Chemical Weathering of Granite in Ice and Its Implication for Weathering in Polar Regions


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Pretreatment and Batch Experiments
2.3. Chemical and Optical Analyses
3. Results
3.1. Filtrate Analysis
3.2. XRD Analysis and Amorphous Silica Formation
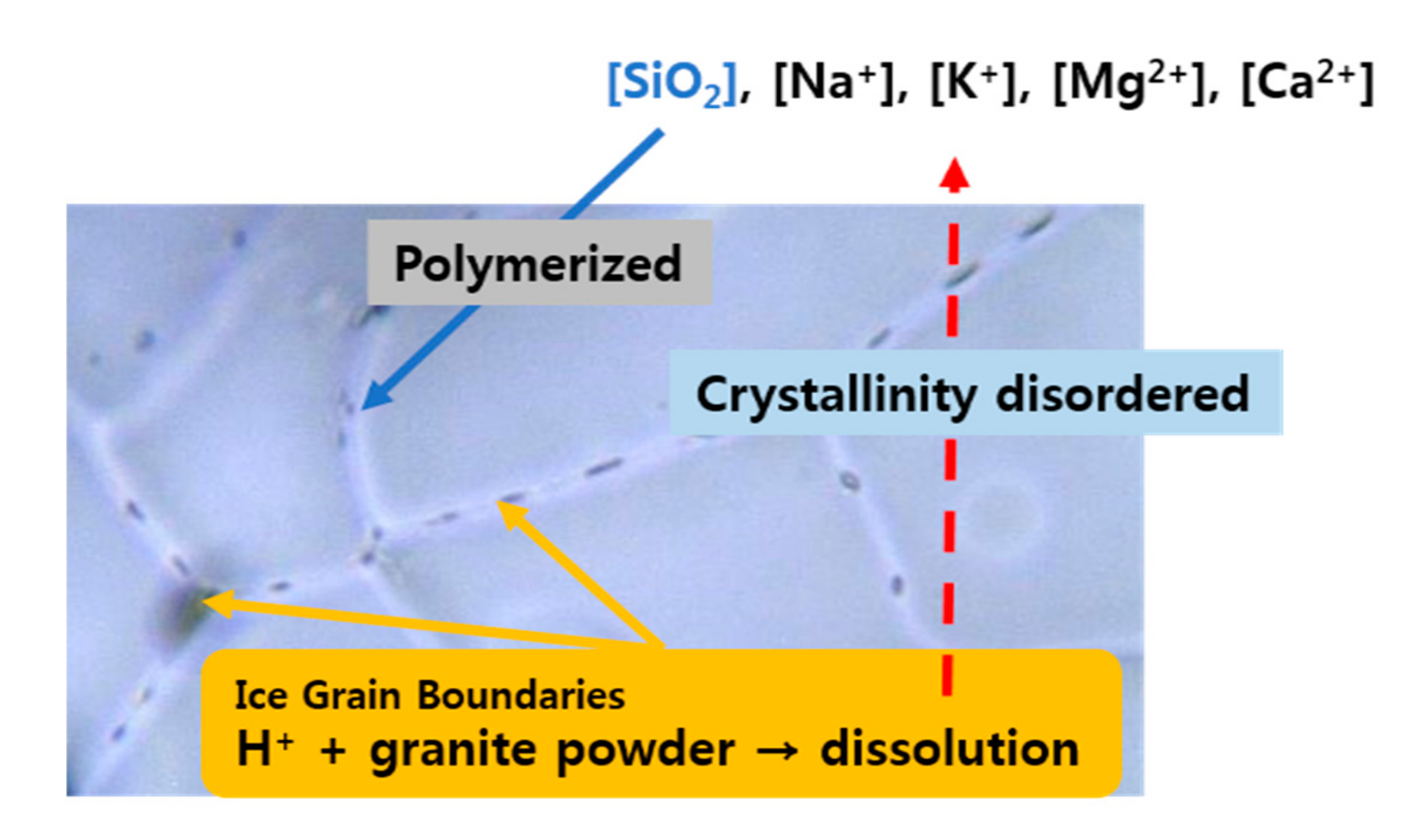
3.3. Optical Images of Ice Grain Boundaries
4. Discussion
4.1. The Mechanism behind the Dissolution of Granite in Ice
4.2. Silica Polymerization in Ice and Its Implication for Polar Regions
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berner, R.A. Weathering, plants, and the long-term carbon cycle. Geochim. Cosmochim. Acta 1992, 56, 3225–3231. [Google Scholar] [CrossRef]
- Schlesinger, W.H.; Bernhardt, E.S. Biogeochemistry: An Analysis of Global Change; Academic Press: New York, NY, USA, 2013. [Google Scholar]
- White, A.F.; Blum, A.E. Effects of climate on chemical_ weathering in watersheds. Geochim. Cosmochim. Acta 1995, 59, 1729–1747. [Google Scholar] [CrossRef]
- Gaillardet, J.; Dupre, B.; Louvat, P.; Allègre, C. Global silicate weathering and CO2 consumption rates deduced from the chemistry of large rivers. Chem. Geol. 1999, 159, 3–30. [Google Scholar] [CrossRef]
- Drever, J.; Stillings, L. The role of organic acids in mineral weathering. Colloids Surf. A Physicochem. Eng. Asp. 1997, 120, 167–181. [Google Scholar] [CrossRef]
- Bockheim, J.G. Properties and Classification of Cold Desert Soils from Antarctica. Soil Sci. Soc. Am. J. 1997, 61, 224–231. [Google Scholar] [CrossRef]
- Gooseff, M.N.; McKnight, D.; Lyons, W.B.; Blum, A.E. Weathering reactions and hyporheic exchange controls on stream water chemistry in a glacial meltwater stream in the McMurdo Dry Valleys. Water Resour. Res. 2002, 38. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.; Drever, J.I.; Humphrey, N.F. Chemical weathering in glacial environments. Geology 1997, 25, 399. [Google Scholar] [CrossRef]
- Nezat, C.A.; Lyons, W.B.; Welch, K. Chemical weathering in streams of a polar desert (Taylor Valley, Antarctica). GSA Bull. 2001, 113, 1401–1408. [Google Scholar] [CrossRef]
- Huh, Y. Chemical weathering and climate—A global experiment: A review. Geosci. J. 2003, 7, 277–288. [Google Scholar] [CrossRef]
- Bisson, K.; Welch, K.; Welch, S.; Sheets, J.M.; Lyons, W.B.; Levy, J.; Fountain, A.G. Patterns and Processes of Salt Efflorescences in the McMurdo region, Antarctica. Arct. Antarct. Alp. Res. 2015, 47, 407–425. [Google Scholar] [CrossRef] [Green Version]
- Maurice, P.A.; McKnight, D.; Leff, L.; Fulghum, J.E.; Gooseff, M.N. Direct observations of aluminosilicate weathering in the hyporheic zone of an Antarctic Dry Valley stream. Geochim. Cosmochim. Acta 2002, 66, 1335–1347. [Google Scholar] [CrossRef]
