
Synthesis and Structural Elucidation of P-stereogenic Coumarins


Abstract
:1. Introduction
2. Materials and Methods
2.1. Instrumentation
2.1.1. General
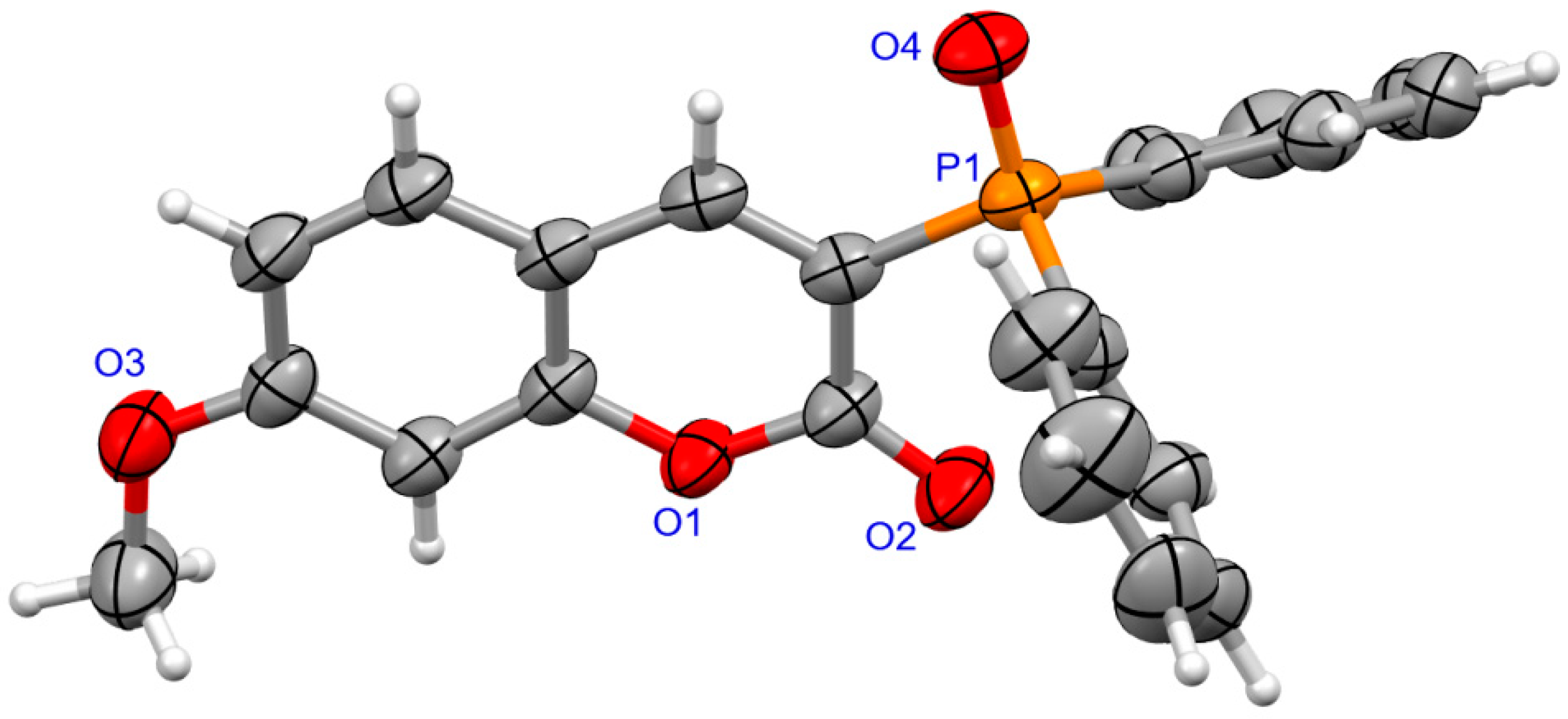
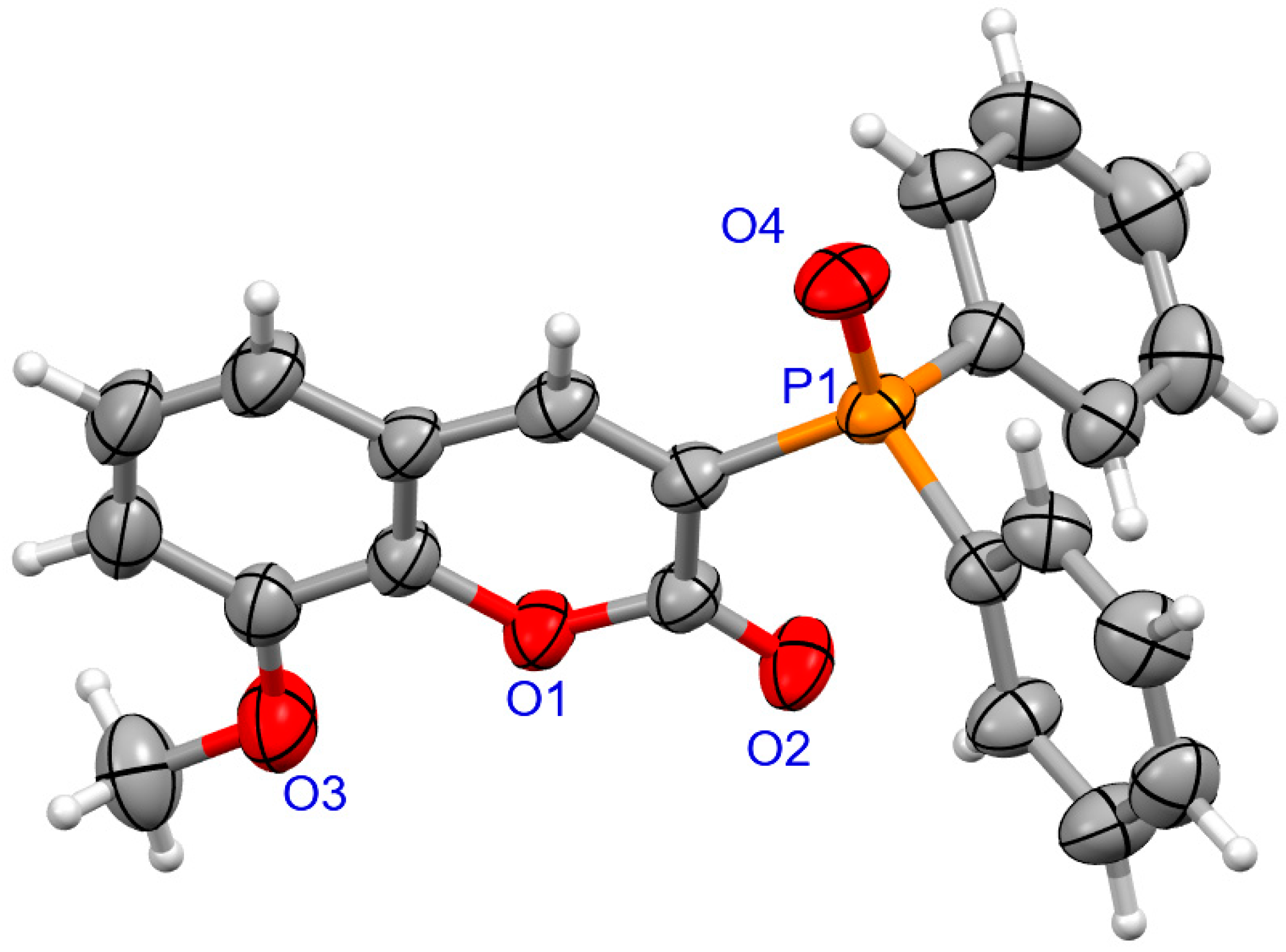
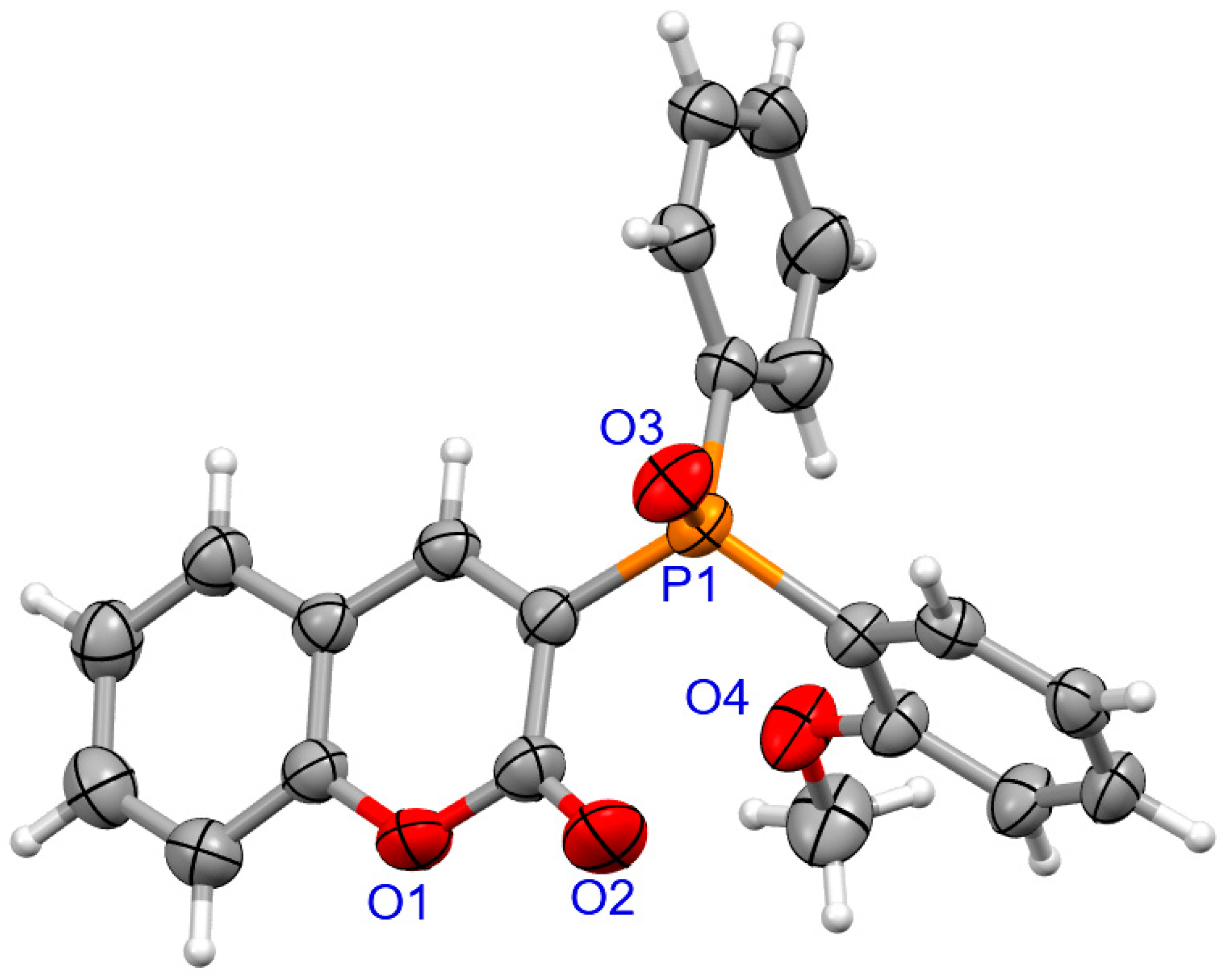
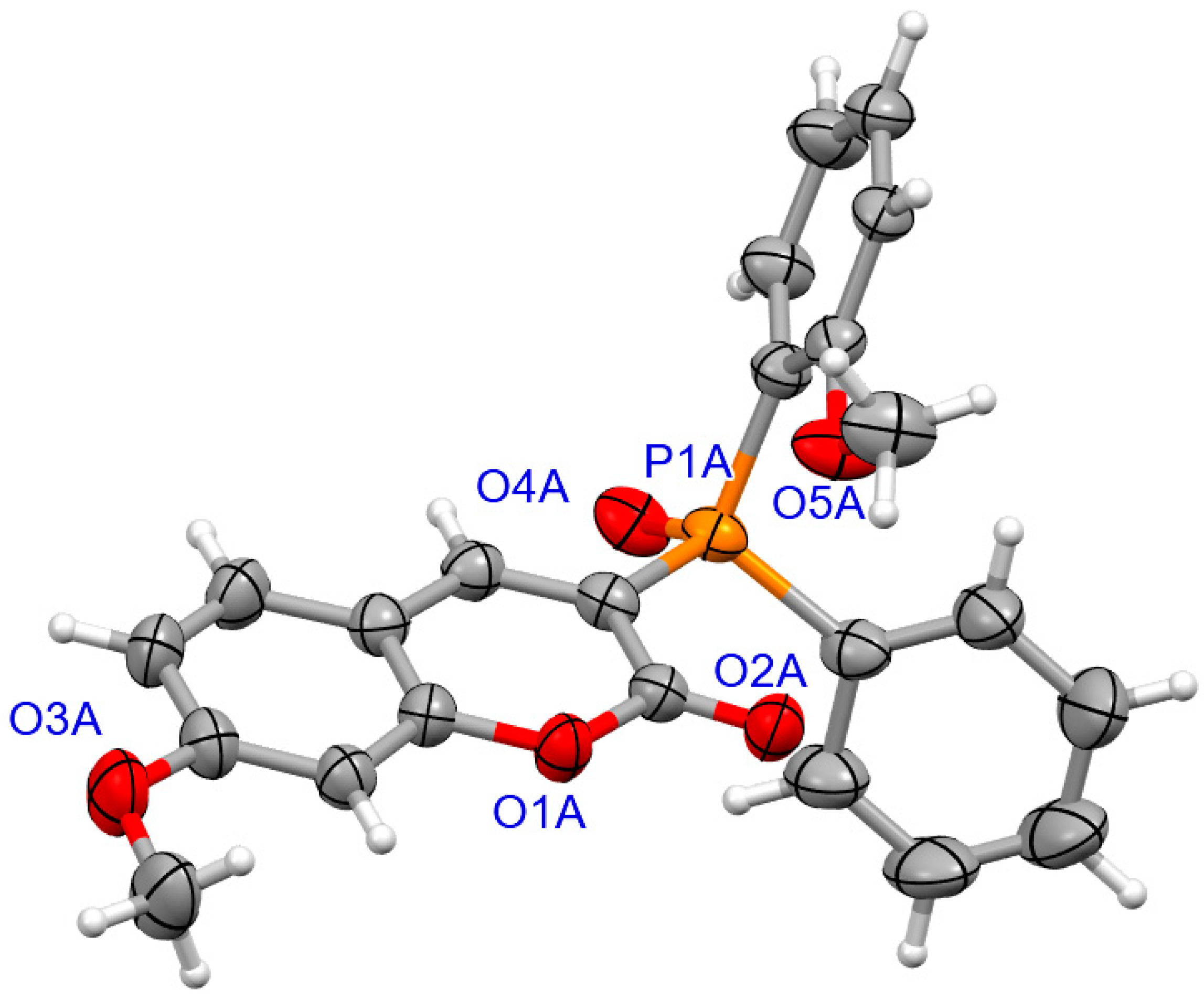
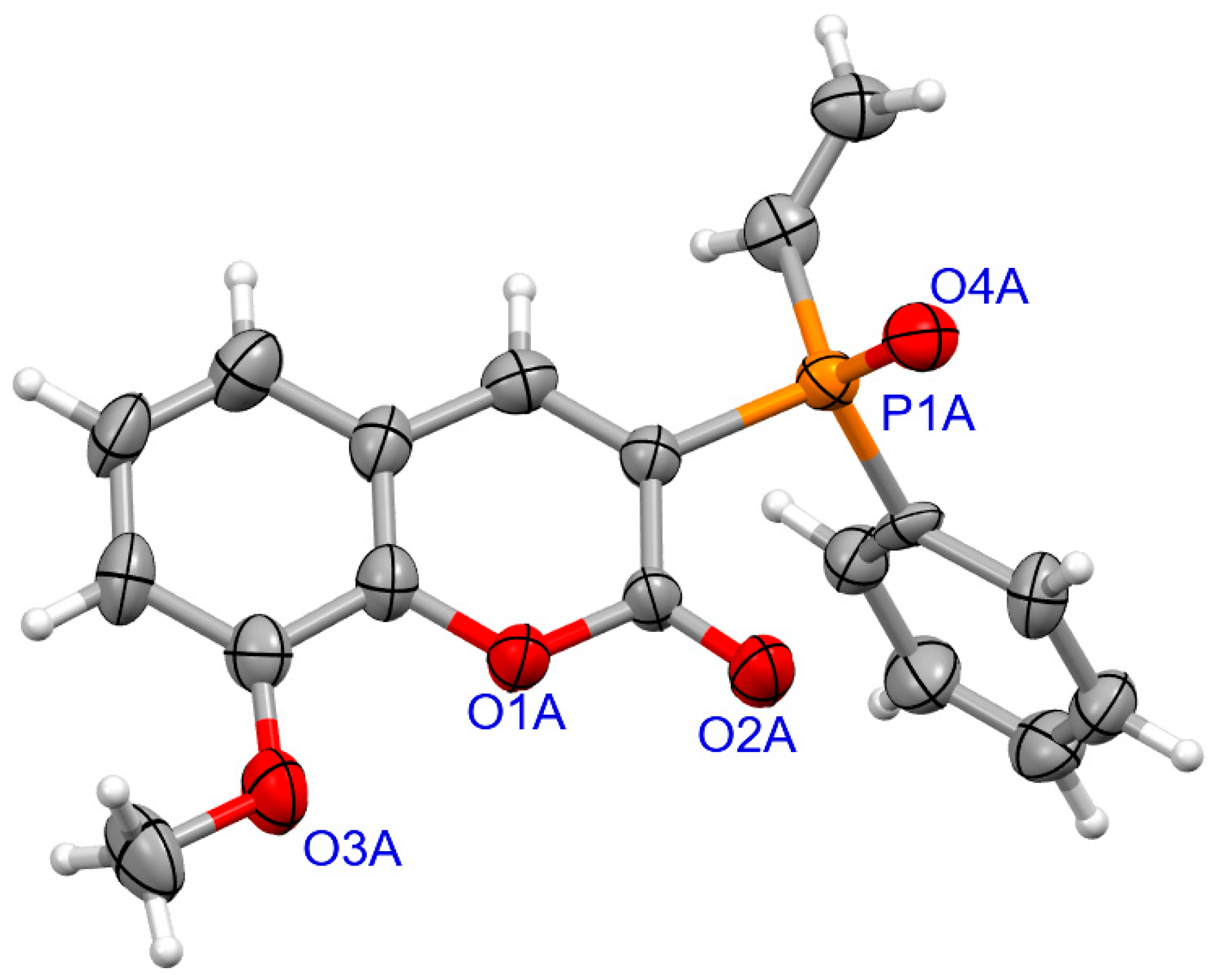
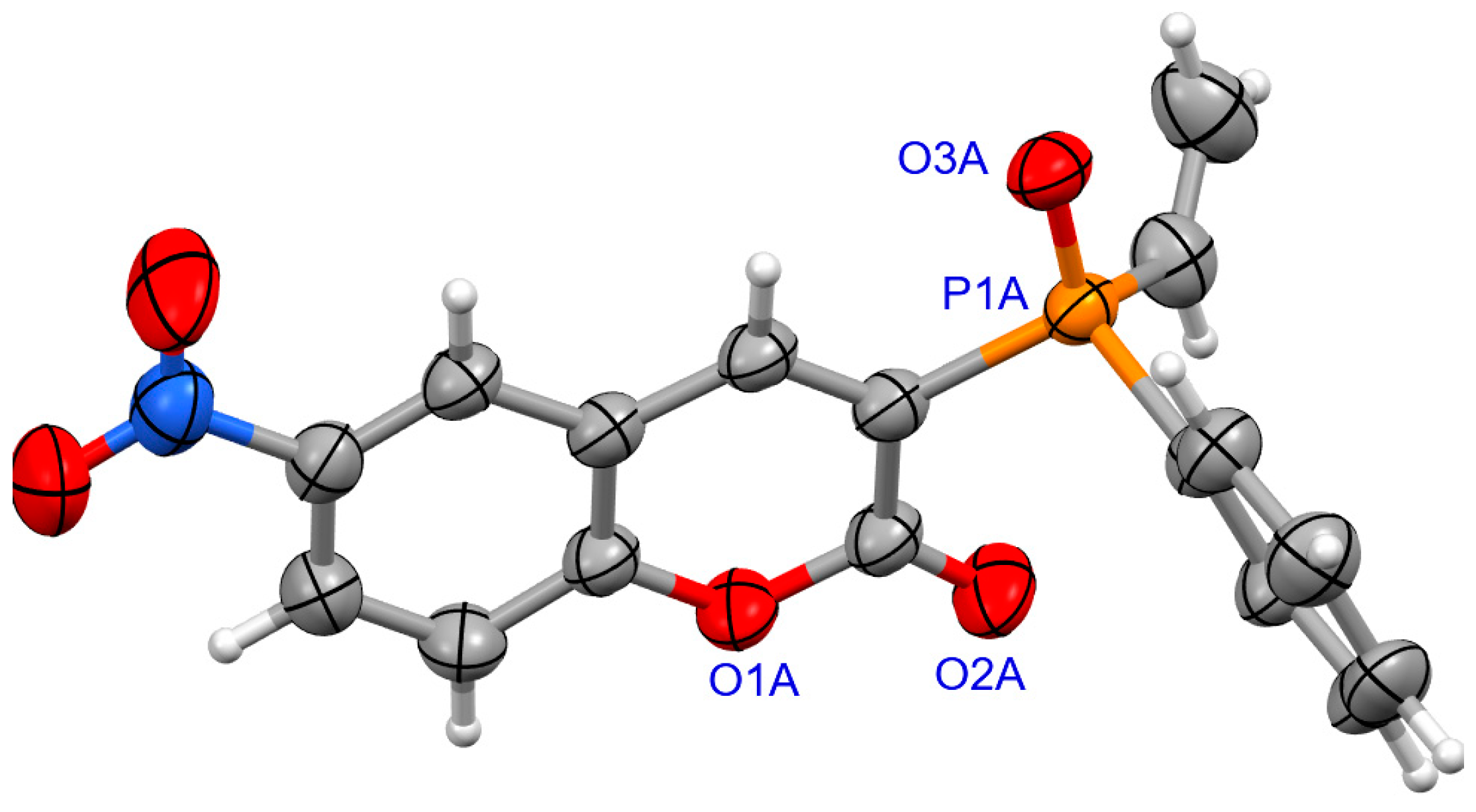
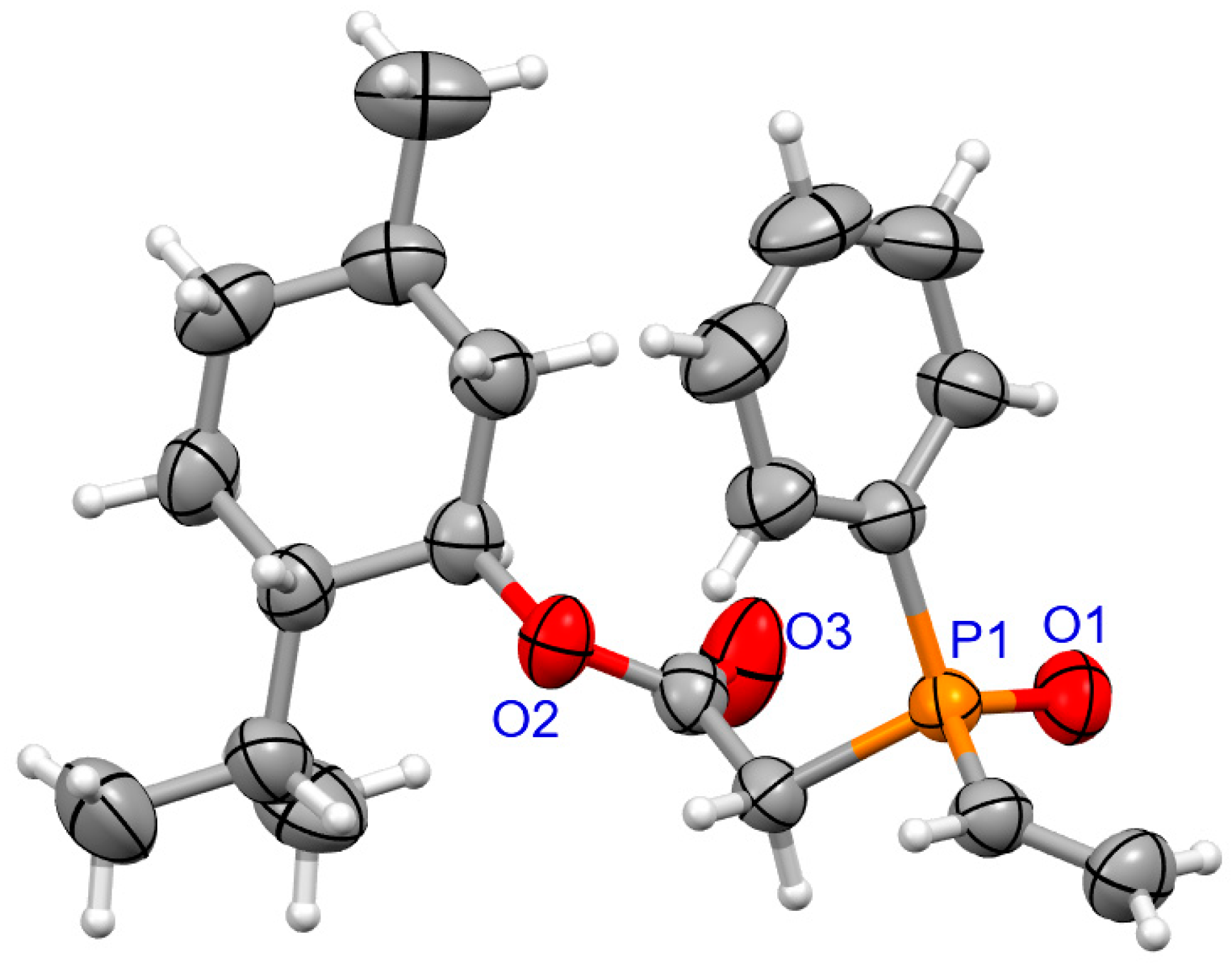
2.1.2. X-ray Crystallography
2.2. Procedure for Synthesis of L-menthyl Diphenylphosphinylacetate (1)
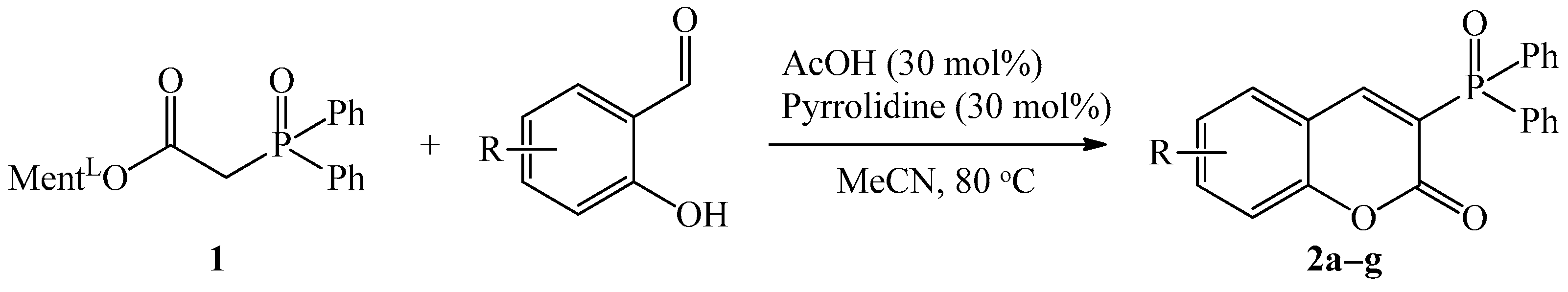
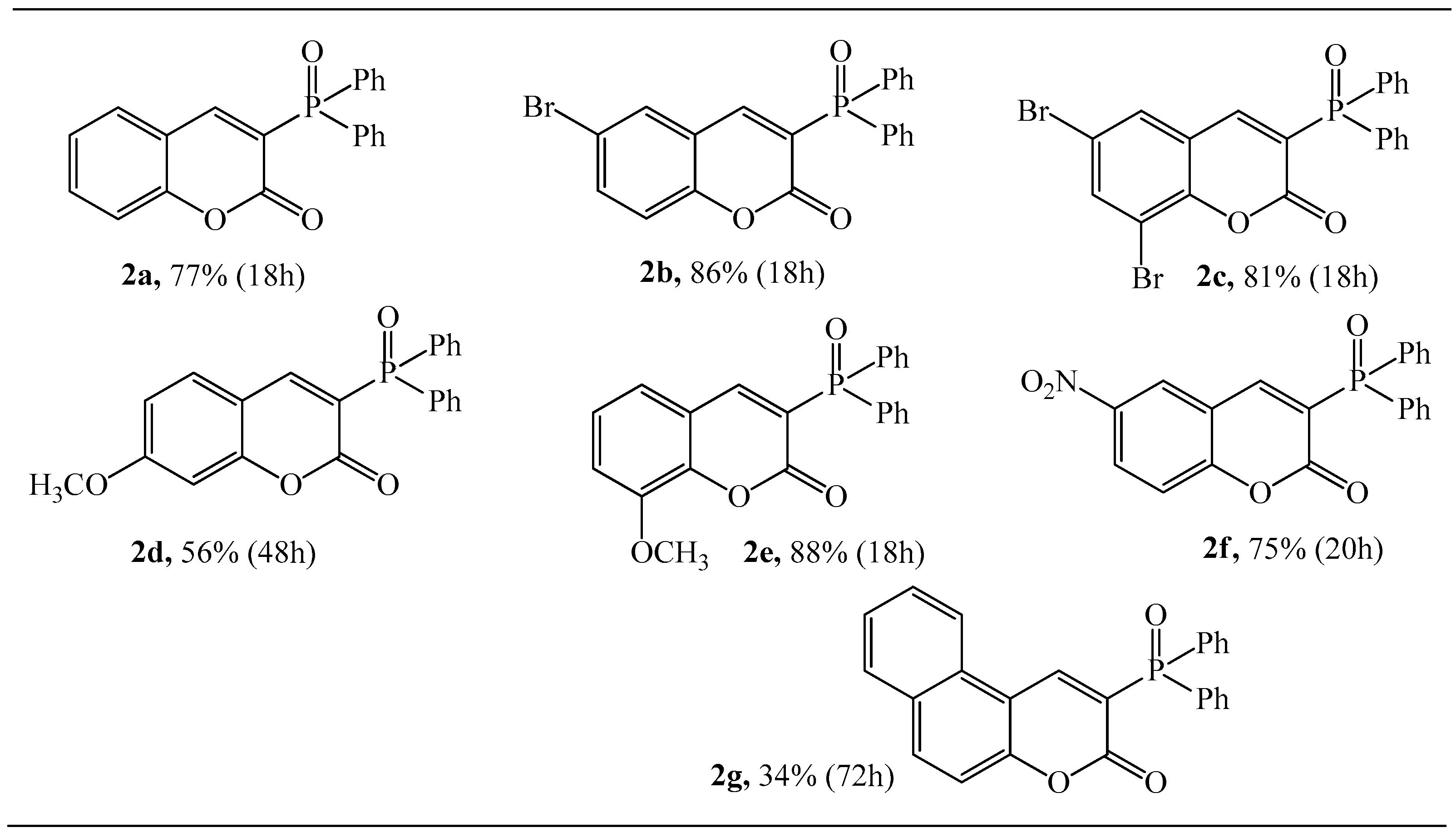
General Procedure for Synthesis of Coumarins 2a–g
2.3. Procedure for Synthesis of (SP)-L-Menthyl (2-methoxyphenyl)phenylphosphinylacetate ((SP)-3)
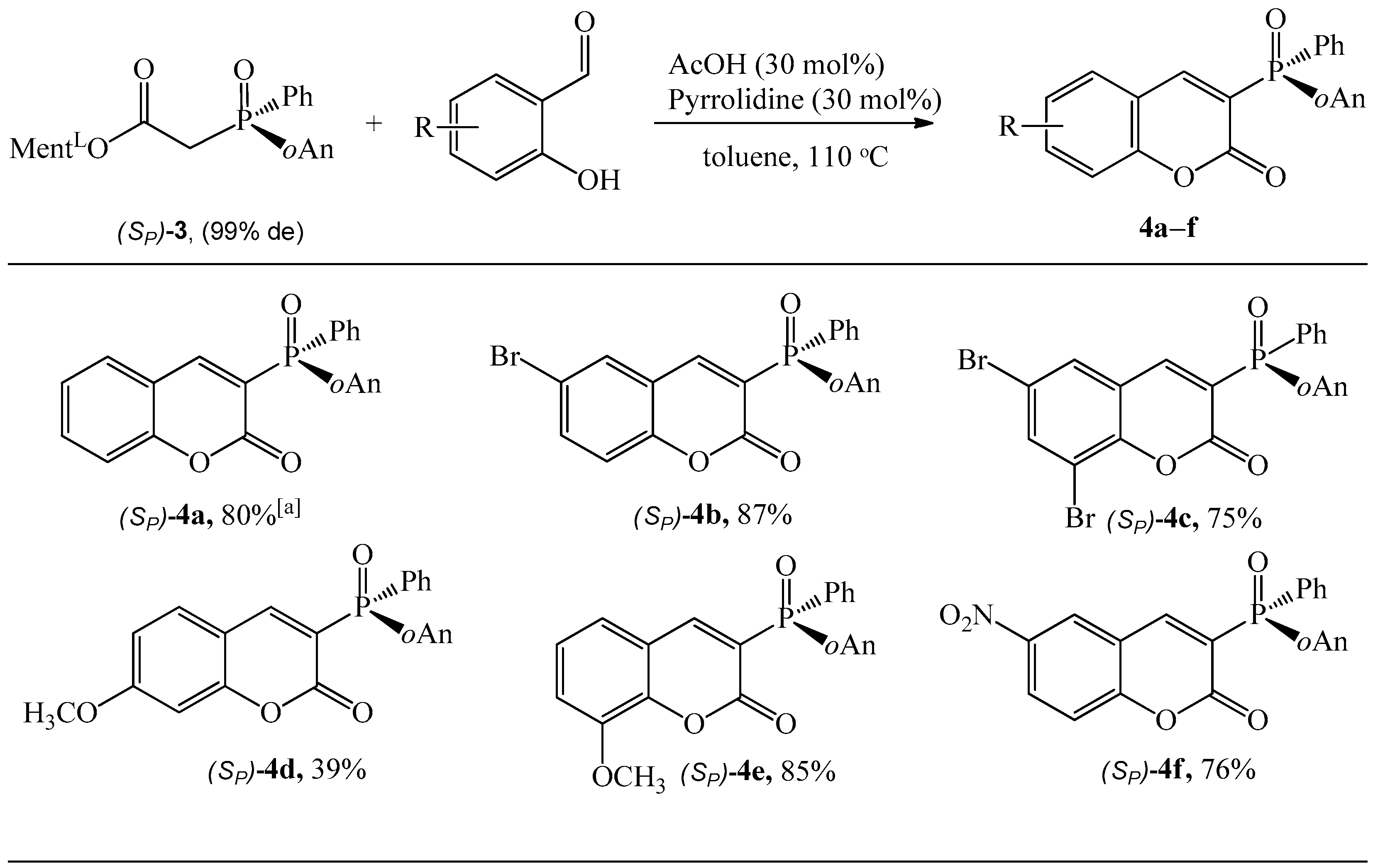
General Procedure for Synthesis of Coumarins (Sp)-4a–e
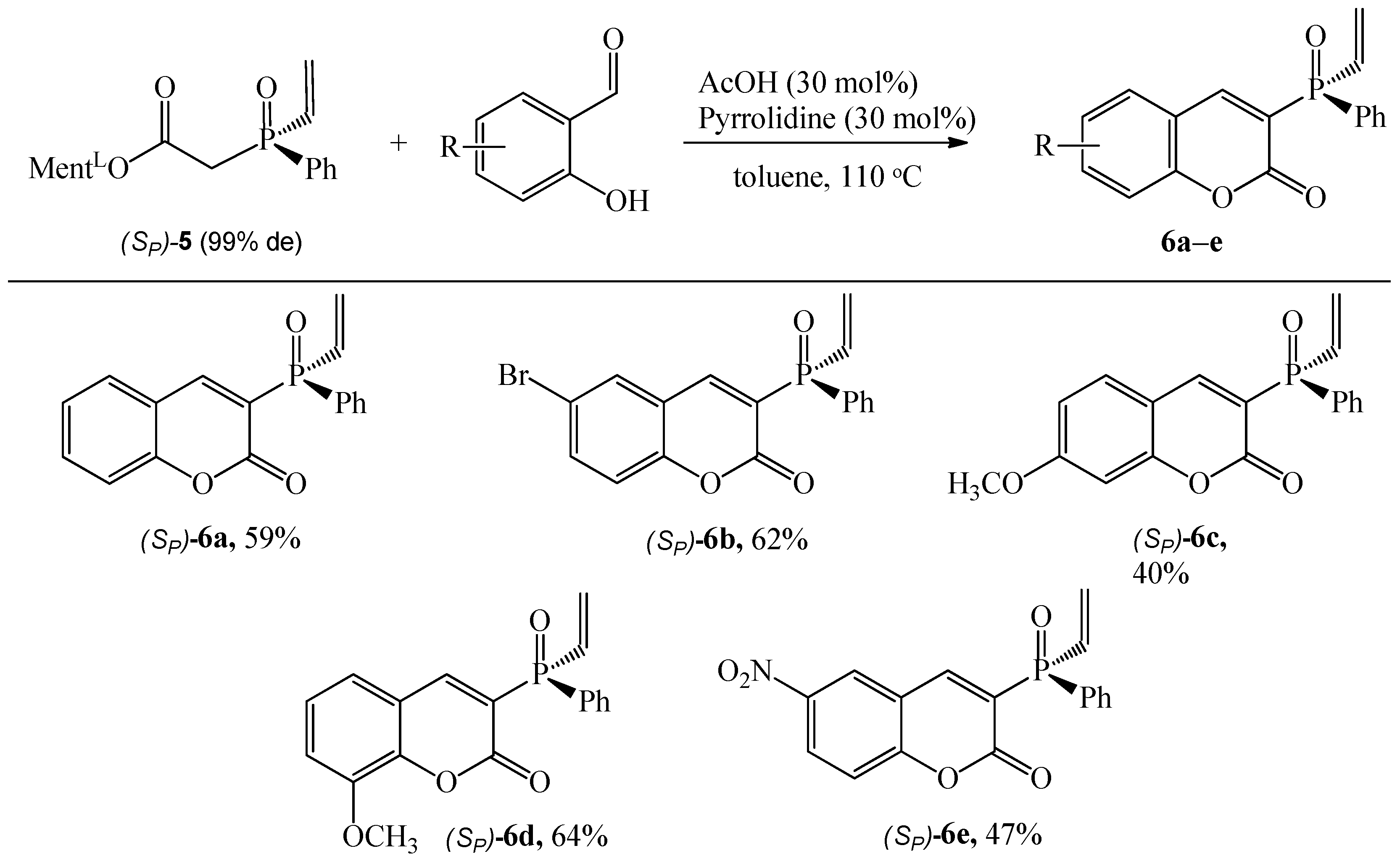
2.4. Procedure for Synthesis of (SP)-L-menthyl phenylvinylphosphinylacetate ((SP)-5)
General Procedure for Synthesis of Coumarins (SP)-6a–e
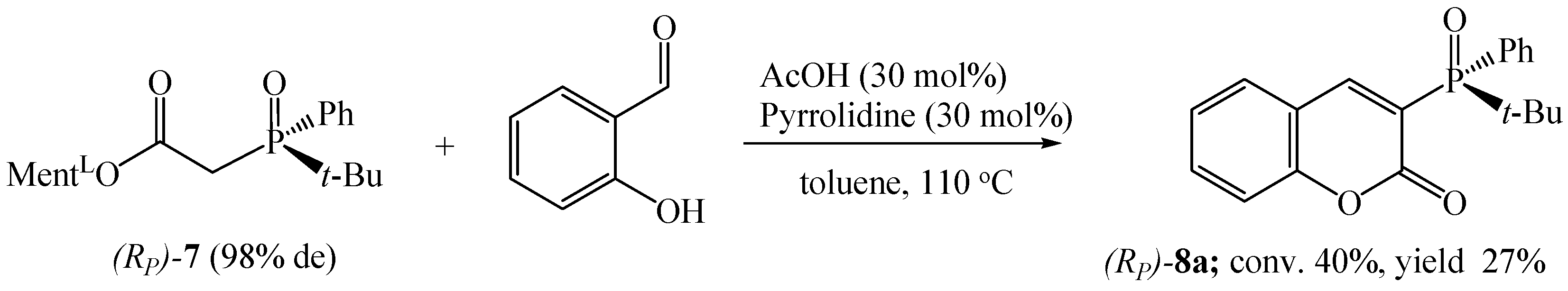
2.5. Procedure for Synthesis of (RP)-tert-butylphenylphosphinyl Acetic Acid Menthyl Ester ((RP)-7)
Synthesis of 3-(RP)-(tert-butylphenylphosphinyl)-2H-chromen-2-one ((RP)-8a)
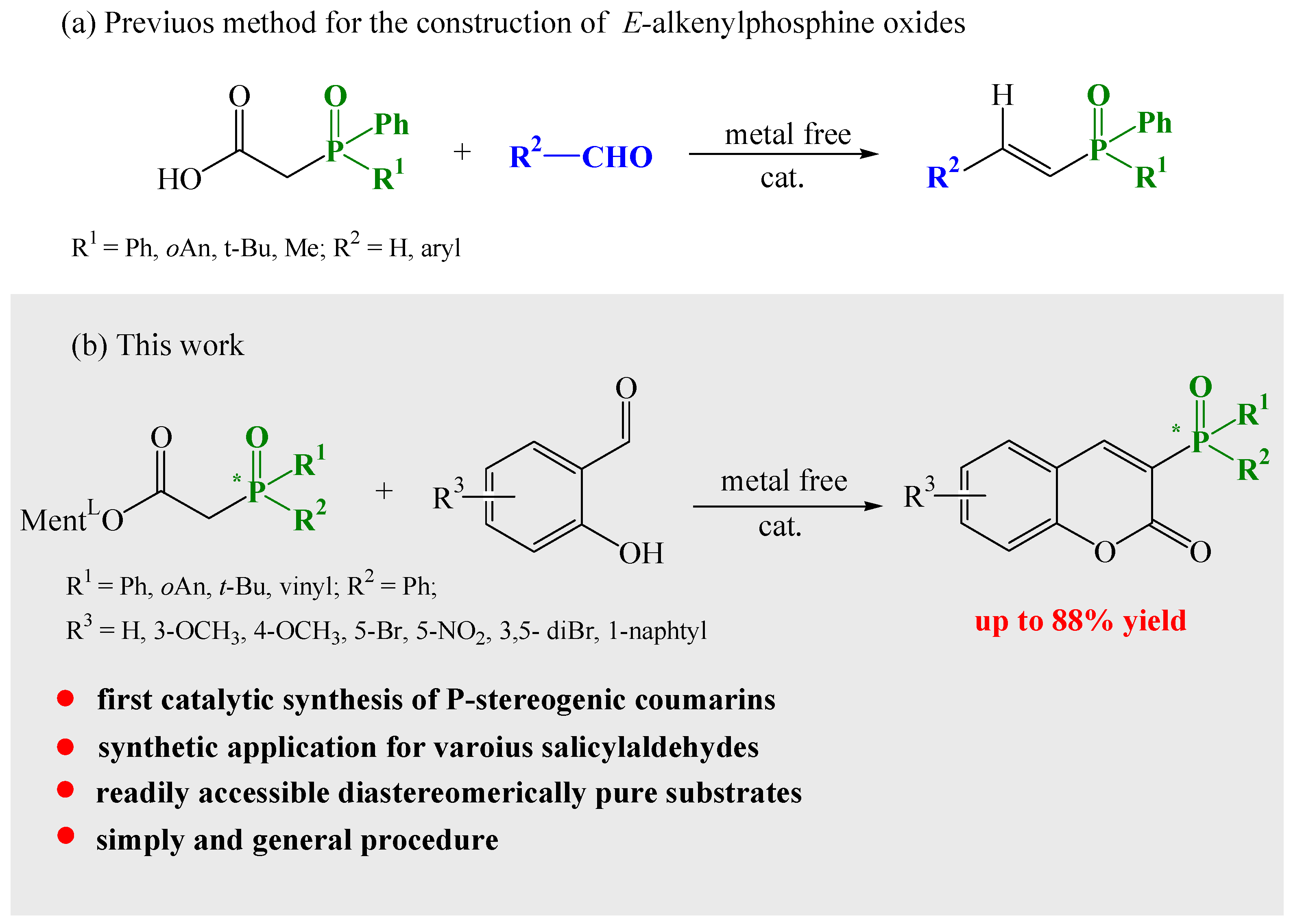
3. Results and Discussion
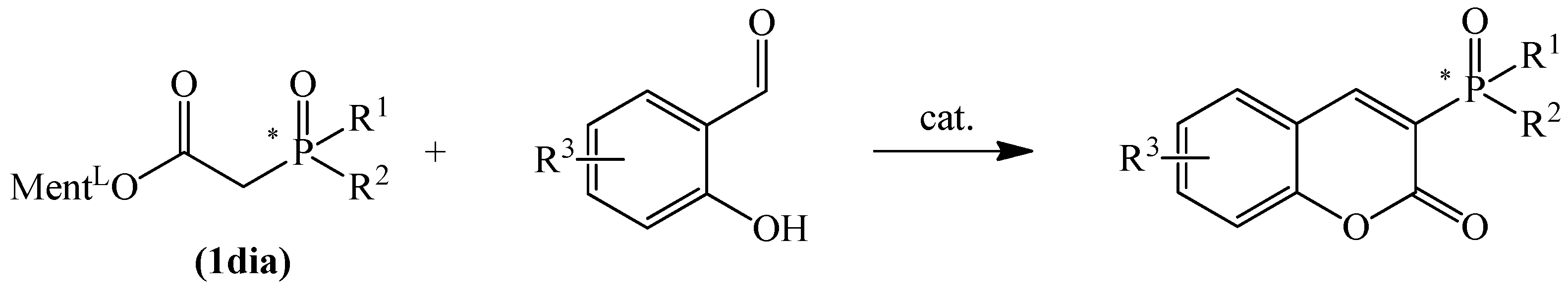
3.1. Synthesis of Phosphinylated Coumarins
3.1.1. Synthesis of 3-Diphenylphosphoryl Coumarins
3.1.2. Synthesis of Coumarins with P-stereogenic Moiety
