
Stereoselectivity in Butadiene Polymerization Promoted by Using Ziegler–Natta Catalysts Based on (Anilidomethyl)pyridine Group (IV) Complexes


Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
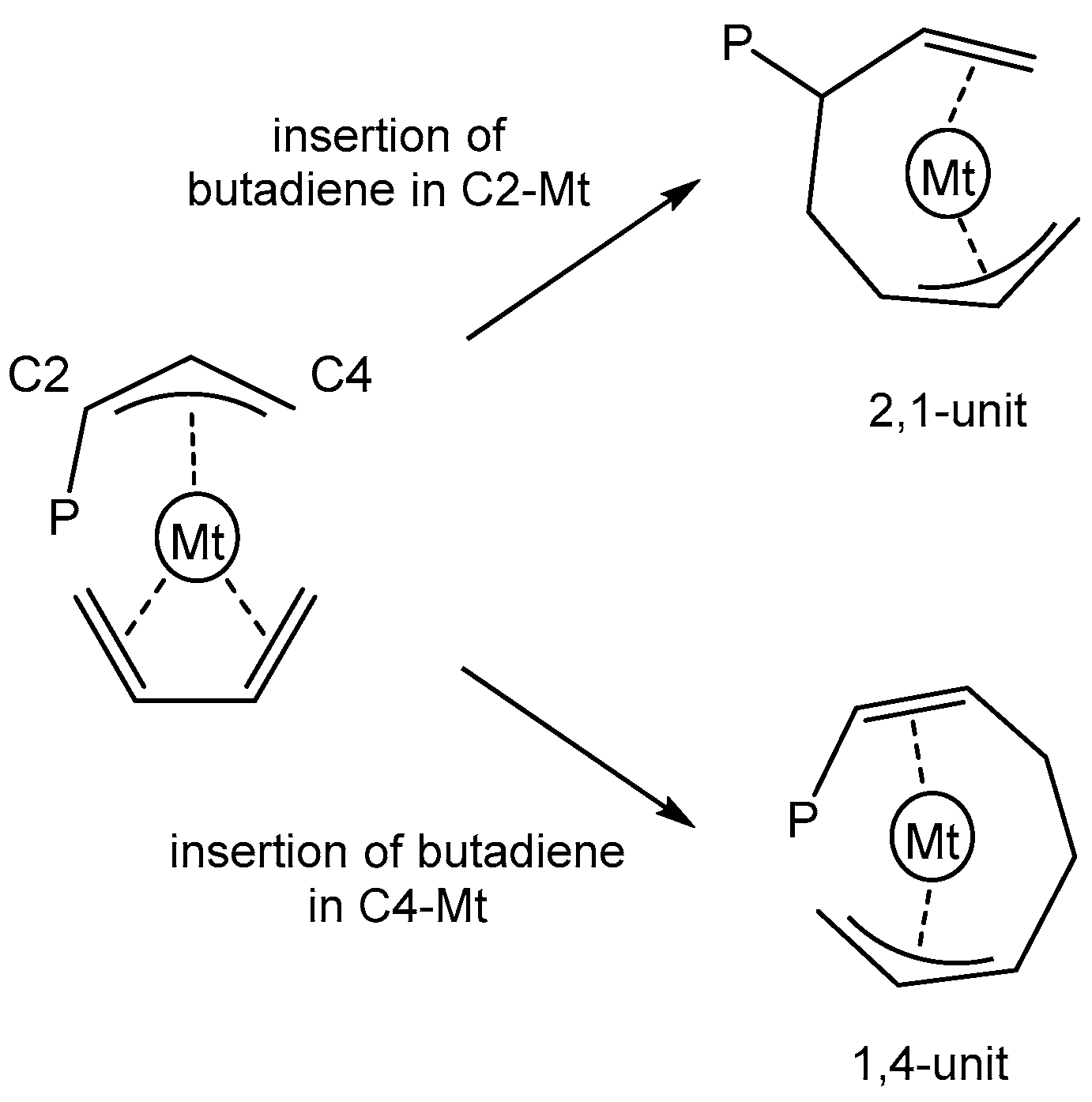
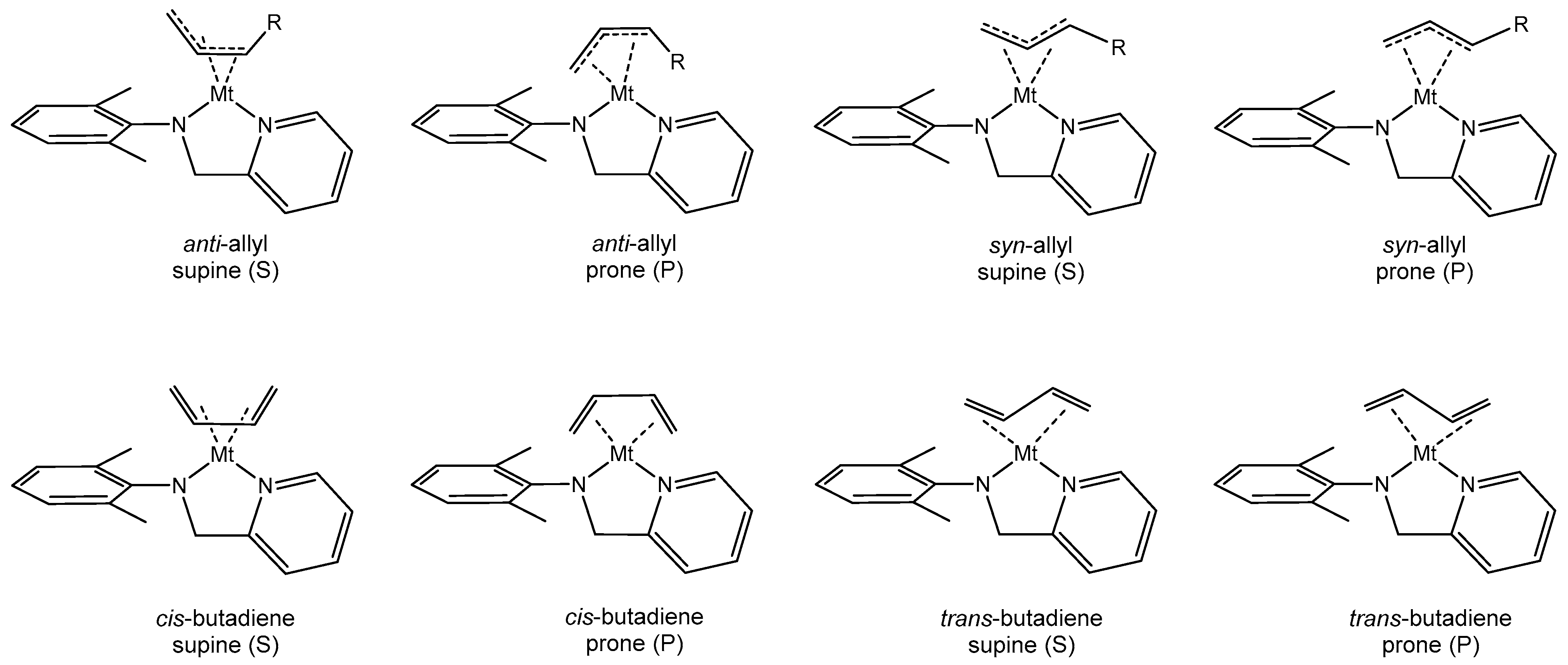
3.1. General Considerations
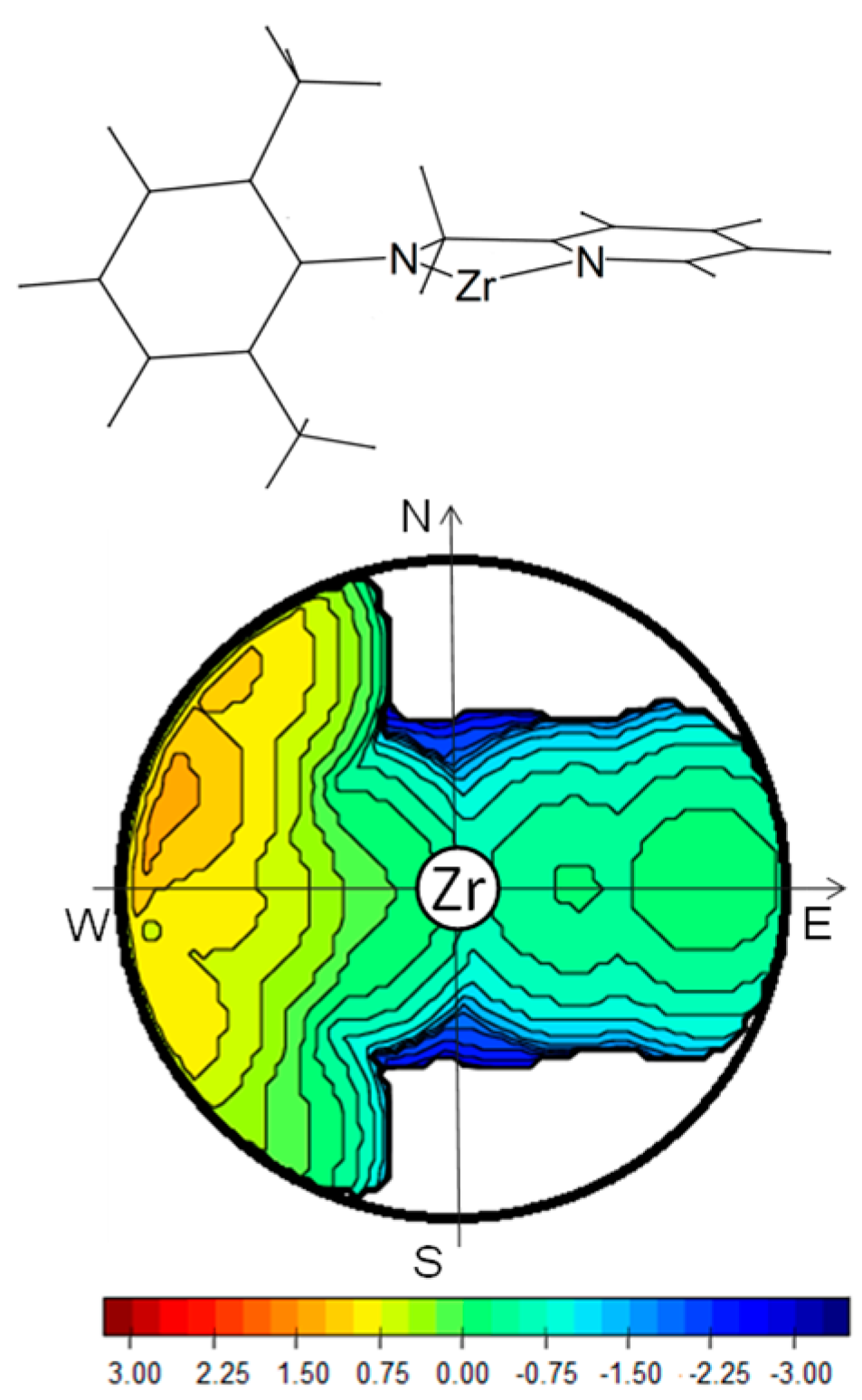
3.2. Model of the Active Species
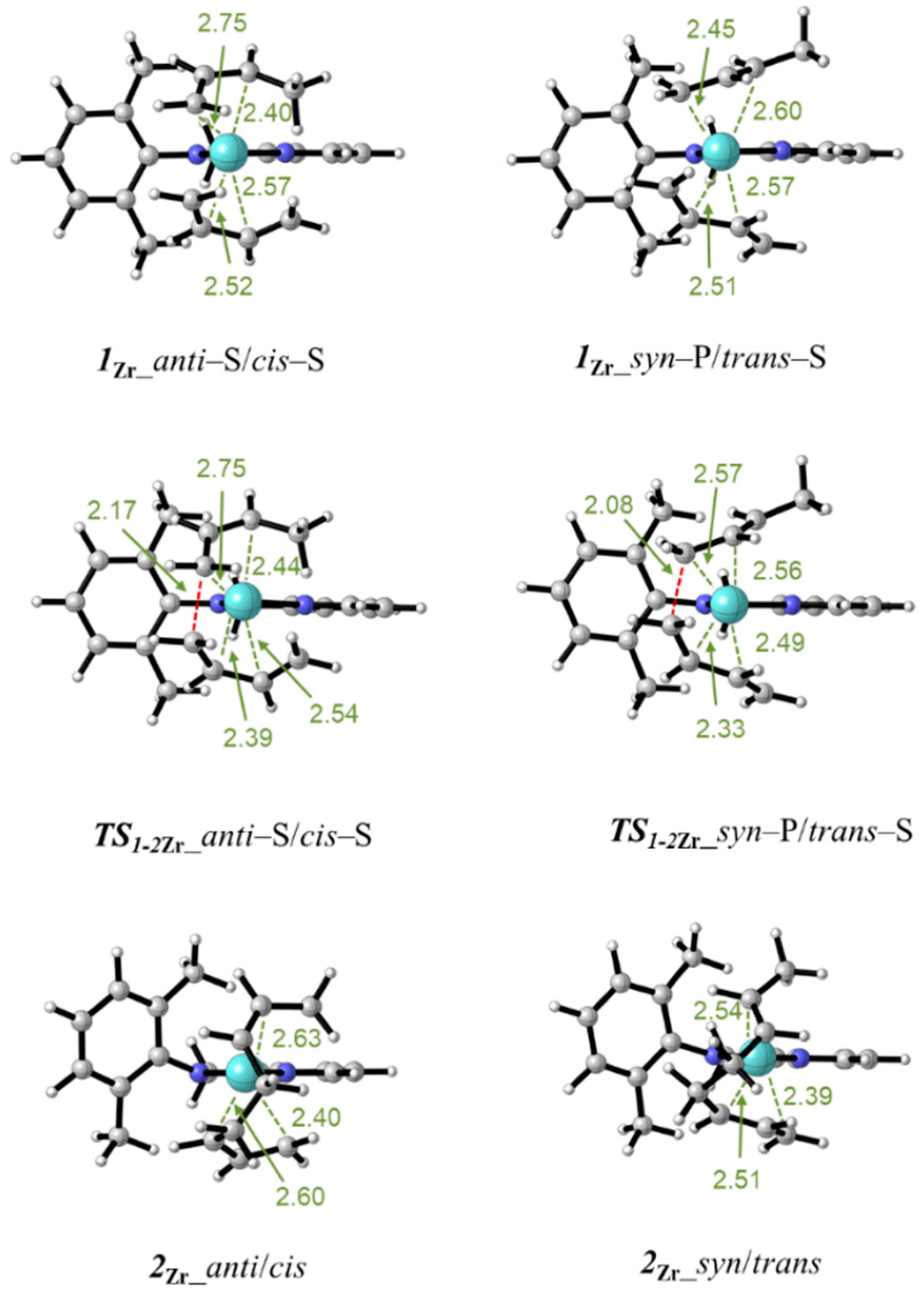
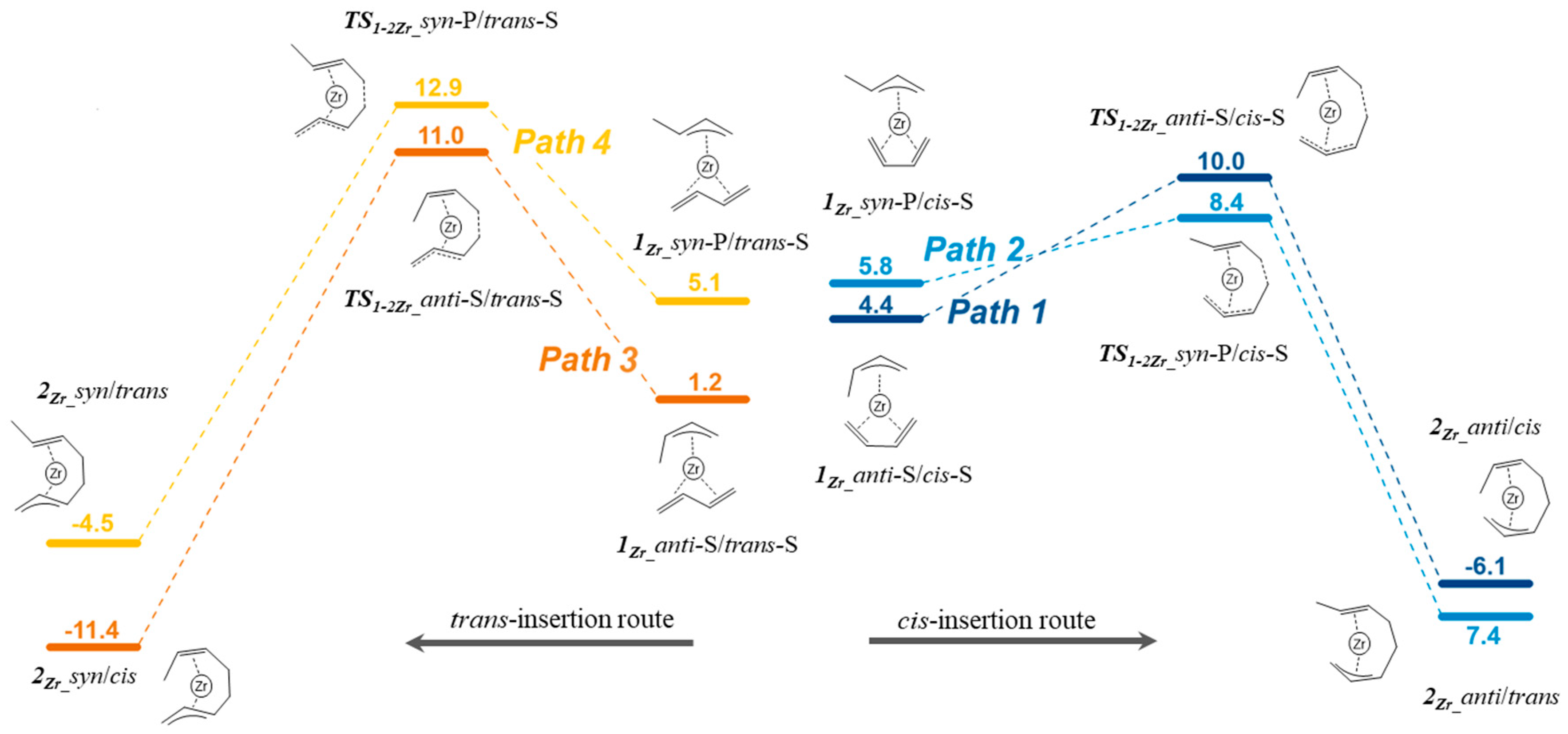
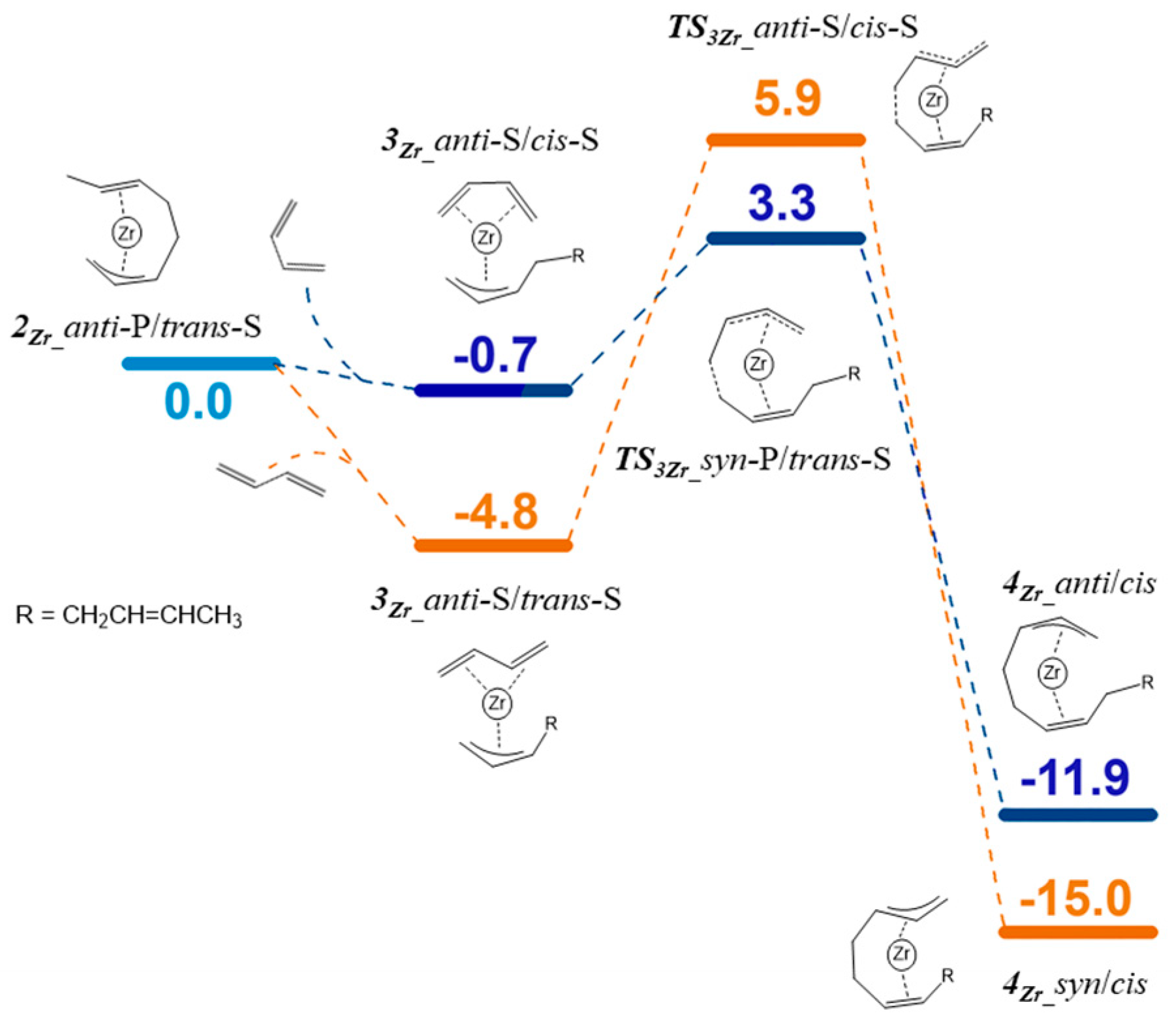
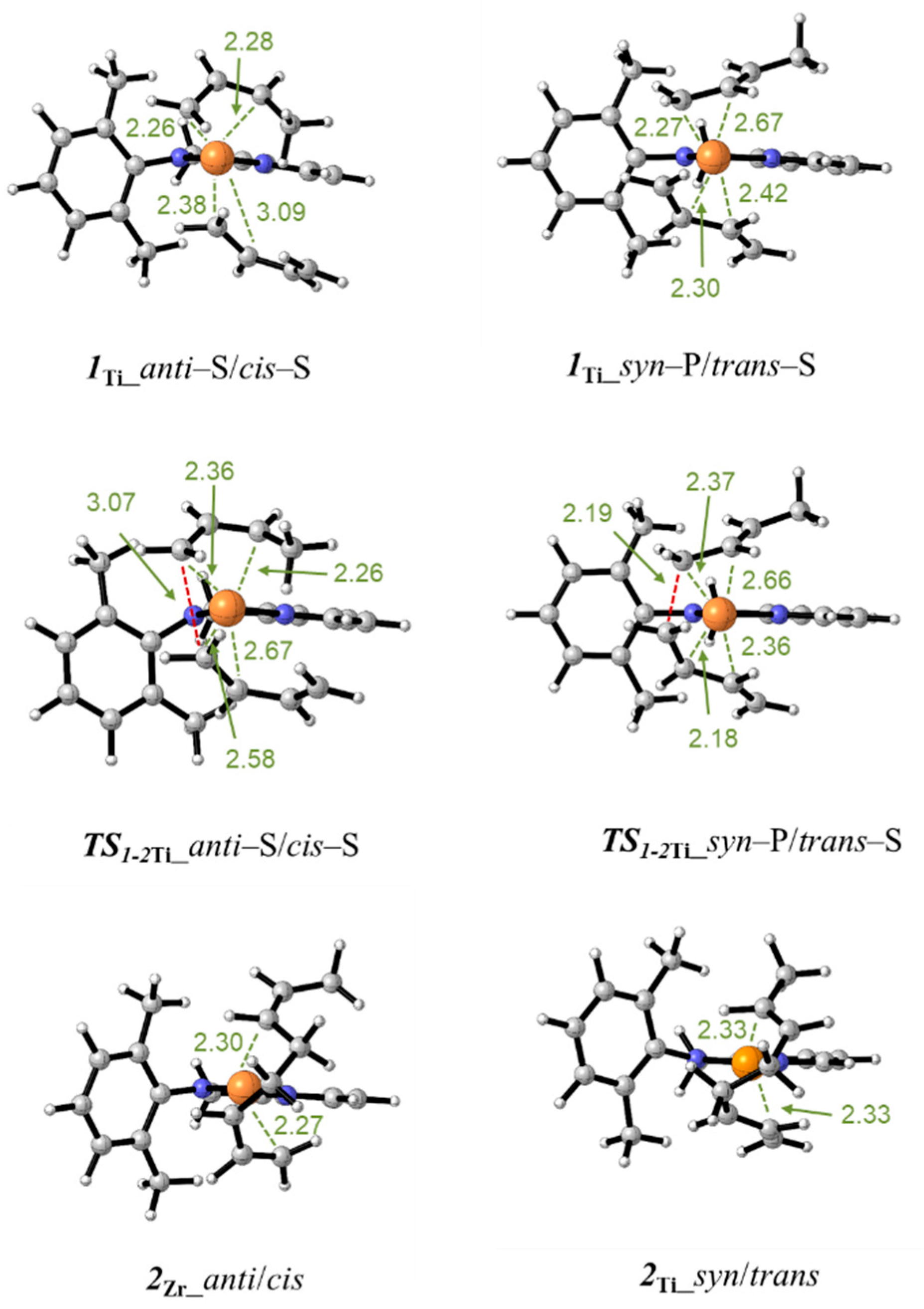
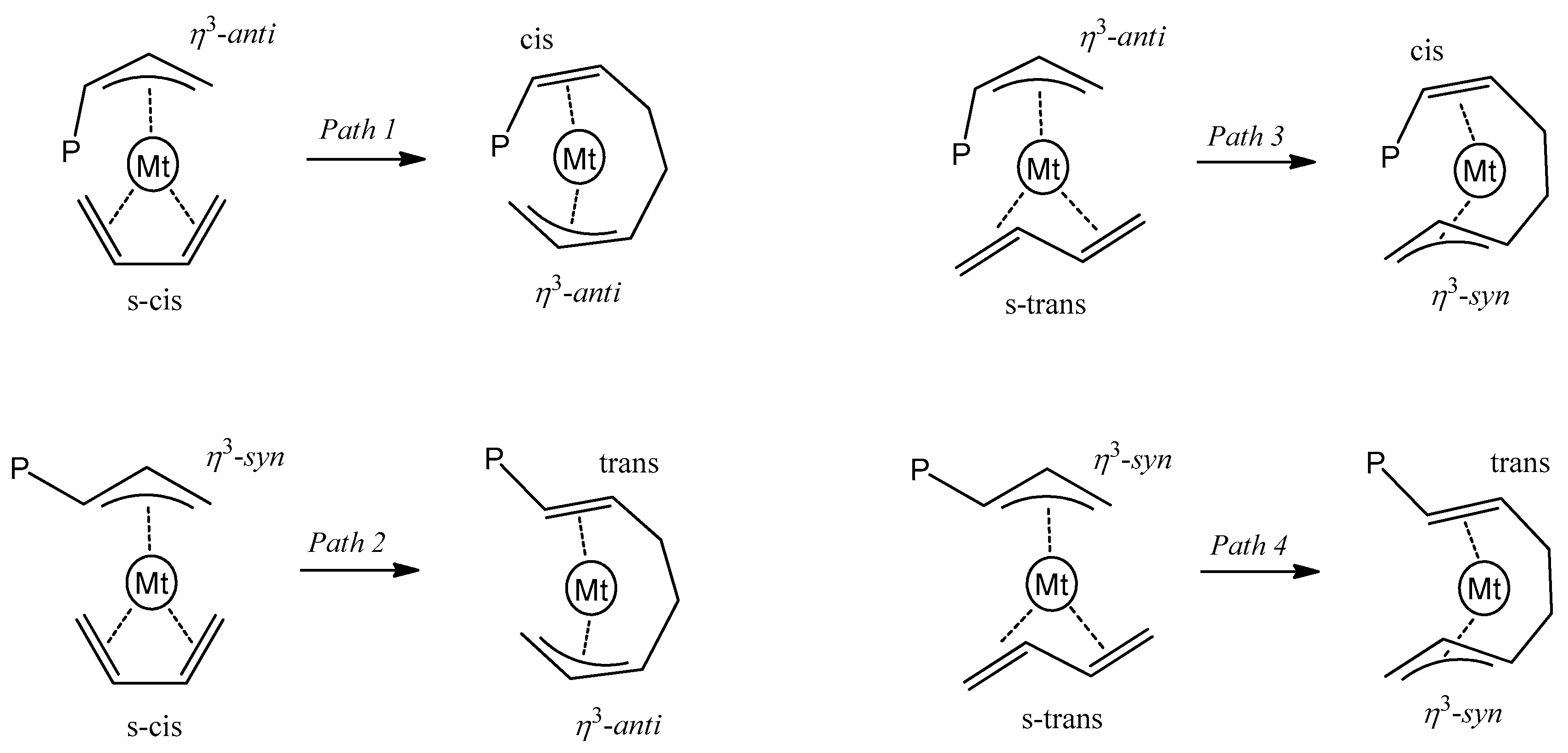
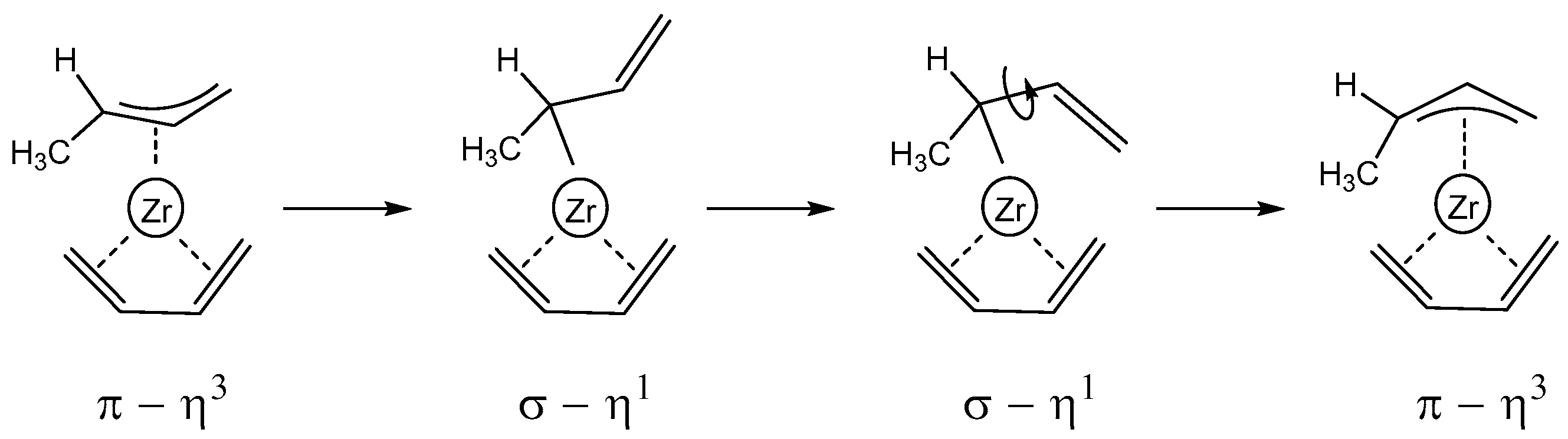
3.3. Polymerization Paths Promoted by Using the Zirconium-Based Catalyst
3.4. Polymerization Paths Promoted by Using the Titanium-Based Catalyst
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, A.; Mohanty, S.; Gupta, V.R. Butadiene Rubber: Synthesis, Microstructure, and Role of Catalysts. Rubber Chem. Technol. 2021, 94, 393–409. [Google Scholar] [CrossRef]
- Porri, L.; Giarrusso, A. Conjugated Diene Polymerization. In Comprehensive Polymer Science; Eastmon, G.C., Ledwith, A., Russo, S., Sigwalt, P., Eds.; Elsevier: Oxford, UK, 1989; Volume 4, pp. 53–108. [Google Scholar]
- Takeuchi, D. Stereoselective polymerization of conjugated dienes. In Encyclopedia of Polymer Science and Technology, 4th ed.; Mark, H.F., Ed.; Wiley: New York, NY, USA, 2014; Volume 13, pp. 126–150. [Google Scholar]
- Ricci, G.; Leone, G. Polymerization of 1,3-butadiene with organometallic complexes-based catalysts. In Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Four Volumes, 3rd ed.; Cornils, B., Hermann, W.A., Beller, M., Paciello, R., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2017; Volume 1, p. 251. [Google Scholar]
