
Chiral Organophosphorus Pharmaceuticals: Properties and Application


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
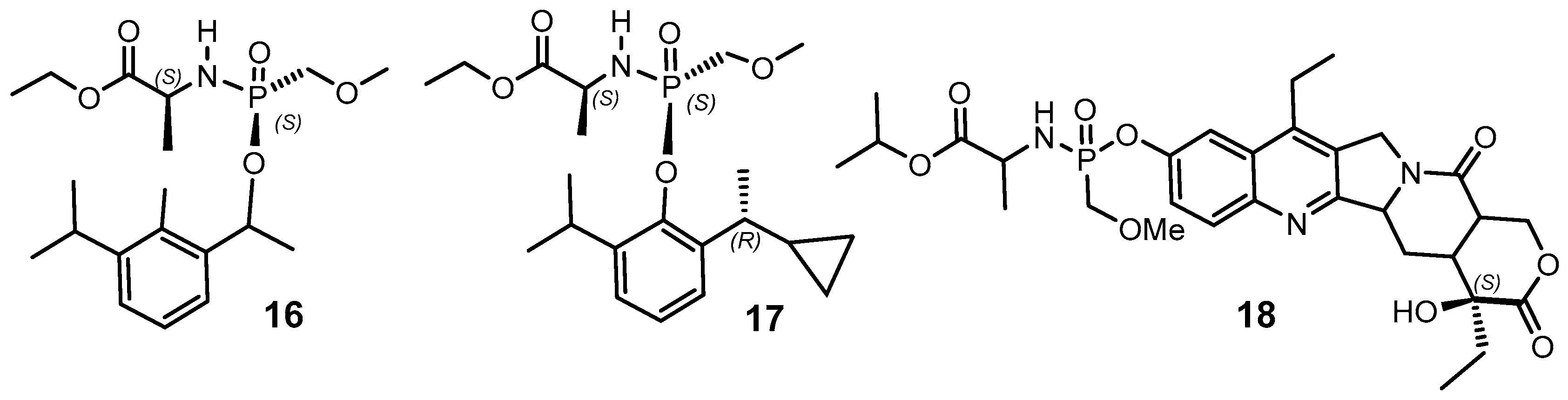{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
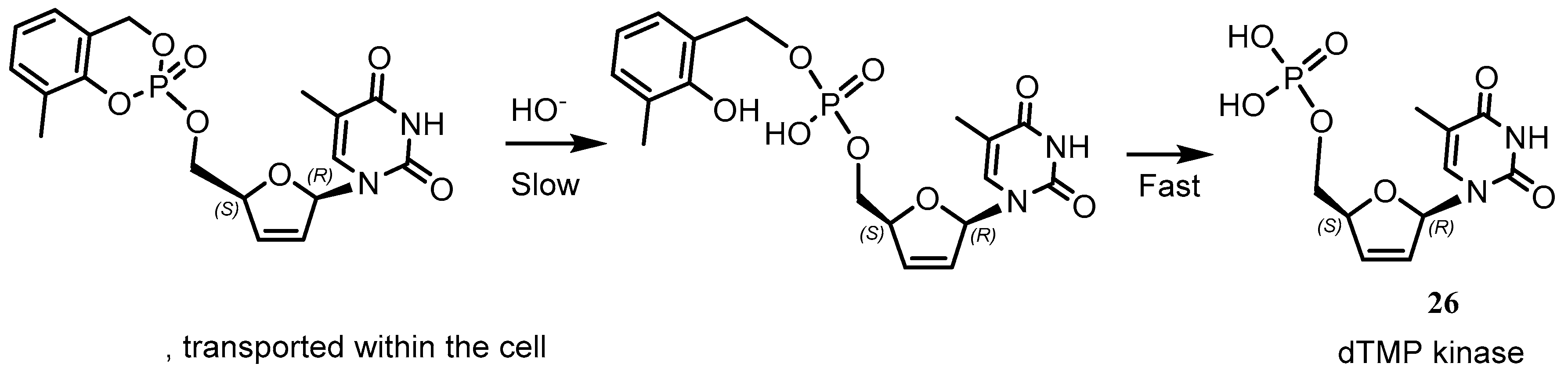{kind=link}
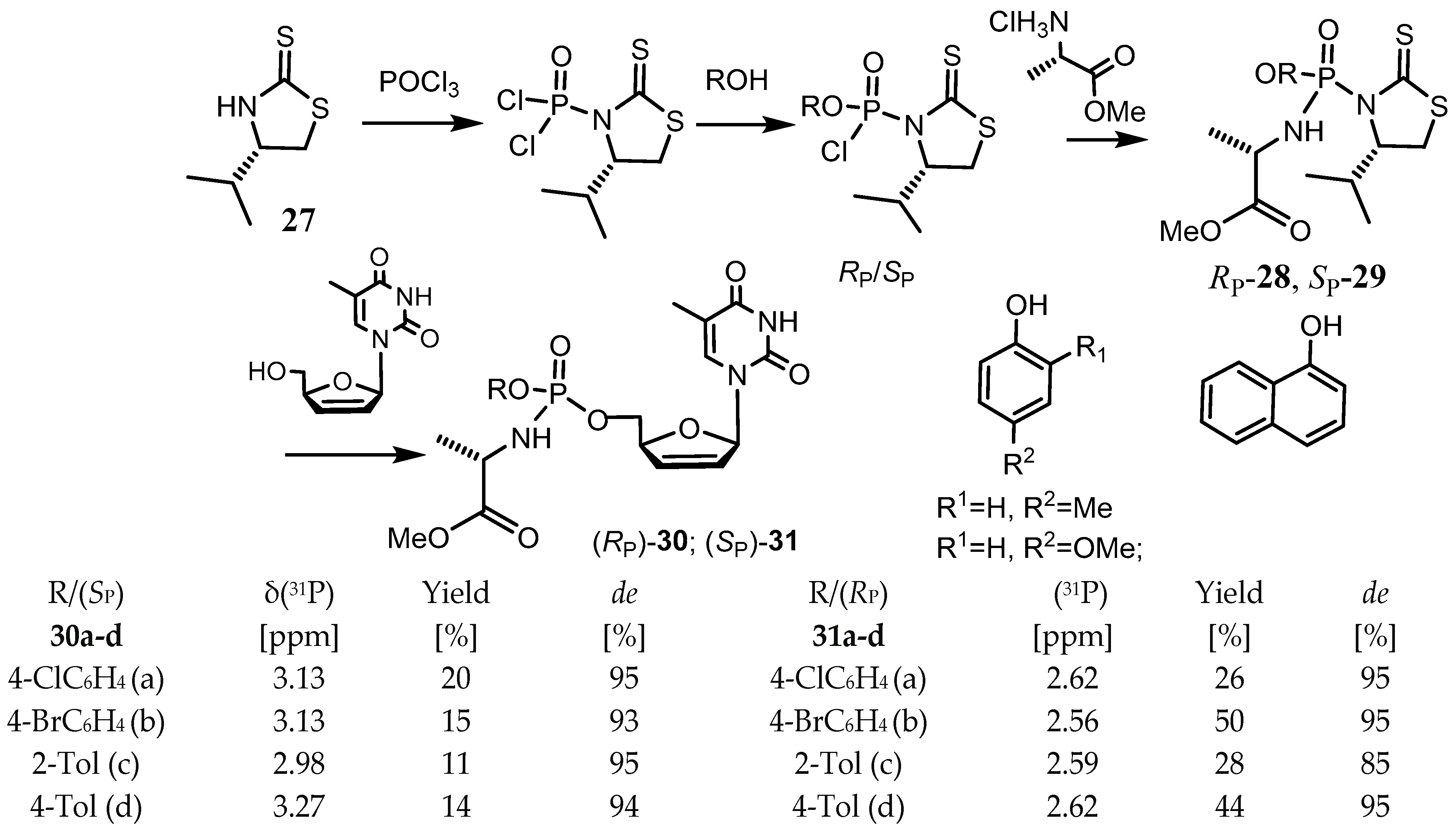{kind=link}
Abstract
:1. Introduction
2. Discussion
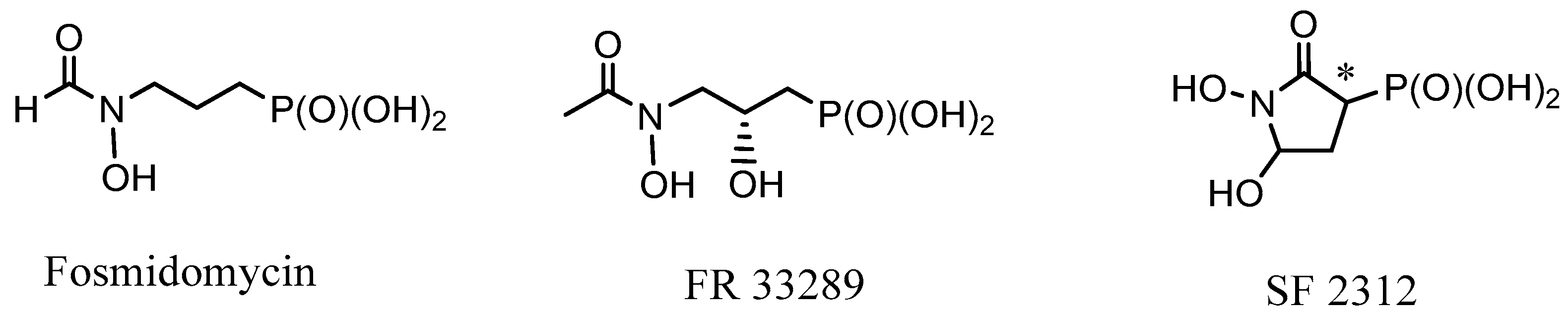
2.1. Chiral Natural Phosphorus Compounds
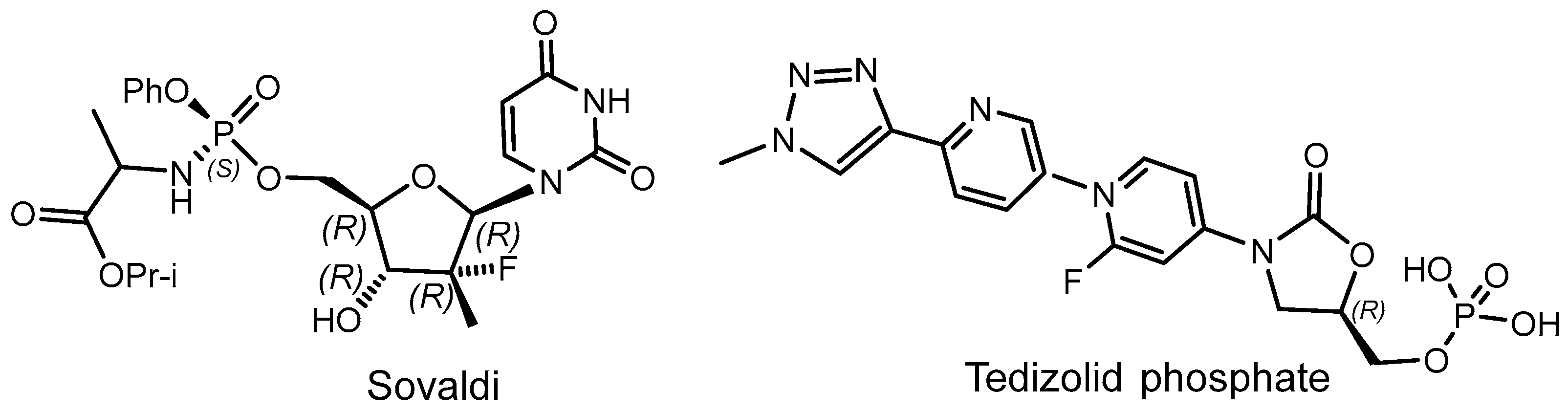
2.2. Synthetic Chiral Phosphorus Drugs
2.3. Phosphorus Prodrugs
3. Conclusions and Future Directions
4. Recommendations for Future Research
- (1)
- According to the FDA regulations, drug discovery and development researchers must determine at an early stage of research whether racemates or enantiomerically pure compounds should be sought.
- (2)
- Virtual screening and molecular modeling methods make it possible to identify leading compounds using calculated free binding energies. However, due to synthetic difficulties in obtaining enantiomerically pure stereomers and the methods for linking them, it is important to start by determining the exact stereochemistry of compounds in virtual drug candidate libraries.
- (3)
- In the approval process, justification must be provided for the development of a racemic mixture or an enantiomerically pure compound. Molecular modeling and virtual screening are indispensable tools in the early stages of new drug development in initial screening and design.
- (4)
- The effect of chirality in drugs is the main goal of the intensive research on the active principle of the drug being developed; the other enantiomer can be considered as “isomeric ballast”. However, it is not uncommon for the second enantiomer to exhibit significantly different biological properties, ranging from agonistic or antagonistic binding to the same receptor to interaction with other biological targets, which can lead to unwanted adverse effects.
- (5)
- In some cases, the presence of a distomer in a racemic mixture may interfere with the results due to the detrimental effect of the distomer or its conversion to the eutomeric configuration.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Food and Drug Administration. Development of New Stereoisomeric Drugs. 6 July 2005. Available online: www.fda.gov/Drugs/GuidanceComplianceRegulatorylnformation/Guidances/ucm122883.htm (accessed on 23 April 2010).
- Caldwell, J.; Wainer, I.W. Stereochemistry: Definitions and a note on nomenclature. Hum. Psychopharmacol. 2001, 16, S105–S107. [Google Scholar] [CrossRef]
- Sahajwalla, C. Regulatory Considerations in Drug Development of Stereoisomers, Chiralitv in Drug Design and Development; Reddy, I., Mehvar, R., Eds.; CRC Press: New York, NY, USA, 2004; ISBN 9780429215537. [Google Scholar]
- Agency, E.M. Investigation of Chiral Active Substances; 1994. Available online: www.ema.europa.eu/pdfs/human/qwp/3cc29aeu.pdf (accessed on 25 April 2010).
- Agranat, I.; Caner, H.; Caldwell, J. Putting chirality to work: The strategy of chiral switches. Nat. Rev. Drug Discov. 2002, 7, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Kolodiazhnyi, O.I. Asymmetric Synthesis in Organophosphorus Chemistry: Synthetic Methods, Catalysis and Application; Wiley VCH: Weinheim, Germany, 2016; ISBN 978-3-527-34150-4. [Google Scholar]
- Calcaterra, A.D.; Acquarica, I. The market of chiral drugs: Chiral switches versus de novo enantiomerically pure compounds. J. Pharm. Biomed. Anal. 2018, 147, 323–340. [Google Scholar] [CrossRef] [PubMed]
