
Azulene, Reactivity, and Scientific Interest Inversely Proportional to Ring Size; Part 2: The Seven-Membered Ring

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
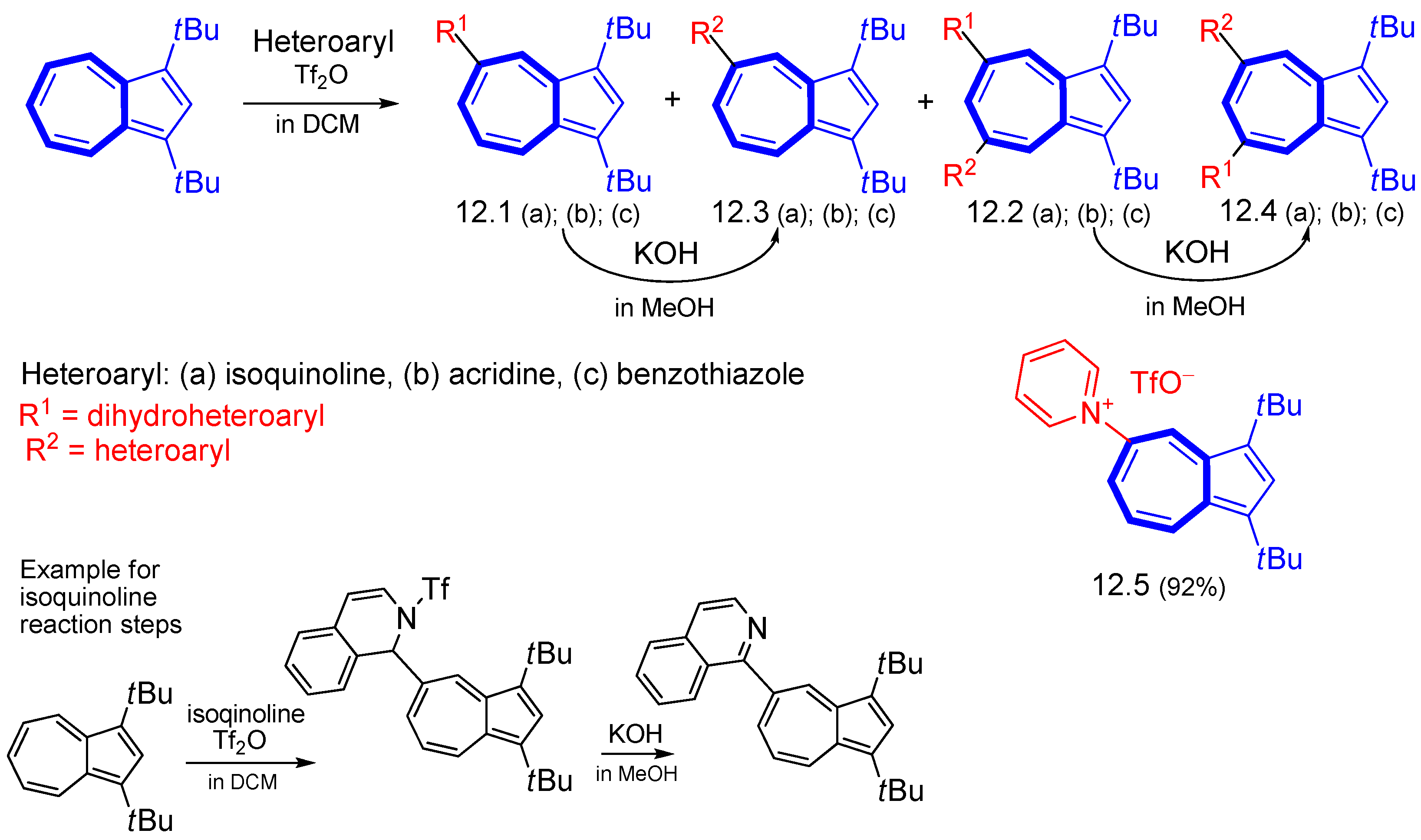{kind=link}
{kind=link}
{kind=link}
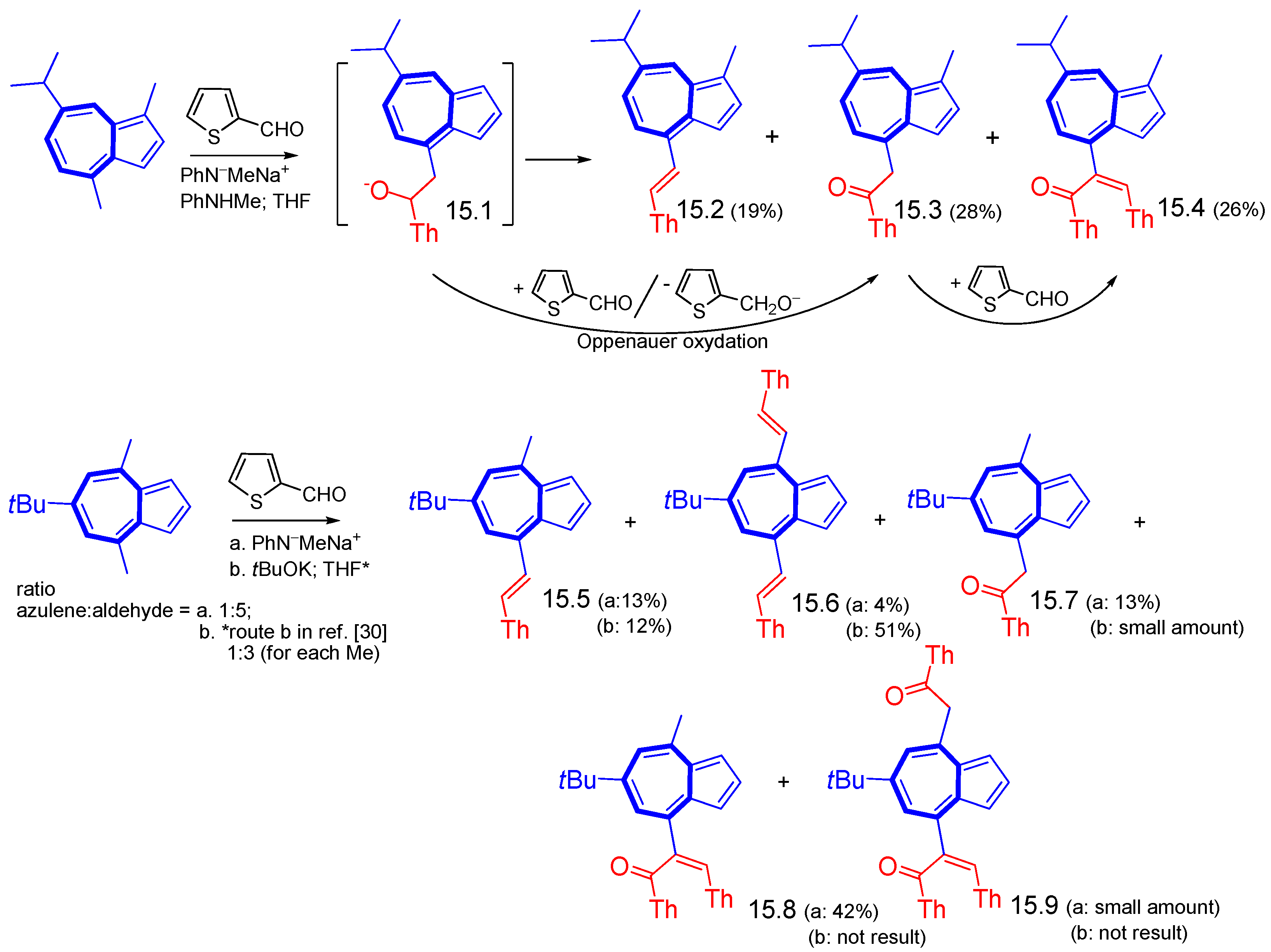