- Ricci, G.; Pampaloni, G.; Sommazzi, A.; Masi, F. Dienes Polymerization: Where We Are and What Lies Ahead. Macromolecules 2021, 54, 5879–5914. [Google Scholar] [CrossRef]
- Ricci, G.; Leone, G. Recent advances in the polymerization of butadiene over the last decade. Polyolefins J. 2014, 1, 43–60. [Google Scholar]
- Pino, P.; Giannini, U.; Porri, L. Insertion Polymerization. In Encyclopedia of Polymer Science and Engineering, 2nd ed.; Mark, H.F., Bikales, N.M., Overberger, C.G., Menges, G., Eds.; Wiley: New York, NY, USA, 1987; Volume 8, pp. 147–220. [Google Scholar]
- Porri, L.; Giarrusso, A.; Ricci, G. On the Mechanism of Formation of Isotactic and Syndiotactic Polydiolefins. Macromol. Symp. 2002, 178, 55–68. [Google Scholar] [CrossRef]
- Porri, L.; Ricci, G.; Shubin, N. Polymerization of 1,3-dienes with neodymium catalysts. Macromol. Symp. 1998, 128, 53–61. [Google Scholar] [CrossRef]
- Porri, L.; Giarrusso, A.; Ricci, G. Recent views on the mechanism of diolefin polymerization with transition metal initiator systems. Prog. Polym. Sci. 1991, 16, 405–441. [Google Scholar] [CrossRef]
- Pragliola, S.; Botta, A.; Longo, P. Solvent effect in 1,3-butadiene polymerization by cyclopentadienyl titanium trichloride (CpTiCl3)/methylaluminoxane (MAO) and pentamethylcyclopentadienyl titanium trichloride (Cp*TiCl3)/MAO catalysts. Eur. Polym. J. 2019, 111, 20–27. [Google Scholar] [CrossRef]
- Pragliola, S.; Costabile, C.; Venditto, V. Ethylene/1,3-butadiene cyclocopolymerization catalyzed by zirconocene systems. Eur. Polym. J. 2014, 58, 157–163. [Google Scholar] [CrossRef]
- Nath, D.C.D.; Shiono, T.; Ikeda, T. Copolymerization of 1,3-butadiene and isoprene with cobalt dichloride/methylaluminoxane in the presence of triphenylphosphine. J. Polym. Sci. A Polym. Chem. 2002, 40, 3086–3092. [Google Scholar] [CrossRef]