- Caner, H.; Groner, E.; Levy, L. Trends in the development of chiral drugs. Drug Discov. Today. 2004, 9, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Stinson, S. Chiral drugs. Chem. Eng. News 2000, 78, 55–78. [Google Scholar] [CrossRef]
- Núñez, M.C.; García-Rubiño, M.E.; Conejo-García, A.; Cruz-López, O.; Kimatrai, M.; Gallo, M.A.; Espinosa, A.; Campos, J.M. Homochiral drugs: A demanding tendency of the pharmaceutical industry. Curr. Med. Chem. 2009, 16, 2064–2074. [Google Scholar] [CrossRef]
- D’Acquarica, I.; Agranat, I. The Quest for Secondary Pharmaceuticals: Drug Repurposing/Chiral-Switches Combination Strategy. ACS Pharmacol. Transl. Sci. 2023, 6, 201–219. [Google Scholar] [CrossRef]
- Mentel, M.; Blankenfeldt, W.; Breinbauer, R. The active site of an enzyme can host both enantiomers of a racemic ligand simultaneously. Angew Chem. Int. Ed. Engl. 2009, 48, 9084–9087. [Google Scholar] [CrossRef]
- Takatsu, T.; Horiuchi, N.; Ishikawa, M.; Wanibuchi, K.; Moriguchi, T.; Takahashi, S. 1100-50, a novel nematocide from Streptomyces lavendulae SANK 64297. J. Antibiot. 2003, 56, 306–309. [Google Scholar] [CrossRef] [Green Version]
- Baba, M.; Okamoto, M.; Takeuchi, H. Inhibition of human immunodeficiency virus type 1 replication in acutely and chronically infected cells by EM2487, a novel substance produced by a Streptomyces species. Antimicrob. Agents Chemother. 1999, 43, 2350–2355. [Google Scholar] [CrossRef]
- Monroy-Noyola, A.; Sogorb, M.A.; Vilanova, E. Stereospecific hydrolysis of a phosphoramidate as a model to understand the role of biotransformation in the neurotoxicity of chiral organophosphorus compounds. Toxicol. Lett. 2007, 170, 157–164. [Google Scholar] [CrossRef] [PubMed]
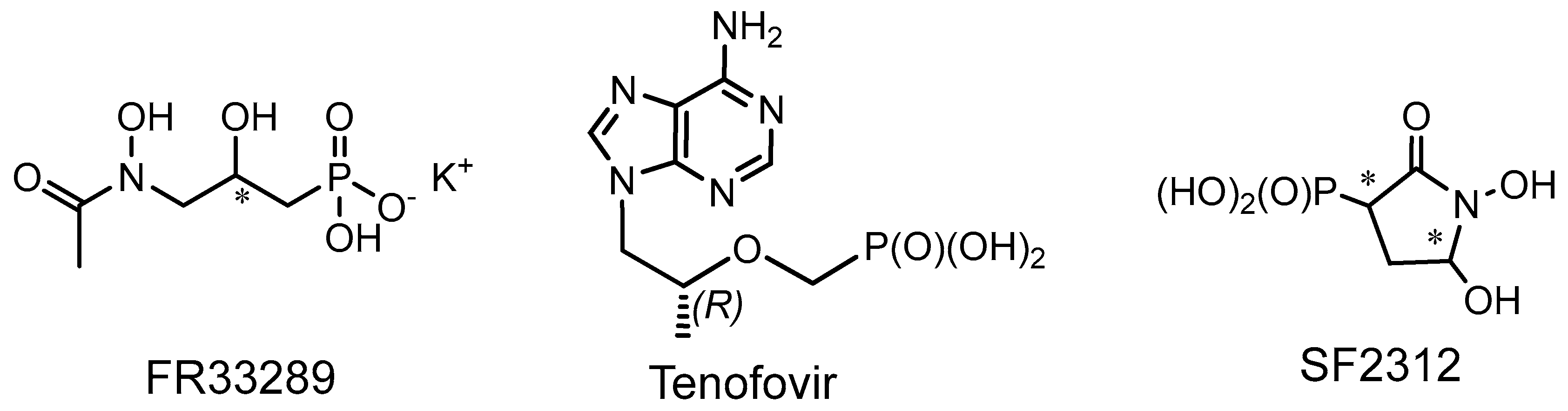
- Kolodiazhnyi, O.I. Phosphorus Compounds of Natural Origin: Prebiotic, Stereochemistry, Application. Symmetry 2021, 13, 889. [Google Scholar] [CrossRef]
- Kolodiazhna, A.O.; Kolodiazhnyi, O.I. Catalytic asymmetric synthesis of C-chiral phosphonate. Symmetry 2022, 14, 1758–1825. [Google Scholar] [CrossRef]
- Kolodiazhna, A.O.; Kolodiazhnyi, O.I. Asymmetric Electrophilic Reactions in Phosphorus Chemistry. Symmetry 2020, 12, 108. [Google Scholar] [CrossRef] [Green Version]
- Horsman, G.P.; Zechel, D.L. Phosphonate Biochemistry. Chem. Rev. 2017, 117, 5704–5783. [Google Scholar] [CrossRef]
- Hudson, H.; Kukhar, V.P. Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity; John Wiley: New York, NY, USA, 2000; 660p, ISBN 978-0-471-89149-9. [Google Scholar]
- Crooke, S.T.; Seth, P.P.; Vickers, T.A.; Liang, X.-H. The Interaction of Phosphorothioate-Containing RNA Targeted Drugs with Proteins Is a Critical Determinant of the Therapeutic Effects of These Agents. J. Am. Chem. Soc. 2020, 142, 14754–14771. [Google Scholar] [CrossRef]
- Falagas, M.E.; Vouloumanou, K.; Samonis, G.; Vardakas, K.Z. Fosfomycin. Clin. Microbiol. Rev. 2016, 29, 321–347. [Google Scholar] [CrossRef] [Green Version]
- Omura, S.; Murata, M.; Hanaki, H.; Hinotozawa, K.; Oiwa, R.; Tanaka, H. Phosalacine, a new herbicidal antibiotic containing phosphinothricin. Fermentation, isolation, biological activity and mechanism of action. J. Antibiot. 1984, 37, 829–835. [Google Scholar] [CrossRef]
- Lell, B.; Ruangweerayut, R.; Wiesner, J.; Missinou, M.A.; Schindler, A.; Baranek, T.; Hintz, M.; Hutchinson, D.; Jomaa, H.; Kremsner, P.G. Fosmidomycin, a novel chemotherapeutic agent for malaria. Antimicrob. Agents Chemother. 2003, 47, 735–738. [Google Scholar] [CrossRef] [Green Version]
