | ||
| dav. (Å) | 0.1480 | 0.1258 | ||
| Δ | 0.041 | 0.043 | ||
| Pb(H2AsO4)2 | Pb(H2PO4)2 [19] | |u| | Ba(H2AsO4)2 [30] (1) | |u| |
| a = 7.9497, b = 8.6137, c = 5.9984 Å, α = 108.888, β = 96.128, γ = 108.449° | a = 7.823, b = 8.315, c = 5.856 Å, α = 108.24, β = 96.90, γ = 108.61° | a = 7.2453, b = 7.4341, c = 8.1890 Å, α = 104.685, β = 96.210, γ = 110.276° | ||
| Pb1 | Pb1 | 0.0082 | Ba1 | 0.9896 |
| As2 | P2 | 0.0125 | As2 | 0.5838 |
| As1 | P1 | 0.0226 | As1 | 0.4551 |
| O3 | O3 | 0.0767 | O3 | 0.7901 |
| O8 | O8 | 0.0795 | O8 | 0.9794 |
| O7 | O7 | 0.1049 | O7 | 1.9663 |
| O4 | O4 | 0.1160 | O2 | 0.7304 |
| O1 | O1 | 0.1284 | O4 | 0.8412 |
| O6 | O6 | 0.1309 | O5 | 1.7444 |
| O2 | O2 | 0.1328 | O1 | 0.4070 |
| O5 | O5 | 0.1340 | O6 | 0.6014 |
| S | 0.0151 | 0.1107 | ||
| dmax. (Å) | 0.1340 | 1.9663 | ||
| dav. (Å) | 0.0860 | 0.9172 | ||
| Δ | 0.047 | 0.703 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weil, M. Crystal Structure Refinements of the Lead(II) Oxoarsenates(V) Pb2As2O7, Pb(H2AsO4)2, Pb5(AsO4)3OH and NaPb4(AsO4)3 from Single-Crystal X-ray Data. Minerals 2021, 11, 1156. https://doi.org/10.3390/min11111156
Weil M. Crystal Structure Refinements of the Lead(II) Oxoarsenates(V) Pb2As2O7, Pb(H2AsO4)2, Pb5(AsO4)3OH and NaPb4(AsO4)3 from Single-Crystal X-ray Data. Minerals. 2021; 11(11):1156. https://doi.org/10.3390/min11111156
Chicago/Turabian StyleWeil, Matthias. 2021. "Crystal Structure Refinements of the Lead(II) Oxoarsenates(V) Pb2As2O7, Pb(H2AsO4)2, Pb5(AsO4)3OH and NaPb4(AsO4)3 from Single-Crystal X-ray Data" Minerals 11, no. 11: 1156. https://doi.org/10.3390/min11111156