- Kaita, S.; Takeguchi, Y.; Hou, Z.; Nishiura, M.; Doi, Y.; Wakatsuki, Y. Pronounced Enhancement Brought in by Substituents on the Cyclopentadienyl Ligand: Catalyst System (C5Me4R)2Sm(THF)x/MMAO (R = Et, iPr, nBu, TMS; MMAO = Modified Methylaluminoxane) for 1,4-Cis Stereospecific Polymerization of 1,3-Butadiene in Cyclohexane Solvent. Macromolecules 2003, 36, 7923–7926. [Google Scholar]
- Maiwald, S.; Sommer, C.; Müller, G.; Taube, R. Highly Active Single-Site Catalysts for the 1,4-cis Polymerization of Butadiene from Allylneodymium(III) Chlorides and Trialkylaluminiums–A Contribution to the Activation of Tris(allyl)neodymium(III) and the Further Elucidation of the Structure-Activity Relationship. Macromol. Chem. Phys. 2002, 203, 1029–1039. [Google Scholar]
- Milione, S.; Cuomo, C.; Capacchione, C.; Zannoni, C.; Grassi, A.; Proto, A. Stereoselective Polymerization of Conjugated Dienes and Styrene−Butadiene Copolymerization Promoted by Octahedral Titanium Catalyst. Macromolecules 2007, 40, 5638–5643. [Google Scholar] [CrossRef]
- Pires, N.M.T.; Ferreira, A.A.; de Lira, C.H.; Coutinho, P.L.A.; Nicolini, L.F.; Soares, B.G.; Coutinho, F.M.B. Performance Evaluation of High-cis 1,4-Polybutadienes. J. Appl. Polym. Sci. 2006, 99, 88–99. [Google Scholar] [CrossRef]
- Pires, N.M.T.; Coutinho, F.M.B.; Costa, M.A.S. Synthesis and characterization of high cis-polybutadiene: Influence of monomer concentration and reaction temperature. Eur. Polym. J. 2004, 40, 2599–2603. [Google Scholar] [CrossRef]
- Gargani, L.; Bruzzone, M. Fatigue Resistance of Polybutadienes and Effect of Microstructure. In Advances in Elastomers and Rubber Elasticity; Lal, J., Mark, J.E., Eds.; Springer US: Boston, MA, USA, 1986; pp. 233–251. [Google Scholar]