- Gahungu, M.; Arguelles-Arias, A.; Fickers, P.; Zervosen, A.; Joris, B.; Damblon, C. Synthesis and biological evaluation of potential threonine synthase inhibitors: Rhizocticin A and Plumbemycin, A. Bioorg. Med. Chem. 2013, 21, 4958–4967. [Google Scholar] [CrossRef]
- Takeuchi, M.; Nakajima, M.; Ogita, T.; Inukai, M.; Kodama, K.; Furuya, K.; Nagaki, H.; Haneishi, T. Fosfonochlorin, a new antibiotic with spheroplast forming activity. J. Antibiot. 1989, 42, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, K.; Kohno, T.; Uno, T.; Morita, T.; Tsukamoto, G. Organic phosphorus compounds 1.4-(Benzothiazol-2-yl) benzylphosphonate as potent calcium antagonistic vasodilator. J. Med. Chem. 1986, 29, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, R.; Zucchelli, P.; Caspi, A.; Nouriel, H.; Paz, R.; Sclarovsky, S. Comparison of the pharmacokinetics of fosinoprilat with enalaprilat and lisinopril in patients with congestive heart failure and chronic renal insufficiency. Br. J. Clin. Pharmacol. 2000, 49, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchin, K.L.; Waclawki, A.P.; Tu, J.I.; Manning, J.; Frantz, M.; Willard, D.A. Pharmacokinetics, safety, and pharmacologic effects of fosinopril sodium, an angiotensin-converting enzyme inhibitor in healthy subjects. J. Clin. Pharmacol. 1991, 31, 58–64. [Google Scholar] [CrossRef]
- Cohen, P. The origins of protein phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130. [Google Scholar] [CrossRef]
- Mita, M.M.; Gong, J.; Chawla, S.P. Ridaforolimus in advanced or metastatic soft tissue and bone sarcomas. Expert Rev. Clin. Pharmacol. 2013, 6, 465–482. [Google Scholar] [CrossRef]
- Nakamura, T.; Hashimoto, I.; Sawada, Y.; Mikami, J.; Bekki, E. Clinical studies on fosfomycin sodium following intravenous administration (tissue concentration and clinical efficacy). Jpn. J. Antibiot. 1985, 38, 2057–2067. [Google Scholar] [PubMed]
- García-Rodríguez, J.A.; Martín, I.T.; Baquero, F.; Cisterna, R.; Gobernado, M.; Liñares, F.L. In vitro activity of fosfomycin trometamol against pathogens from urinary tract infections: A Spanish multicenter study. J. Chemother. 1997, 9, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Cappello, A.R.; Madeo, M.; Martello, E.; Iacopetta, D.; Dolce, V. Interaction of fosfomycin with the glycerol 3-phosphate transporter of Escherichia coli. Biochim. Et Biophys. Acta (BBA) 2011, 1810, 1323–1329. [Google Scholar] [CrossRef]
- Patel, S.S.; Balfour, J.A.; Bryson, H.M. Fosfomycin tromethamine. A review of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy as a single-dose oral treatment for acute uncomplicated lower urinary tract infections. Drugs 1997, 53, 637–656. [Google Scholar] [CrossRef]
- Rieusset, J.; Touri, F.; Michalik, L.; Escher, P.; Desvergne, B.; Niesor, E.; Wahli, W. A New Selective Peroxisome Proliferator-Activated Receptor γ Antagonist with Antiobesity and Antidiabetic Activity. Mol. Endocrinol. 2002, 16, 2628–2644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilote, L.; Abrahamowicz, M.; Eisenberg, M.; Humphries, K.; Behlouli, H.; Tu, J.V. Effect of different angiotensin-converting-enzyme inhibitors on mortality among elderly patients with congestive heart failure. CMAJ 2008, 178, 1303–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuhara, M.; Kuroda, Y.; Goto, T.; Okamoto, M.; Terano, H.; Kohsaka, M. Studies on a new phosphonic acid antibiotic III.Isolation and characterisation of FRFR-31564, FR-32863 and FR-33289. J. Antibiot. 1980, 33, 24–28. [Google Scholar] [CrossRef] [Green Version]
- Bayer, E.; Gugel, K.H.; Hägele, K.; Hagenmaier, H.; Jessipow, S.; König, W.A.; Zähner, H. Metabolic Products of Microorganisms.98. Phosphinothricin and Phosphinothricyl-Alanyl-Alanine. Helv. Chim. Acta 1972, 55, 224–239. [Google Scholar] [CrossRef]