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Perkin, W.H. On the artificial production of coumarin and formation of its homologues. J. Chem Soc. 1868, 21, 53–63. [Google Scholar] [CrossRef]
- Patel, G.; Banerjee, S. Review on Synthesis of Bio-Active Coumarins Fused Heterocyclic Molecules. Curr. Org. Chem. 2020, 24, 2566–2587. [Google Scholar] [CrossRef]
- Todorov, L.; Saso, L.; Kostova, I. Antioxidant Activity of Coumarins and Their Metal Complexes. Pharmaceuticals 2023, 16, 651. [Google Scholar] [CrossRef] [PubMed]
- Flores-Morales, V.; Villasana-Ruíz, A.P.; Garza-Veloz, I.; González-Delgado, S.; Martinez-Fierro, M.L. Therapeutic Effects of Coumarins with Different Substitution Patterns. Molecules 2023, 28, 2413. [Google Scholar] [CrossRef] [PubMed]
- Cubukcu, B.; Bray, D.H.; Warhurst, D.C.; Mericli, A.H.; Ozhatay, N.; Sariyar, G. In vitro antimarlarial activity of crude extracts and compounds from Artemisia abrotanum L. Phytother. Res. 1990, 4, 203–204. [Google Scholar] [CrossRef]
- Song, X.F.; Fan, J.; Liou, L.; Liu, X.F.; Gao, F. Coumarin derivatives with anticancer activities: An update. Arch. Pharm. 2020, 353, 2000025. [Google Scholar] [CrossRef]
- Stasi, L.C. Natural Coumarin Derivatives Activating Nrf2 Signaling Pathway as Lead Compounds for the Design and Synthesis of Intestinal Anti-Inflammatory Drugs. Pharmaceuticals 2023, 16, 511. [Google Scholar] [CrossRef]
- Berzina, L.; Mierina, I. Antiradical and Antioxidant Activity of Compounds Containing 1,3-Dicarbonyl Moiety: An Overview. Molecules 2023, 28, 6203. [Google Scholar] [CrossRef]