- Ricci, G.; Sommazzi, A.; Masi, F.; Forni, A. Polymerization of 1,3-Butadiene with Catalysts Based on Cobalt Dichloride Complexes with Aminophosphines: Switching the Regioselectivity by Varying the MAO/Co Molar Ratio. Chin. J. Polym. Sci. 2023, in press. [Google Scholar] [CrossRef]
- Cass, P.; Pratt, K.; Mann, T.; Laslett, B.; Rizzardo, E.; Burford, R. Investigation of methylaluminoxane as a cocatalyst for the polymerization of 1,3-butadiene to high cis-1,4-polybutadiene. J. Polym. Sci. A Polym. Chem. 1999, 37, 3277–3284. [Google Scholar] [CrossRef]
- Endo, K.; Hatakeyama, N. Stereospecific and molecular weight-controlled polymerization of 1,3-butadiene with Co (acac) 3-MAO catalyst. J. Polym Sci. Part A Polym. Chem. 2001, 39, 2793–2798. [Google Scholar] [CrossRef]
- Sivaram, S.; Upadhyay, V.K. Synthesis of High–cis–Polybutadiene Using Cobalt (II)-2-Ethylhexoate Modified Triethylaluminum Catalyst. J. Macromol. Sci. Pure Appl. Chem. 1992, 29, 13–19. [Google Scholar] [CrossRef]
- Wang, B.; Liu, H.; Tang, T.; Zhang, T. cis-1,4 Selective Coordination Polymerization of 1,3-Butadiene and Copolymerization with Polar 2-(4-Methoxyphenyl)-1,3-butadiene by Acenaphthene-Based α-Diimine Cobalt Complexes Featuring Intra-Ligand π-π Stacking Interactions. Polymers 2021, 13, 3329. [Google Scholar] [CrossRef]
- Gong, D.; Jia, X.; Wang, B.; Zhang, X.; Jiang, L. Synthesis, characterization, and butadiene polymerization of iron(III), iron(II) and cobalt(II) chlorides bearing 2,6-bis(2-benzimidazolyl)pyridyl or 2,6-bis(pyrazol)pyridine ligand. J. Organomet. Chem. 2011, 702, 10–18. [Google Scholar] [CrossRef]