- Hall, K.; Thorn, C.E.; Matsuoka, N.; Prick, A. Weathering in cold regions: Some thoughts and perspectives. Prog. Phys. Geogr. Earth Environ. 2002, 26, 577–603. [Google Scholar] [CrossRef]
- Hall, K. Evidence for freeze–thaw events and their implications for rock weathering in northern Canada: II. The temperature at which water freezes in rock. Earth Surf. Process. Landf. 2007, 32, 249–259. [Google Scholar] [CrossRef]
- Kim, K.; Yoon, H.-I.; Choi, W. Enhanced Dissolution of Manganese Oxide in Ice Compared to Aqueous Phase under Illuminated and Dark Conditions. Environ. Sci. Technol. 2012, 46, 13160–13166. [Google Scholar] [CrossRef]
- Kim, K.; Choi, W.; Hoffmann, M.R.; Yoon, H.-I.; Park, B.-K. Photoreductive Dissolution of Iron Oxides Trapped in Ice and Its Environmental Implications. Environ. Sci. Technol. 2010, 44, 4142–4148. [Google Scholar] [CrossRef] [Green Version]
- Takenaka, N.; Ueda, A.; Daimon, T.; Bandow, H.; Dohmaru, T.; Maeda, Y. Acceleration Mechanism of Chemical Reaction by Freezing: The Reaction of Nitrous Acid with Dissolved Oxygen. J. Phys. Chem. 1996, 100, 13874–13884. [Google Scholar] [CrossRef]
- Kim, K.; Menacherry, S.P.M.; Kim, J.; Chung, H.Y.; Jeong, D.; Saiz-Lopez, A.; Choi, W. Simultaneous and Synergic Production of Bioavailable Iron and Reactive Iodine Species in Ice. Environ. Sci. Technol. 2019, 53, 7355–7362. [Google Scholar] [CrossRef]
- Bockheim, J.G. The Soils of Antarctica; Springer: New York, NY, USA, 2015. [Google Scholar]
- Coradin, T.; Eglin, D.; Livage, J. The silicomolybdic acid spectrophotometric method and its application to silicate/biopolymer interaction studies. J. Spectrosc. 2004, 18, 567–576. [Google Scholar] [CrossRef]
- Jung, J.; Yoo, K.-C.; Lee, K.-H.; Park, Y.K.; Lee, J.I.; Kim, J. Clay Mineralogical Characteristics of Sediments Deposited during the Late Quaternary in the Larsen Ice Shelf B Embayment, Antarctica. Minerals 2019, 9, 153. [Google Scholar] [CrossRef] [Green Version]
- Heger, D.; Klanova, J.; Klán, P. Enhanced Protonation of Cresol Red in Acidic Aqueous Solutions Caused by Freezing. J. Phys. Chem. B 2006, 110, 1277–1287. [Google Scholar] [CrossRef]
- Furrer, G.; Stumm, W. The coordination chemistry of weathering: I. Dissolution kinetics of δ-Al2O3 and BeO. Geochim. Cosmochim. Acta 1986, 50, 1847–1860. [Google Scholar] [CrossRef]
- Anderson, S. Biogeochemistry of Glacial Landscape Systems. Annu. Rev. Earth Planet. Sci. 2007, 35, 375–399. [Google Scholar] [CrossRef]
- Burton, J.D.; Leatherland, T.M.; Liss, P.S. The Reactivity of Dissolved Silicon in Some Natural Waters. Limnol. Oceanogr. 1970, 15, 473–476. [Google Scholar] [CrossRef]