- Balewski, Ł.; Szulta, S.; Jalińska, A.; Kornicka, A. A Mini-Review: Recent Advances in Coumarin-Metal Complexes With Biological Properties. Front. Chem. 2021, 9, 781779. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Y.; Li, G.; Liu, Z.; Ma, J.; Liu, M.; Li, D.; Han, J.; Wang, B. Synthesis and Evaluation of Bi-functional 7-hydroxycoumarin Platinum(IV) Complexes as Antitumor Agents. Bioorg. Med. Chem. 2019, 27, 2112–2121. [Google Scholar] [CrossRef]
- Sarkar, T.; Kumar, A.; Sahoo, S.; Hussain, A. Mixed-ligand Cobalt(III) Complexes of a Naturally Occurring Coumarin and Phenanthroline Bases as Mitochondria-Targeted Dual-Purpose Photochemotherapeutics. Inorg. Chem. 2021, 60, 6649–6662. [Google Scholar] [CrossRef] [PubMed]
- Yernale, N.G.; Bennikallu Hire Mathada, M. Preparation of Octahedral Cu(II), Co(II), Ni(II) and Zn(II) Complexes Derived from 8- Formyl-7-Hydroxy-4-Methylcoumarin: Synthesis, Characterization and Biological Study. J. Mol. Struct. 2020, 1220, 128659. [Google Scholar] [CrossRef]
- Stojanović, M.; Flores-Diaz, N.; Ren, Y.; Vlachopoulos, N.; Pfeifer, L.; Shen, Z.; Liu, Y.; Zakeeruddin, S.M.; Milić, J.V.; Hagfeldt, A. The Rise of Dye-Sensitized Solar Cells: From Molecular Photovoltaics to Emerging Solid-State Photovoltaic Technologies. Helv. Chim. Acta 2021, 104, e2000230. [Google Scholar] [CrossRef]
- Kumar, A.; Baccoli, R.; Fais, A.; Cincotti, A.; Pilia, L.; Gatto, G. Substitution Effects on the Optoelectronic Properties of Coumarin Derivatives. Appl. Sci. 2019, 10, 144. [Google Scholar] [CrossRef]
- Safavi-Mirmahalleh, S.A.; Golshan, M.; Gheitarani, B.; Hosseini, M.S.; Salami-Kalajahi, M. A review on applications of coumarin and its derivatives in preparation of photo-responsive polymers. Eur. Polym. J. 2023, 199, 112430. [Google Scholar] [CrossRef]