- Liu, W.; Pan, W.; Wang, P.; Li, W.; Mu, J.; Mu, J.; Weng, G.; Jia, X.; Gong, D.; Huang, K.W. Synthesis of mixed-ligand cobalt complexes and their applications in high cis-1,4-selective butadiene polymerization. Inorganica Chim. Acta 2015, 436, 132–138. [Google Scholar] [CrossRef]
- Gong, D.; Liu, W.; Pan, W.; Chen, T.; Jia, X.; Huang, K.W.; Zhang, X. Tunable regioselectivity in 1,3-butadiene polymerization by using 2,6-bis(dimethyl-2-oxazolin-2-yl)pyridine incorporated transition metal (Cr, Fe and Co) catalysts. J. Mol. Catal. A Chem. 2015, 406, 78–84. [Google Scholar] [CrossRef]
- Liu, J.; Fan, X.; Min, X.; Zhu, X.; Zhao, N.; Wang, Z. Synthesis of high cis-1,4 polybutadiene with narrow molecular weight distribution via a neodymium-based binary catalyst. RSC Adv. 2018, 8, 21926–21932. [Google Scholar] [CrossRef] [PubMed]
- Friebe, L.; Nuyken, O.; Obrecht, W. Ziegler/Natta Catalysts and their Application in Diene Polymerization. In Neodymium Based Ziegler Catalysts–Fundamental Chemistry. Advances in Polymer Science; Nuyken, O., Ed.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 204, pp. 5–7. [Google Scholar]
- Annunziata, L.; Pragliola, S.; Pappalardo, D.; Tedesco, C.; Pellecchia, C. New (Anilidomethyl)pyridine Titanium(IV) and Zirconium(IV) Catalyst Precursors for the Highly Chemo- and Stereoselective cis-1,4-Polymerization of 1,3-Butadiene. Macromolecules 2011, 44, 1934–1941. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. (Eds.) Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Becke, A. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Falivene, L.; Cao, Z.; Petta, A.; Serra, L.; Poater, A.; Oliva, R.; Scarano, V.; Cavallo, L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11, 872–879. [Google Scholar] [CrossRef]