- Macdonald, R.; McLaughlin, F. The effect of storage by freezing on dissolved inorganic phosphate, nitrate and reactive silicate for samples from coastal and estuarine waters. Water Res. 1982, 16, 95–104. [Google Scholar] [CrossRef]
- Bates, S.; Zografi, G.; Engers, D.; Morris, K.; Crowley, K.; Newman, A.W. Analysis of Amorphous and Nanocrystalline Solids from Their X-Ray Diffraction Patterns. Pharm. Res. 2006, 23, 2333–2349. [Google Scholar] [CrossRef]
- Biswas, R.K.; Khan, P.; Mukherjee, S.; Mukhopadhyay, A.K.; Ghosh, J.; Muraleedharan, K. Study of short range structure of amorphous Silica from PDF using Ag radiation in laboratory XRD system, RAMAN and NEXAFS. J. Non-Crystalline Solids 2018, 488, 1–9. [Google Scholar] [CrossRef]
- Hartmann, J.; West, A.J.; Renforth, P.; Köhler, P.; De La Rocha, C.L.; Dürr, H.; Scheffran, J.; Wolf-Gladrow, D.A. Enhanced chemical weathering as a geoengineering strategy to reduce atmospheric carbon dioxide, supply nutrients, and mitigate ocean acidification. Rev. Geophys. 2013, 51, 113–149. [Google Scholar] [CrossRef] [Green Version]
- Hawkings, J.; Wadham, J.L.; Benning, L.G.; Hendry, K.R.; Tranter, M.; Tedstone, A.J.; Nienow, P.; Raiswell, R. Ice sheets as a missing source of silica to the polar oceans. Nat. Commun. 2017, 8, 14198. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxide Composition | SiO2 | Al2O3 | K2O | Na2O | Fe2O3 | CaO | MgO | TiO2 | MnO |
|---|---|---|---|---|---|---|---|---|---|
| Weight Percentage (%) | 72.50 | 13.59 | 7.56 | 1.83 | 1.65 | 1.04 | 0.58 | 0.37 | 0.10 |
| DW (aq) | DW (ice) | pH 3 (aq) | pH 3 (ice) | pH 2 (aq) | pH 2 (ice) | |
|---|---|---|---|---|---|---|
| [Na+] | 0.27 | n.a. | 0.68 | 0.39 | 1.19 | 0.46 |
| [K+] | 1.21 | 0.01 | 3.44 | 2.61 | 9.63 | 4.22 |
| [Mg2+] | 0.18 | n.a. | 1.17 | 0.98 | 7.50 | 3.34 |
| [Ca2+] | 2.65 | 0.09 | 5.59 | 4.56 | 13.45 | 9.43 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, H.Y.; Jung, J.; Lee, D.H.; Kim, S.; Lee, M.K.; Lee, J.I.; Yoo, K.-C.; Lee, Y.I.; Kim, K. Chemical Weathering of Granite in Ice and Its Implication for Weathering in Polar Regions. Minerals 2020, 10, 185. https://doi.org/10.3390/min10020185
Chung HY, Jung J, Lee DH, Kim S, Lee MK, Lee JI, Yoo K-C, Lee YI, Kim K. Chemical Weathering of Granite in Ice and Its Implication for Weathering in Polar Regions. Minerals. 2020; 10(2):185. https://doi.org/10.3390/min10020185
Chicago/Turabian StyleChung, Hyun Young, Jaewoo Jung, Du Hyeong Lee, Sunghan Kim, Min Kyung Lee, Jae Il Lee, Kyu-Cheul Yoo, Yong Il Lee, and Kitae Kim. 2020. "Chemical Weathering of Granite in Ice and Its Implication for Weathering in Polar Regions" Minerals 10, no. 2: 185. https://doi.org/10.3390/min10020185