- Donn, G.; Köcher, H. Inhibitors of Glutamine Synthetase. In Herbicide Classes in Development; Springer: Berlin/Heidelberg, Germany, 2002; pp. 87–101. [Google Scholar]
- Mowbray, S.L.; Kathiravan, M.K.; Pandey, A.A.; Odell, L.R. Inhibition of Glutamine Synthetase: A Potential Drug Target in Mycobacterium Tuberculosis. Molecules 2014, 19, 13161–13176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, H.; Ohkubo, A.; Sekine, M.; Seio, K.; Kakeya, H.; Osada, H. Synthesis and Biological Properties of New Phosmidosine Analogs Having an N-Acylsulfamate. Nucleosides Nucleotides Nucleic Acids 2006, 25, 647–654. [Google Scholar] [CrossRef]
- Sekine, M.; Okada, K.; Seio, K.; Kakeya, H.; Osada, H.; Obata, T.; Sasaki, T. Synthesis of Chemically Stabilized Phosmidosine Analogues and the Structure-Activity Relationship of Phosmidosine. J. Org. Chem. 2004, 69, 314–326. [Google Scholar] [CrossRef]
- Sekine, M.; Okada, K.; Seio, K.; Obata, T.; Sasaki, T.; Kakeya, H.; Osada, H. Synthesis of a biotin-conjugate of phosmidosine O-ethyl ester as a G1 arrest antitumor drug. Bioorg. Med. Chem. 2004, 12, 6343–6349. [Google Scholar] [CrossRef]
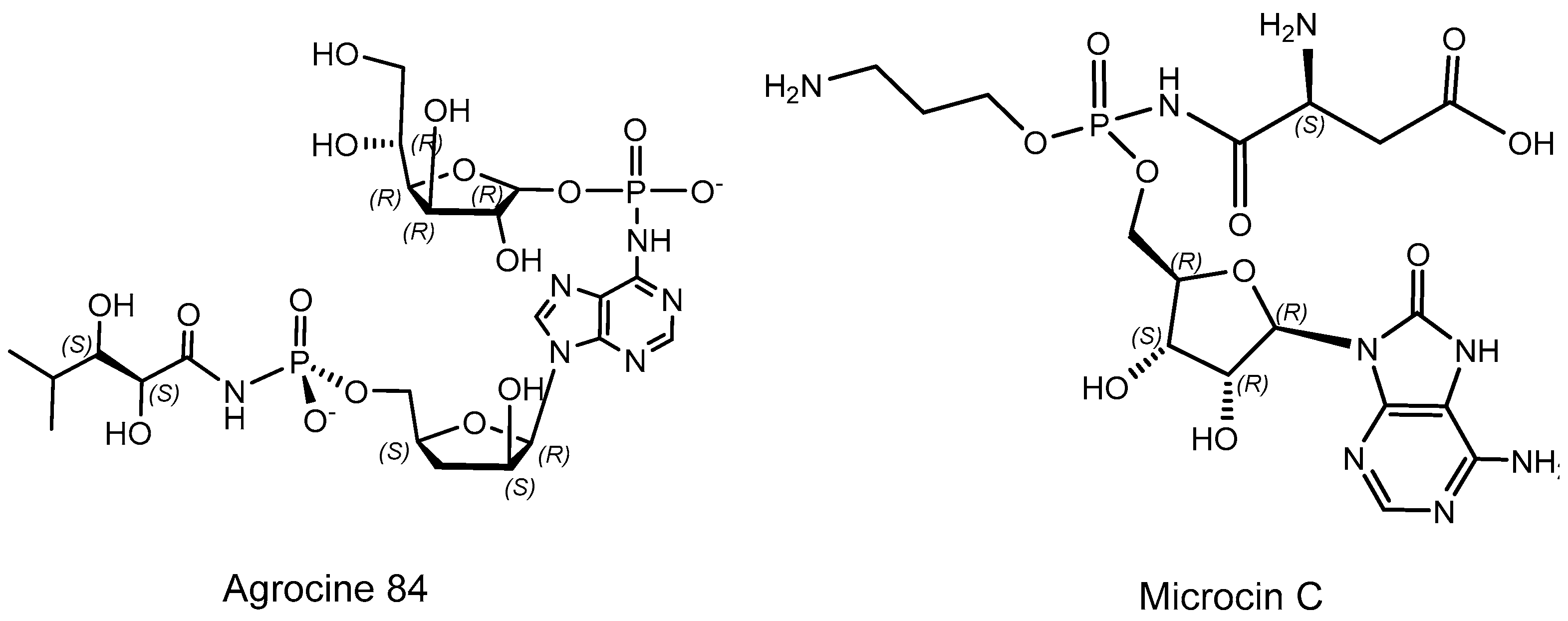
- Roberts, W.P.; Tate, M.E.; Kerr, A. Agrocin 84 is a 6-N-phosphoramidate of an adenine nucleotide analogue. Nature 1977, 265, 379–381. [Google Scholar] [CrossRef]
- Kim, J.-G.; Park, B.K.; Kim, S.-U.; Choi, D.; Nahm, B.H.; Moon, J.S. Bases of biocontrol: Sequence predicts synthesis and mode of action of agrocin 84, the Trojan Horse antibiotic that controls crown gall. Proc. Natl. Acad. Sci. USA 2006, 103, 8846–8851. [Google Scholar] [CrossRef]
- Pertusati, F.; McGuigana, C. Diastereoselective synthesis of P-chirogenic phosphoramidate prodrugs of nucleoside analogues (ProTides) via copper catalysed reaction. Chem. Commun. 2015, 51, 8070–8073. [Google Scholar] [CrossRef] [Green Version]
- Tate, M.E.; Murphy, P.J.; Roberts, W.P.; Kerr, A. Adenine N6-substituent of agrocin 84 determines its bacteriocin-like specificity. Nature 1979, 280, 697–699. [Google Scholar] [CrossRef] [PubMed]
- Reader, J.S.; Ordoukhanian, P.T.; Kim, J.-G.; de Crecy-Lagard, V.; Hwang, I.; Farrand, S.; Schimmel, P. Major Biocontrol of Plant Tumors Targets tRNA Synthetase. Science 2005, 309, 1533 LP–1553 LP. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz-Yepes, J.; Brahamsha, B.; Palenik, B. Role of a microcin-C-like biosynthetic gene cluster in allelopathic interactions in marine Synechococcus. Proc. Natl. Acad. Sci. USA 2013, 110, 12030–12035. [Google Scholar] [CrossRef] [PubMed]
- Eliot, A.C.; Griffin, B.M.; Thomas, P.M.; Johannes, T.W.; Kelleher, N.L.; Zhao, H.; Metcalf, W.W. Cloning, expression, and biochemical characterization of Streptomyces rubellomurinus genes required for biosynthesis of antimalarial compound FR900098. Chem. Biol. 2008, 15, 765–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baquero, F.; Lanza, V.F.; Baquero, M.-R.; del Campo1, R.; Bravo-Vázquez, D.A. Microcins in Enterobacteriaceae: Peptide Antimicrobials in the Eco-Active Intestinal Chemosphere. Front. Microbiol. 2019, 10, 2261–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga, P.R.; Szabó, R.O.; Dormán, G.; Bősze, S.; Keglevich, G. Cytotoxic Activity of α-Aminophosphonic Derivatives Coming from the Tandem Kabachnik–Fields Reaction and Acylation. Pharmaceuticals 2023, 16, 506. [Google Scholar] [CrossRef]