- Taube, R.; Schmidt, U.; Gehrke, J.-P.; Böhme, P.; Langlotz, J.; Wache, S. New mechanistic aspects and structure activity relationships in the allyl nickel complex catalysed butadiene polymerization. Makromol. Chem. Makromol. Symp. 1993, 66, 245–260. [Google Scholar] [CrossRef]
- Taube, R.; Windisch, H.; Maiwald, S. The catalysis of the stereospecific butadiene polymerization by allyl nickel and allyl lanthanide complexes–A mechanistic comparison. Macromol. Symp. 1995, 89, 393–409. [Google Scholar] [CrossRef]
- Tobisch, S.; Bögel, H.; Taube, R. Mechanistic studies of the 1,4-polymerization of butadiene according to the π-allyl-insertion mechanism. 2. Density functional study of the C-C bond formation reaction in cationic and neutral (η3-crotyl)(η2-/η4-butadiene)nickel(II) complexes [Ni(C4H7)(C4H6)]+, [Ni(C4H7)]. Organometallics 1996, 15, 3563–3571. [Google Scholar] [CrossRef]
- Taube, R.; Sylvester, G. Applied Homogeneous Catalysis with Organometallic Complexes; Cornils, B., Herrmann, W.A., Eds.; VCH: Weinheim, Germany, 1996; pp. 280–317. [Google Scholar]
- Peluso, A.; Improta, R.; Zambelli, A. Mechanism of Isoprene and Butadiene Polymerization in the Presence of CpTiCl3−MAO Initiator: A Theoretical Study. Macromolecules 1997, 30, 2219–2227. [Google Scholar] [CrossRef]
- Guerra, G.; Cavallo, L.; Corradini, P.; Fusco, R. Molecular mechanics and stereospecificity in Ziegler-Natta 1,2 and cis-1,4 polymerizations of conjugated dienes. Macromolecules 1997, 30, 677–684. [Google Scholar] [CrossRef]
- Tobisch, S.; Taube, R. Mechanistic studies of the 1,4-polymerization of butadiene according to the π-allyl-insertion mechanism. 3. Density functional study of the C-C bond formation reaction in cationic “ligand-free” (η3:η2-heptadienyl) (η2-/η4-butadiene)nickel(II) complexes [Ni(C7H11)(C4H6)]+. Organometallics 1999, 18, 5204–5218. [Google Scholar]