- Sun, X.; Liu, T.; Sun, J.; Wang, X. Synthesis and application of coumarin fluorescence probes. RSC Adv. 2020, 10, 10826–10847. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Liu, Z.; Verwilst, P.; Koo, S.; Jangjili, P.; Kim, J.S.; Lin, W. Coumarin-Based Small-Molecule Fluorescent Chemosensors. Chem. Rev. 2019, 119, 10403–10519. [Google Scholar] [CrossRef]
- Szwaczko, K. Fluorescent Coumarin-based Probe for Detection of Biological Thiols. Curr. Org. Chem. 2023, 15, 1329–1335. [Google Scholar] [CrossRef]
- Yu, H.; Yang, H.; Shi, E.; Tang, W. Development and Clinical Application of Phosphorus-Containing Drugs. Med. Drug Discov. 2020, 8, 100063. [Google Scholar] [CrossRef]
- Huang, W.S.; Liu, S.; Zou, D.; Thomas, M.; Wang, Y.; Zhou, T.; Romero, J.; Kohlmann, A.; Li, F.; Qi, J.; et al. Discovery of Brigatinib (AP26113), a Phosphine Oxide-Containing, Potent, Orally Active Inhibitor of Anaplastic Lymphoma Kinase. J. Med. Chem. 2016, 59, 4948. [Google Scholar] [CrossRef]
- Val’dman, A.; Kozlovskaia, M.M.; Zaikonnikova, I.V.; Bravkov, M.F.; Rzhevskaia, G.F. Vliianie gidifena na povedenie i gemodinamicheskie proiavleniia émotsional’no-stressovoĭ reaktsii [Effect of gidifen on behavior and hemodynamic signs of an emotionally stressful reaction]. Biull. Eksp. Biol. Med. 1980, 89, 310–312. [Google Scholar] [PubMed]