- Karpus, A.; Yesypenko, O.; Cherenok, S.; Boiko, V.; Kalchenko, O.; Voitenko, Z.; Tribrat, O.; Poli, R.; Daran, J.-C.; Manoury, E.; et al. Chiral phosphorus-containing calixarenes. Phosph. Sulf. Silicon Relat. Elem. 2019, 194, 471–475. [Google Scholar] [CrossRef]
- Buldenko, V.M.; Trush, V.V.; Kobzar, O.L.; Drapailo, A.B.; Kalchenko, V.I.; Vovk, A.I. Calixarene-based phosphinic acids as inhibitors of protein tyrosine phosphatases. Bioorg. Med. Chem. Lett. 2019, 29, 797–801. [Google Scholar] [CrossRef]
- Rodriguez, J.B.; Falcone, B.N.; Szajnman, S.H. Approaches for Designing new Potent Inhibitors of Farnesyl Pyrophosphate Synthase. Expert Opin. Drug Discov. 2016, 11, 307–320. [Google Scholar] [CrossRef]
- Russell, R.G.G. Bisphosphonates: The first 40 years. Bone 2011, 49, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.J.; Frith, J.C.; Luckman, S.P.; Coxon, F.P.; Benford, H.L.; Mçnkkçnen, A.S.; Chilton, K.M.; Russell, R.G.G. Molecular mechanisms of action of bisphosphonates. Bone 1999, 24, 73S–79S. [Google Scholar] [CrossRef] [PubMed]
- Kolodiazhnyi, O.I.; Kolodiazhna, O.O. New Catalyst for Phosphonylation of C=X Electrophiles. Synth. Comm. 2012, 42, 1637–1649. [Google Scholar] [CrossRef]
- Kolodiazhna, O.O.; Kolodiazhna, A.O.; Kolodiazhnyi, O.I. Highly effective catalyst for the reaction of trialkylphosphites with C=X electrophiles. Phosph. Sulf. Silicon Relat. Elem. 2011, 186, 796–798. [Google Scholar] [CrossRef]
- Haratipour, P.; Minard, C.; Nakhjiri, M.; Negahbani, A.; Kashemirov, B.A.; McKenna, C.E. New chirally modified bisphosphonates for synthesis of individual beta,gamma-CHX-deoxynucleotide diastereomers Phosph. Sulf. Silicon Relat. Elem. 2019, 194, 329–330. [Google Scholar] [CrossRef] [PubMed]
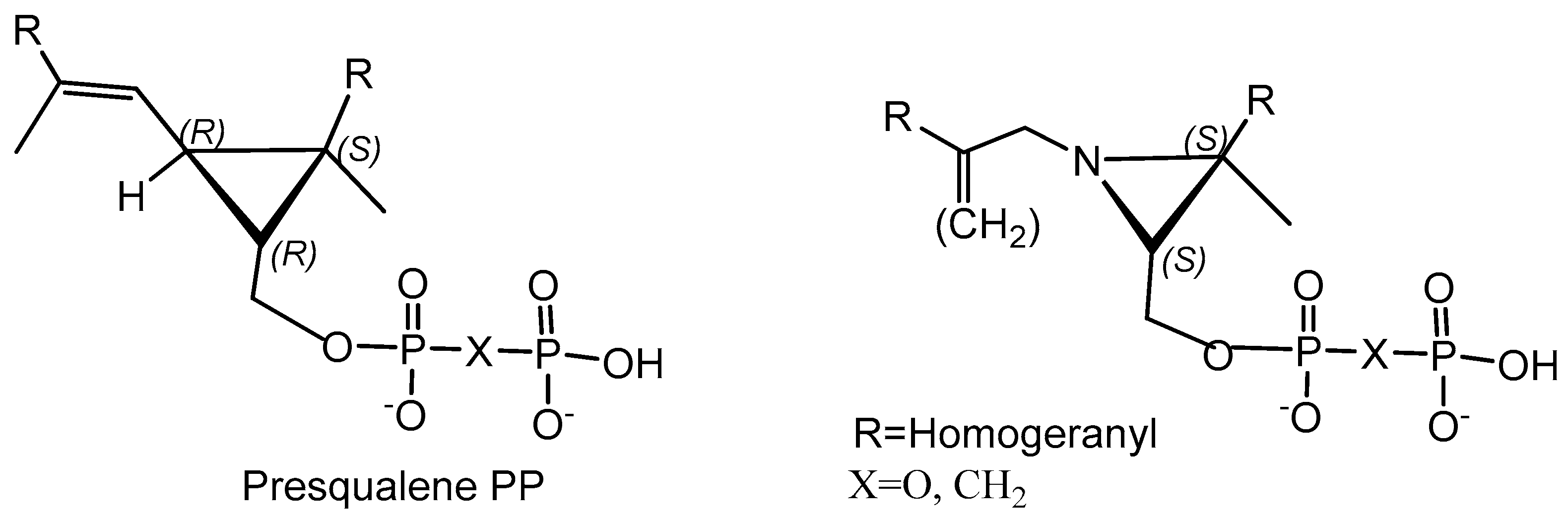
- Koohang, A.; Bailey, J.L.; Coates, R.M.; Erickson, H.K.; Owen, D.; Doulter, C.J. Enantioselective Inhibition of Squalene Synthase by Aziridine Analogues of Presqualene Diphosphate. J. Org. Chem. 2010, 75, 4769–4777. [Google Scholar] [CrossRef] [Green Version]
- Wasko, B.M.; Smits, J.P.; Shull, L.W.; Wiemer, D.F.; Hohl, R.J. A novel bisphosphonate inhibitor of squalene synthase combined with a statin or a nitrogenous bisphosphonate in vitro. J. Lipid Res. 2011, 52, 1957–1964. [Google Scholar] [CrossRef] [Green Version]
- Magnin, D.R.; Biller, S.A.; Dickson, J.K., Jr.; Logan, J.V.; Lawrence, R.M.; Chen, Y.; Sulsky, R.B.; Ciosek, C.P., Jr.; Harrity, T.W.; Jolibois, K.G.; et al. 1,l-Bisphosphonate Squalene Synthase Inhibitors: Interplay Between the Isoprenoid Subunit and the Diphosphate Surrogate. J. Med. Chem. 1995, 38, 2596–2605. [Google Scholar] [CrossRef]
- Magnin, D.R.; Biller, S.A.; Chen, Y.; Dickson, J.K.; Fryszman, O.M.; Lawrence, R.M.; Logan, J.V.H.; Sieber-McMmaster, E.S.; Sulsky, R.B.; Traeger, S.C.; et al. r-Phosphonosulfonic Acids: Potent and Selective Inhibitors of Squalene Synthase. J. Med. Chem. 1996, 39, 657–660. [Google Scholar] [CrossRef]
- Lawrence, R.M.; Biller, S.A.; Dickson, J.K.; Logan, J.V.H.; Magnin, D.R.; Sulsky, R.B.; DiMarco, J.D.; Gougoutas, J.Z.; Beyer, B.D.; Taylor, S.C.; et al. Enantioselective Synthesis of r-Phosphono Sulfonate Squalene Synthase Inhibitors: Chiral Recognition in the Interactions of an r-Phosphono Sulfonate Inhibitor with Squalene Synthase. J. Am. Chem. Soc. 1996, 118, 11668–11669. [Google Scholar] [CrossRef]
- Ciosek, C.P., Jr.; Magnin, D.R.; Harrity, T.W.; Logan, J.V.; Dickson, J.K., Jr.; Gordon, E.M.; Hamilton, K.A.; Jolibois, K.G.; Kunselman, L.K.; Lawrence, R.M. Lipophilic 1,1-bisphosphonates are potent squalene synthase inhibitors and orally active cholesterol lowering agents in vivo. J. Biol. Chem. 1993, 268, 24832–24837. [Google Scholar] [CrossRef]