- Costabile, C.; Milano, G.; Cavallo, L.; Guerra, G. Stereoselectivity and Chemoselectivity in Ziegler-Natta Polymerizations of Conjugated Dienes. 1. Monomers with Low-Energy s-Cis h4 Coordination. Macromolecules 2001, 34, 7952–7960. [Google Scholar] [CrossRef]
- Taube, R.; Sylvester, G. Stereospecific polymerization of butadiene or isoprene. In Applied Homogeneous Catalysis with Organometallic Compounds, 2nd ed.; Cornils, B., Herrmann, W.A., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2002; Volume 1, pp. 285–315. [Google Scholar]
- Tobisch, S. Reaction Mechanism in the Stereospecific 1,4-Polymerization of Butadiene with Ziegler-Natta Type Catalysts of Early Transition Metals: Comprehensive Density Functional Investigation for the Cationic [TiIIICp(polybutadienyl)(butadiene)]+ Active catalyst. Organometallics 2003, 22, 2729–2740. [Google Scholar] [CrossRef]
- Tobisch, S. The stereospecific polymerization of 1,3-butadiene mediated by early and late transition-metal catalysts. Towards a deeper understanding of the catalytic structure–reactivity relationships from computational-mechanistic studies. J. Mol. Struct. Theochem 2006, 771, 171–179. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Path 1 | Path 3 | ||||||
|---|---|---|---|---|---|---|---|
| 1Zr | TS1-2Zr | 2Zr | 1Zr | TS1-2Zr | 2Zr | ||
| anti–S/cis–S | 4.4 | 10.0 | −6.1 | anti–S/trans–P | 0.0 | 21.4 | −5.8 |
| anti–S/cis–P | 5.2 | 22.9 | −5.6 | anti–S/trans–S | 1.2 | 11.0 | −8.6 |
| anti–P/cis–S | 9.7 | 12.3 | −3.9 | anti–P/trans–P | 4.5 | 23.0 | −0.5 |
| anti–P/cis–P | 6.6 | 25.0 | −4.0 | anti–P/trans–S | 4.3 | 16.3 | −0.3 |
| Path 2 | Path 4 | ||||||
| 1Zr | TS1-2Zr | 2Zr | 1Zr | TS1-2Zr | 2Zr | ||
| syn–S/cis–S | 6.2 | 11.0 | −7.2 | syn–S/trans–P | 2.5 | 22.6 | −4.3 |