- Nicholson, A.; Wright, C. Activity of fosazepam, a soluble analogue of diazepam. Br. J. Clin. Pharmacol. 1977, 4, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Stambirskyi, M.V.; Kostiuk, T.; Sirobaba, S.I.; Rudnichenko, A.; Titikaiev, D.L.; Dmytriv, Y.V.; Kuznietsova, H.; Pishel, I.; Borysko, P.; Mykhailiuk, P.K. Phosphine Oxides (-POMe2) for Medicinal Chemistry: Synthesis, Properties, and Applications. J. Org. Chem. 2021, 86, 12783–12801. [Google Scholar] [CrossRef] [PubMed]
- Szwaczko, K. Coumarins Synthesis and Transformation via C–H Bond Activation—A Review. Inorganics 2022, 10, 23. [Google Scholar] [CrossRef]
- Mi, X.; Huang, M.; Zhang, J.; Wang, C.; Wu, Y. Regioselective palladium-catalyzed phosphonation of coumarins with dialkyl H-phosphonates via C-H functionalization. Org. Lett. 2013, 15, 6266–6269. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, X.; Yuan, J.; Xiao, Y.; Mao, P. Catalytic activity of chelating N-heterocyclic carbene palladium complexes towards selective phosphorylation of coumarins. J. Organomet. Chem. 2016, 818, 179. [Google Scholar] [CrossRef]
- Yuan, J.; Li, Y.; Yang, L.; Mai, W.; Mao, P.; Xiao, Y.; Qu, L. Silver-catalyzed direct Csp2-H radical phosphorylation of coumarins with H-phosphites. Tetrahedron 2015, 71, 8178–8186. [Google Scholar] [CrossRef]
- Mi, X.; Wang, C.; Huang, M.; Zhang, J.; Wu, Y.; Wu, Y. Silver-Catalyzed Synthesis of 3-Phosphorated Coumarins via Radical Cyclization of Alkynoates and Dialkyl H-Phosphonates. Org. Lett. 2014, 12, 3356–3359. [Google Scholar] [CrossRef]
- Zhou, P.; Jiang, Y.J.; Zou, J.P.; Zhang, W. Manganese(III) Acetate Mediated Free-Radical Phosphonylation of Flavones and Coumarins. Synthesis 2012, 7, 1043. [Google Scholar] [CrossRef]