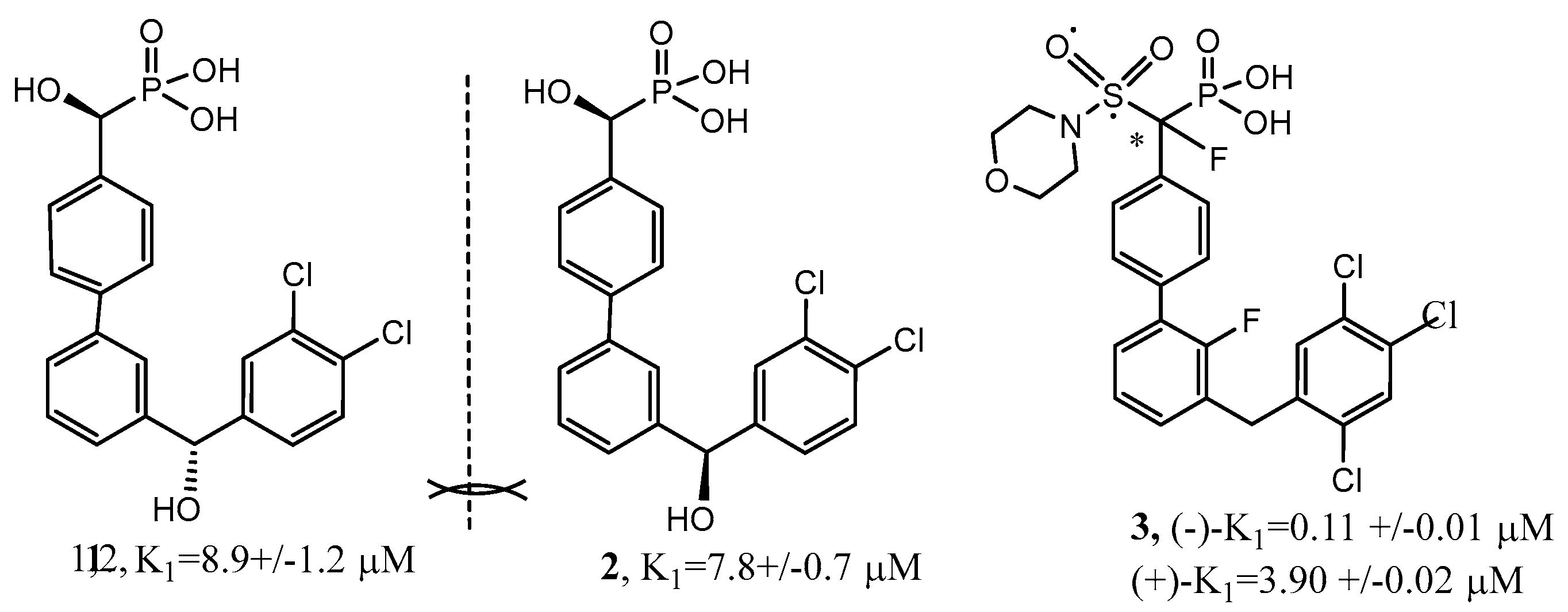
- Johnson, S.A.; Hunter, T. Kinomics: Methods for deciphering the kinome. Nat. Methods 2005, 2, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Baguley, T.D.; Xu, H.-D.; Chatterjee, M.; Nairn, A.C.; Lombroso, P.J.; Ellman, J.A. Substrate-Based Fragment Identification for the Development of Selective, Nonpeptidic Inhibitors of Striatal-Enriched Protein Tyrosine Phosphatase. J. Med. Chem. 2013, 56, 7636–7650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witten, M.R.; Wissler, L.; Snow, M.; Geschwindner, S.; Read, J.A.; Brandon, N.J.; Nairn, A.C.; Lombroso, P.J.; Kack, H.; Ellman, J.A. X-ray Characterization and Structure-Based Optimization of Striatal-Enriched Protein Tyrosine Phosphatase Inhibitors. J. Med. Chem. 2017, 60, 9299–9319. [Google Scholar] [CrossRef]
- Barr, A.J.; Ugochukwu, E.; Lee, W.H.; King, O.N.F.; Filippakopoulos, P.; Alfano ISavitsky, P.; Burgess-Brown, N.A.; Muller, S.; Knapp, S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 2009, 136, 352–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chofor, R.; Sooriyaarachchi, S.; Risseeuw, M.D.P.; Bergfors, T.; Pouyez, J.; Johny, C.; Haymon, A.; Everaert, A.; Dowd, C.S.; Maes, L.; et al. Synthesis and Bioactivity of β-Substituted Fosmidomycin Analogues Targeting 1-Deoxy-D-xylulose-5-phosphate Reductoisomerase. J. Med. Chem. 2015, 58, 2988–3001. [Google Scholar] [CrossRef] [Green Version]
- Kafarski, P. Phosphonopeptides containing free phosphonic groups: Recent advances. RSC Adv. 2020, 10, 25898–25910. [Google Scholar] [CrossRef]
- Blodgett, J.A.V.; Zhang, J.K.; Yu, X.; Metcalf, W.W. Conserved biosynthetic pathways for phosalacine, bialaphos and newlydiscovered phosphonic acid natural products. J. Antibiot. 2016, 69, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Peck, S.C.; van der Donk, W. Phosphonate biosynthesis and catabolism: A treasure trove for unusual enzymology. Curr. Opin. Chem. Biol. 2013, 17, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.T. The Chemical Biology of Phosphorus; RSC: Cambridge, UK, 2020; 546p. [Google Scholar] [CrossRef]
- Rautio, J.; Meanwell, N.A.; Li, D.; Hageman, M.J. The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 2018, 17, 559–587. [Google Scholar] [CrossRef]
- Sofosbuvir (Sovaldi)—Treatment—Hepatitis C Online. Available online: www.hepatitisc.uw.edu (accessed on 8 January 2017).
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Hecker, S.J.; Erion, M.D. Prodrugs of phosphates and phosphonates. J. Med. Chem. 2008, 51, 2328–2345. [Google Scholar] [CrossRef]
- Wiemer, A.J.; Wiemer, D.F. Prodrugs of phosphonates and phosphates: Crossing the membrane barrier. Top. Curr. Chem. 2015, 360, 115–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuts, P.G.M. (Ed.) Protection for the Phosphate Group. In Greene’s Protective Groups in Organic Synthesis; Wiley: Hoboken, NJ, USA, 2014. [Google Scholar]
- Meier, C.; Gorbig, U.; Muller, C.; Balzarini, J. cycloSal-PMEA and cycloAmb-PMEA: Potentially new phosphonate prodrugs based on the cycloSal-pronucleotide approach. J. Med. Chem. 2005, 48, 8079–8086. [Google Scholar] [CrossRef]
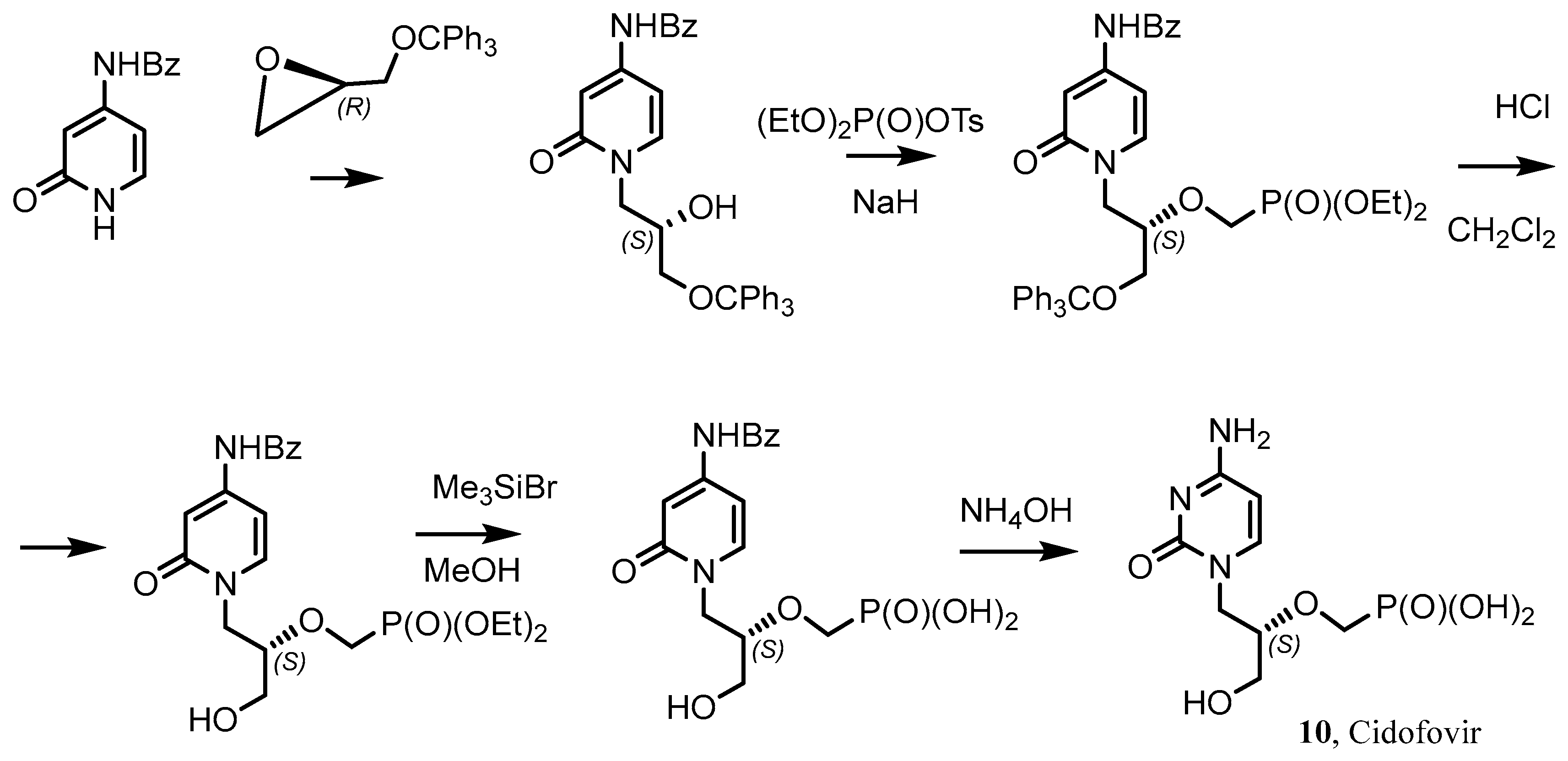
- Cundy, K.C. Clinical Pharmacokinetics of the Antiviral Nucleotide Analogues Cidofovir and Adefovir. Clin. Pharmacokinet. 1999, 36, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Vistide (Cidofovir) Dosing, Indications, Interactions, Adverse Effects, and More. Medscape Reference. WebMD. Available online: https://reference.medscape.com/drug/cidofovir-342606 (accessed on 4 February 2014).