| syn–S/cis–P | 5.5 | 21.1 | −5.8 | syn–S/trans–S | 2.8 | 13.7 | −7.1 |
| syn–P/cis–S | 5.8 | 8.4 | −7.4 | syn–P/trans–P | 4.8 | 20.9 | −3.9 |
| syn–P/cis–P | 5.3 | 21.9 | −5.3 | syn–P/trans–S | 5.1 | 12.9 | −4.5 |
| Path 1 | Path 3 | ||||||
|---|---|---|---|---|---|---|---|
| 1Ti | TS1-2Ti | 2Ti | 1Ti | TS1-2Ti | 2Ti | ||
| anti–S/cis–S | 3.2 | 5.4 | −12.4 | anti–S/trans–P | 0.2 | 15.4 | −12.1 |
| anti–S/cis–P | 4.8 | 15.5 | −11.1 | anti–S/trans–S | 0.8 | 8.0 | −13.2 |
| anti–P/cis–S | 8.6 | 8.7 | −11.7 | anti–P/trans–P | 3.5 | 15.9 | −5.2 |
| anti–P/cis–P | 6.1 | 18.6 | −4.3 | anti–P/trans–S | 3.2 | 8.6 | −8.9 |
| Path 2 | Path 4 | ||||||
| 1Ti | TS1-2Ti | 2Ti | 1Ti | TS1-2Ti | 2Ti | ||
| syn–S/cis–S | 5.2 | - | −13.0 | syn–S/trans–P | 1.0 | 15.8 | −9.7 |
| syn–S/cis–P | 4.2 | 14.2 | −3.7 | syn–S/trans–S | 0.0 | 7.0 | −11.1 |
| syn–P/cis–S | 4.5 | 5.1 | −14.9 | syn–P/trans–P | 2.3 | 13.5 | −7.8 |
| syn–P/cis–P | 4.4 | 15.3 | −5.5 | syn–P/trans–S | 2.5 | 5.4 | −9.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milione, S.; Pragliola, S. Stereoselectivity in Butadiene Polymerization Promoted by Using Ziegler–Natta Catalysts Based on (Anilidomethyl)pyridine Group (IV) Complexes. Symmetry 2024, 16, 18. https://doi.org/10.3390/sym16010018
Milione S, Pragliola S. Stereoselectivity in Butadiene Polymerization Promoted by Using Ziegler–Natta Catalysts Based on (Anilidomethyl)pyridine Group (IV) Complexes. Symmetry. 2024; 16(1):18. https://doi.org/10.3390/sym16010018
Chicago/Turabian StyleMilione, Stefano, and Stefania Pragliola. 2024. "Stereoselectivity in Butadiene Polymerization Promoted by Using Ziegler–Natta Catalysts Based on (Anilidomethyl)pyridine Group (IV) Complexes" Symmetry 16, no. 1: 18. https://doi.org/10.3390/sym16010018