- Li, Q.; Zhao, X.; Li, Y.; Huang, M.; Kim, J.K.; Wu, Y. Regioselective phosphinylation of coumarins under green LED irradiation and its mechanism. Org. Biomol. Chem. 2017, 15, 9775. [Google Scholar] [CrossRef]
- Liu, D.; Chen, J.; Wang, X.; Xu, P. Metal-Free, Visible-Light-Promoted Synthesis of 3-Phosphorylated Coumarins via Radical C−P/C−C Bond Formation. ASC Commun. 2017, 359, 2773–2777. [Google Scholar] [CrossRef]
- Grinenko, V.; Khrizanforov, M.; Strekalova, S.; Khrizanforova, V.; Kholin, K.; Gryaznova, T.; Budnikova, Y.H. Electrooxidative phosphorylation of coumarins by bimetallic catalytic systems Ni(II)/Mn(II) or Co(II)/Mn(II). Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1660–1661. [Google Scholar] [CrossRef]
- Khrizanforov, M.; Strekalova, S.; Kholin, K.; Khrizanforova, V.; Kadirov, M.; Gryaznova, T.; Budnikova, Y.H. Novel approach to metal-induced oxidative phosphorylation of aromatic compounds. Catal. Today 2017, 279, 133–141. [Google Scholar] [CrossRef]
- Robinson, C.N.; Addison, J.F. Condensation of Triethyl Phosphonoacetate with Aromatic Aldehydes. J. Org. Chem. 1966, 31, 4325–4326. [Google Scholar] [CrossRef]
- Bouyssou, P.; Chenault, J. Phosphonates and phosphine oxides as reagents in a one-pot synthesis of coumarins. Tetrahedron Lett. 1991, 32, 5341–5344. [Google Scholar] [CrossRef]
- Bojilova, A.; Nikolova, R.; Ivanov, C.; Rodios, N.; Terzis, A.; Raptopoulou, C. A comparative study of the interaction of salicylaldehydes with phosphonoacetates under Knoevenagel reaction conditions. Synthesis of 1,2-benzoxaphosphorines and their dimers. Tetrahedron 1996, 52, 12597–12612. [Google Scholar] [CrossRef]