- De Gascun, C.F.; Carr, M. Human polyomavirus reactivation: Disease pathogenesis and treatment approaches. Clin. Dev. Immunol. 2013, 2013, 373579. [Google Scholar] [CrossRef] [Green Version]
- Brodfuehrer, P.R.; Howell, H.G.; Sapino, C., Jr.; Vemishetti, P. A practical synthesis of (S)-HPMPC. Tetrahedron Lett. 1994, 35, 3243. [Google Scholar] [CrossRef]
- Florescu, D.F.; Keck, M.A. Development of CMX001 (Brincidofovir) for the treatment of serious diseases or conditions caused by dsDNA viruses. Expert Rev. Anti-Infect. Ther. 2014, 12, 1171–1178. [Google Scholar] [CrossRef]
- Birnkrant, D. Brincidofovir: NDA Approval—Animal Efficacy; Center for Drug Evaluation and Research. U.S. Food and Drug Administration: Silver Spring, MD, USA, 2021. [Google Scholar]
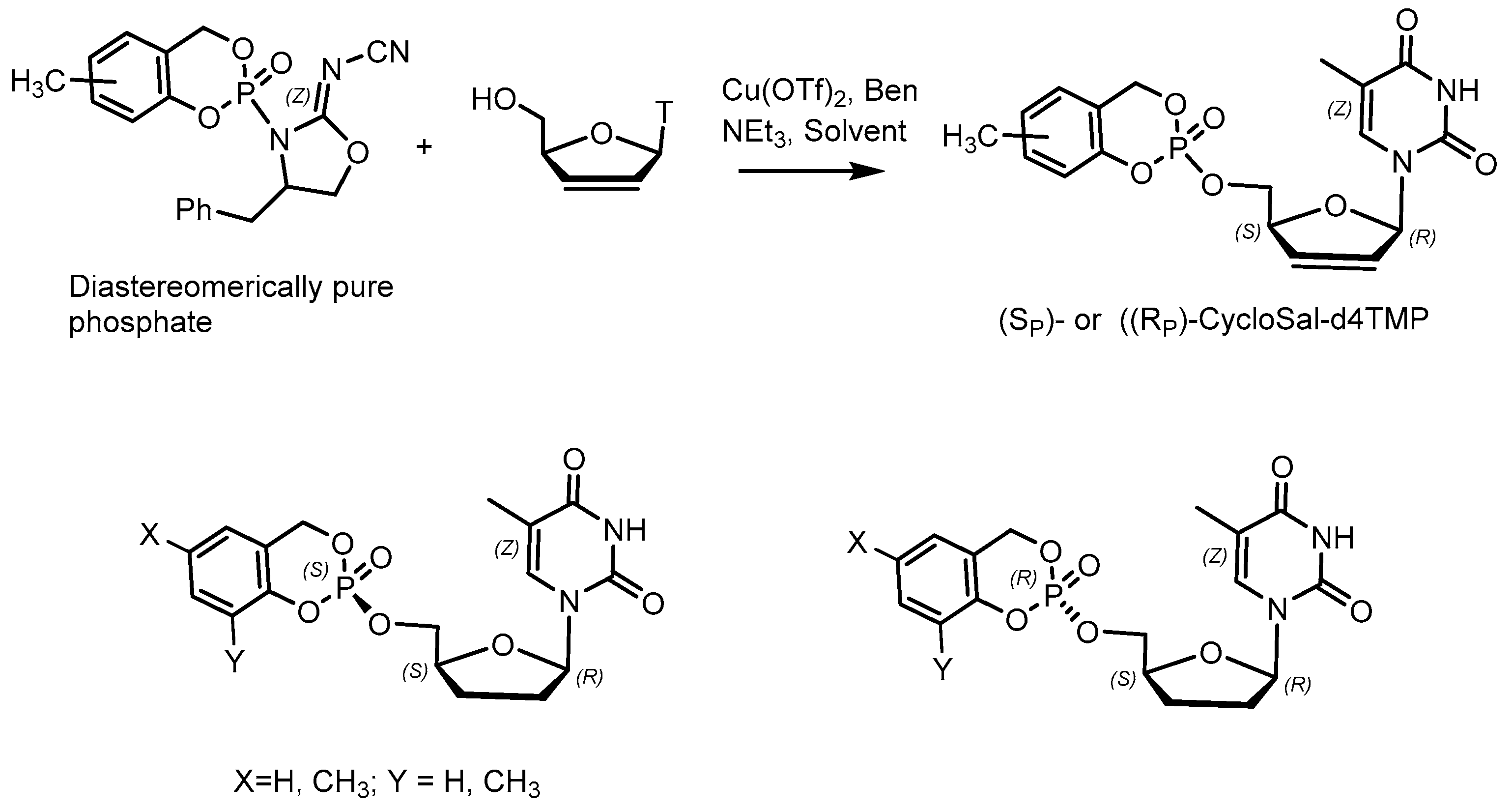
- Meier, C.; Lorey, M.; De Clercq, E.; Balzarini, J. cycloSal-2, 3dideoxy-2,3-didehydrothymidine monophosphate (cycloSal-d4TMP): Synthesis and antiviral evaluation of a new d4TMP delivery system. J. Med. Chem. 1998, 41, 1417–1427. [Google Scholar] [CrossRef]
- Lebeau, I.; Andrei, G.; Dal Pozzo, F. Activities of alkoxvalkyl esters of cidofovir (CDV), cyclic CDV, and (S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine against orthopoxviruses in cell monolayers and in organotypic cultures. Antimicrob. Agents Chemother. 2006, 50, 2525–2529. [Google Scholar] [CrossRef] [Green Version]
- Bischofberger, N.; Hitchcock, M.J.M.; Chen, M.S. 1-((S)-2-hydroxy-2-oxo-1,4,2-dioxaphosphorinan-5-yl)methyl cytosine, an intracellular prodrug for (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine with improved therapeutic index in vivo. Antimicrob. Agents Chemother. 1994, 38, 2387–2391. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, U.; Peterson, L.W.; Kashemirov, B.A. Serine peptide phosphoester prodrugs of cyclic cidofovir: Synthesis, transport, and antiviral activity. Mol. Pharm. 2008, 5, 598–609. [Google Scholar] [CrossRef]
- Peterson, L.W.; Sala-Rabanal, M.; Krylov, I.S.; Serpi, M.; Kashemirov, B.A.; McKenna, C.E. Serine side chain-linked peptidomimetic conjugates of cyclic HPMPC and HPMPA: Synthesis and interaction with hPEPT1. Mol. Pharm. 2010, 7, 2349–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalcharova, V.M.; Serpi, M.; Krylov, I.S. Tyrosine-based 1-(S) 3-Hydroxy-2-(phosphonomethoxy)propyl cytosine and -adenine ((S)-HPMPC and (S)-HPMPA) prodrugs: Synthesis, stability, antiviral activity, and in vivo transport studies. J. Med. Chem. 2011, 54, 5680–5693. [Google Scholar] [CrossRef] [Green Version]
- Keith, K.A.; Wan, W.B.; Ciesla, S.L.; Beadle, J.R.; Hostetler, K.Y.; Kern, E.R. Inhibitory activity of alkoxyalkyl and alkyl esters of cidofovir and cyclic cidofovir against orthopoxvirus replication in vitro. Antimicrob. Agents Chemother. 2004, 48, 1869–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krecmerova, M.; Holy, A.; Andrei, G. Synthesis of Ester Prodrugs of 9-(S)- 3-Hydroxy-2-(phosphonomethoxy)propyl -2,6-diaminopurine (HPMPDAP) as anti-poxvirus agents. J. Med. Chem. 2010, 53, 6825–6837. [Google Scholar] [CrossRef]
- Nack, T.; Dinis de Oliveira, T.; Weber, S.; Schols, D.; Balzarini, J.; Meier, C. γ-Ketobenzyl-Modified Nucleoside Triphosphate Prodrugs as Potential Antivirals. J. Med. Chem. 2020, 63, 13745–13761. [Google Scholar] [CrossRef]
- Morales, E.H.R.; Roman, C.A.; Thomann, J.O.; Meier, C. Linear synthesis of chiral cycloSal pronucleotides. Eur. J. Med. Chem. 2011, 23, 4397–4408. [Google Scholar] [CrossRef]
- Rios Morales, E.H.; Balzarini, J.; Meier, C. Stereoselective synthesis and antiviral activity of methyl-substituted cycloSal-pronucleotides. J. Med. Chem. 2012, 55, 7245–7252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, S.; Warnecke, S.; Ehrit, J.; Freiberger, F.; Gerardy-Schahn, R.; Meier, C. Chemical synthesis and enzymatic testing of CMP-sialic acid derivatives. Chembiochem 2012, 13, 2605–2615. [Google Scholar] [CrossRef]
- Huchting, J.; Ruthenbeck, A.; Meier, C. Synthesis of cycloSal-(glycopyranosyl-6)-phosphates as activated sugar phosphates. Eur. J. Org. Chem. 2013, 2013, 6907–6916. [Google Scholar] [CrossRef]
- Erion, M.D.; van Poelje, P.D.; Mackenna, D.A.; Colby, T.J.; Montag, A.C.; Fujitaki, J.M.; Linemeyer, D.L.; Bullough, D.A. Liver-targeted drug delivery using HepDirect prodrugs. J. Pharmacol. Exp. Ther. 2005, 312, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Boyer, S.H.; Sun, Z.; Jiang, H.; Esterbrook, J.; Gomez-Galeno, J.E.; Craigo, W.; Reddy, K.R.; Ugarkar, B.G.; MacKenna, D.A.; Erion, M.D. Synthesis and characterization of a novel livertargeted prodrug of cytosine-1-beta-D-arabinofuranoside monophosphate for the treatment of hepatocellular carcinoma. J. Med. Chem. 2006, 49, 7711–7720. [Google Scholar] [CrossRef]
- Meier, C.; Lorey, M.; De Clercq, E.; Balzarini, J. Cyclic saligenyl phosphotriesters of 20,30-dideoxy-20,30-didehydrothymidine (d4T)—A new pro-nucleotide approach. Bioorg. Med. Chem. Lett. 1997, 7, 99–104. [Google Scholar] [CrossRef]
- Erion, M.D.; Reddy, K.R.; Boyer, S.H.; Matelich, M.C.; Gomez-Galeno, J.; Lemus, R.H.; Ugarkar, B.G.; Colby, T.J.; Schanzer, J.; Van Poelje, P.D. Design, synthesis, and characterization of a series of cytochrome P(450) 3A-activated prodrugs (HepDirect prodrugs) useful for targeting phosph(on)ate-based drugs to the liver. J. Am. Chem. Soc. 2004, 126, 5154–5163. [Google Scholar] [CrossRef]
- Worek, F.; Wille, T.; Koller, M.; Thiermann, H. Toxicology of organophosphorus compounds in view of an increasing terrorist threat. Arch Toxicol. 2016, 90, 2131–2145. [Google Scholar] [CrossRef]
- Reddy, K.R.; Matelich, M.C.; Ugarkar, B.G.; Gómez-Galeno, J.E.; DaRe, J.; Ollis, K.; Sun, Z.; Craigo, W.; Colby, T.J.; Fujitaki, J.M.; et al. Pradefovir: A prodrug that targets adefovir to the liver for the treatment of hepatitis B. J. Med. Chem. 2008, 51, 666–676. [Google Scholar] [CrossRef]
- Ding, Y.; Zhang, H.; Li, X. Safety, pharmacokinetics and pharmacogenetics of a single ascending dose of pradefovir, a novel liver-targeting, anti-hepatitis B virus drug, in healthy Chinese subjects. Hepatol. Int. 2017, 11, 390–400. [Google Scholar] [CrossRef]
- Erion, M.D.; Cable, E.E.; Ito, B.R. Targeting thyroid hormone receptor-beta agonists to the liver reduces cholesterol and triglycerides and improves the therapeutic index. Proc. Natl. Acad. Sci. USA 2007, 104, 15490–15495. [Google Scholar] [CrossRef]
- Boyer, S.H.; Jiang, H.; Jacintho, J.D. Synthesis and biological evaluation of a series of liver-selective phosphonic acid thyroid hormone receptor agonists and their prodrugs. J. Med. Chem. 2008, 51, 7075–7093. [Google Scholar] [CrossRef]
- Tsukada, T.; Tamaki, K.; Tanaka, J. A prodrug approach towards the development of tricyclic-based FBPase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 2938–2941. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Qiu, G.; Lei, B.; Qin, L.; Chu, H.; Lu, Y.; Zhu, G.; Gao, Q.; Huang, Q.; Qian, G.; et al. Oral Delivery of Propofol with Methoxymethylphosphonic Acid as the Delivery Vehicle. J. Med. Chem. 2017, 60, 8580–8590. [Google Scholar] [CrossRef] [PubMed]
- Chapman, H.; Kernan, M.; Prisbe, E.; Rohloff, J.; Sparacino, M.; Terhorst, T.; Yu, R. Practical synthesis, separation, and stereochemical assignment of the pmpa pro-drug GS-7340. Nucleosides Nucleotides Nucleic Acids 2001, 20, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Sofia, M.J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P.G.; Ross, B.S.; Wang, P.; Zhang, H.R.; et al. Discovery of a β-D-20 -Deoxy-20 -r-fluoro-20 -β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus. J. Med. Chem. 2010, 53, 7202–7218. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, S.; Meier, C. Synthesis of Nucleoside Di- and Triphosphates and Dinucleoside Polyphosphates with cycloSal-Nucleotides. J. Org. Chem. 2009, 74, 3024–3030. [Google Scholar] [CrossRef] [PubMed]
- Balzarini, J.; Aquaro, S.; Knispel, T.; Rampazzo, C.; Bianchi, V.; Perno, C.-F.; De Clercq, E.; Meier, C. Cyclosaligenyl-2′,3′-didehydro-2′, 3′-dideoxythymidine monophosphate: Efficient intracellular delivery of d4TMP. Mol. Pharmacol. 2000, 58, 928–935. [Google Scholar] [CrossRef]
- Ford, A.; Mullins, N.D.; Balzarini, J.; Maguire, A.R. Synthesis and Evaluation of Prodrugs of α-Carboxy Nucleoside Phosphonates. J. Org. Chem. 2022, 87, 14793–14808. [Google Scholar] [CrossRef]
- Cho, A.; Zhang, L.; Xu, J.; Leem, R.; Butler, T.; Metobo, S.; Aktoudianakis, V.; Lew, W.; Ye, H.; Clarke, M. Discovery of the first C-nucleoside HCV polymerase inhibitor (GS-6620) with demonstrated antiviral response in HCV infected patients. J. Med. Chem. 2014, 57, 1812–1825. [Google Scholar] [CrossRef]
- Arbelo Roman, C.; Wasserthal, P.; Balzarini, J.; Meier, C. Diastereoselective synthesis of (aryloxy)phosphoramidate prodrugs. Eur. J. Org. Chem. 2011, 2011, 4899–4909. [Google Scholar] [CrossRef]
- Arbelo Roman, C.; Balzarini, J.; Meier, C. Diastereoselective synthesis of aryloxy phosphoramidate prodrugs of 3’-deoxy-2’,3’-didehydrothymidine monophosphate. J. Med. Chem. 2010, 53, 7675–7681. [Google Scholar] [CrossRef]
- Heidel, K.M.; Dowd, C.S. Phosphonate prodrugs: An overview and recent advances. Future Med. Chem. 2019, 11, 1625–1643. [Google Scholar] [CrossRef]
- Rudge, E.S.; Alex, H.Y.; Leeper, F.J. Prodrugs of pyrophosphates and bisphosphonates: Disguising phosphorus Oxyanions. RSC Med. Chem. 2022, 13, 375–391. [Google Scholar] [CrossRef] [PubMed]
- Krečmerová, M.; Majer, P.; Rais, R.; Slusher, B.S. Phosphonates and Phosphonate Prodrugs in Medicinal Chemistry: Past Successes and Future Prospects. Front. Chem. 2022, 10, 889737. [Google Scholar] [CrossRef] [PubMed]
- Pertusati, F.; Pileggi, E.; Richards, J.; Wootton, M.; Van Leemputte, T.; Persoons, L.; De Coster, D.; Villanueva, X.; Daelemans, D.; Steenackers, H.; et al. Drug repurposing: Phosphate prodrugs of anticancer and antiviral FDA-approved nucleosides as novel antimicrobials. J. Antimicrob. Chemother. 2020, 75, 2864–2878. [Google Scholar] [CrossRef] [PubMed]


































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolodiazhna, A.O.; Kolodiazhnyi, O.I. Chiral Organophosphorus Pharmaceuticals: Properties and Application. Symmetry 2023, 15, 1550. https://doi.org/10.3390/sym15081550
Kolodiazhna AO, Kolodiazhnyi OI. Chiral Organophosphorus Pharmaceuticals: Properties and Application. Symmetry. 2023; 15(8):1550. https://doi.org/10.3390/sym15081550
Chicago/Turabian StyleKolodiazhna, Anastasy O., and Oleg I. Kolodiazhnyi. 2023. "Chiral Organophosphorus Pharmaceuticals: Properties and Application" Symmetry 15, no. 8: 1550. https://doi.org/10.3390/sym15081550