- Janecki, T.; Albrecht, A.; Koszuk, J.; Mondranka, J.; Slowak, D. A simple and effective synthesis of activated vinylphosphonates from 3-methoxy-2-diethoxyphosphorylacrylate. Tetrahedron Lett. 2010, 51, 2274–2276. [Google Scholar] [CrossRef]
- Mondranka, J.; Albrecht, A.; Janecki, T. A Convenient Entry to 3-Methylidenechroman-2-ones and 2-Methylidene- dihydrobenzochromen-3-ones. Synlett 2010, 19, 2867–2870. [Google Scholar] [CrossRef]
- SuperNova Diffractometer (Oxford Diffraction); Agilent Technologies Inc.: Yarnton, UK, 2014.
- CrysAlisPro, 1.171.42.79a; Rigaku Oxford Diffraction: Tokyo, Japan, 2022.
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Kim, I.; Min, M.; Kang, D.; Kim, K.; Hong, S. Direct Phosphonation of Quinolinones and Coumarins Driven by the Photochemical Activity of Substrates and Products. Org. Lett. 2017, 19, 1394–1397. [Google Scholar] [CrossRef] [PubMed]
- Lubańska, M.; Dziuba, K.F.; Pietrusiewicz, K.M. Enantiodivergent Synthesis of Both PAMPO Enantiomers Using L-Menthyl Chloroacetate and Stereomutation at P in Classical Quaternisation Reactions. Synthesis 2020, 52, 909–916. [Google Scholar] [CrossRef]
- Bodalski, R.; Rutkowska-Olma, E.; Pietrusiewicz, K.M. Optically active Phosphine oxides: Synthesis and absolute configuration of (menthoxycarbonylmethyl) phenylvinyl phosphine oxide. Tetrahedron 1980, 36, 2353–2355. [Google Scholar] [CrossRef]
- Cheng, X.; Horton, P.N.; Hursthouse, M.B.; Hii, K.K. Aminohydroxy phosphine oxide ligands in ruthenium catalysed asymmetric transfer hydrogenation reactions. Tetrahedron Asymmetry 2004, 15, 2141–2246. [Google Scholar] [CrossRef]
- Dziuba, K.; Szwaczko, K.; Frynas, S. Knoevenagel Condensation of Phosphinoylacetic Acids with Aldehydes: An Efficient One-Pot Strategy for the Synthesis of P-Functionalized Alkenyl Compounds. Synthesis 2021, 53, 2142–2154. [Google Scholar] [CrossRef]
- Koleva, A.I.; Petkova-Yankova, N.I.; Nikolova, R.D. Synthesis and Chemical Properties of 3-Phosphono-coumarins and 1,2-Benzoxaphosphorins as Precursors for Bioactive Compounds. Molecules 2019, 24, 2030. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst (mol %) | Time, h | Conversion, % [a] |
|---|---|---|---|
| 1 | Piperidine (30) | 48 | - |
| 2 | Pyrrolidine (30) | 48 | - |
| 3 | Piperidine (10)/AcOH (10) | 24 | 80 |
| 4 | Piperidine (30)/AcOH (30) | 24 | 100 |
| 5 | Pyrrolidine (30)/AcOH (30) | 18 | 100 |
| 6 | Pyrrolidine (30)/TCA (30) | 18 | 100 |
| 7 | Pyrrolidine (30)/pTSA (30) | 18 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dziuba, K.F.; Frynas, S.; Kozioł, A.E.; Szwaczko, K. Synthesis and Structural Elucidation of P-stereogenic Coumarins. Symmetry 2024, 16, 73. https://doi.org/10.3390/sym16010073
Dziuba KF, Frynas S, Kozioł AE, Szwaczko K. Synthesis and Structural Elucidation of P-stereogenic Coumarins. Symmetry. 2024; 16(1):73. https://doi.org/10.3390/sym16010073
Chicago/Turabian StyleDziuba, Kamil F., Sławomir Frynas, Anna E. Kozioł, and Katarzyna Szwaczko. 2024. "Synthesis and Structural Elucidation of P-stereogenic Coumarins" Symmetry 16, no. 1: 73. https://doi.org/10.3390/sym16010073