{kind=link}
{kind=link}
{kind=link}
{kind=link}
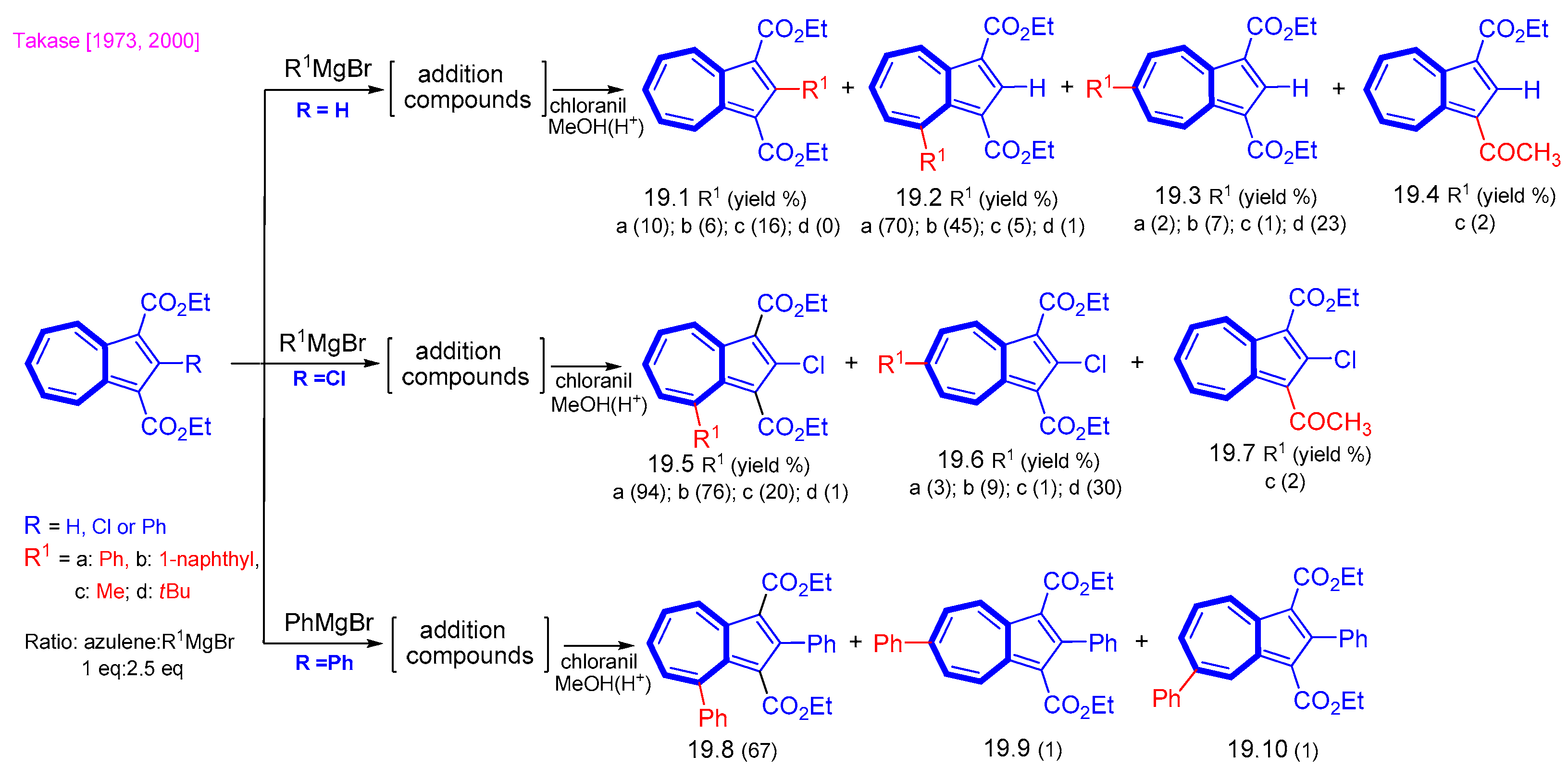{kind=link}
{kind=link}
{kind=link}
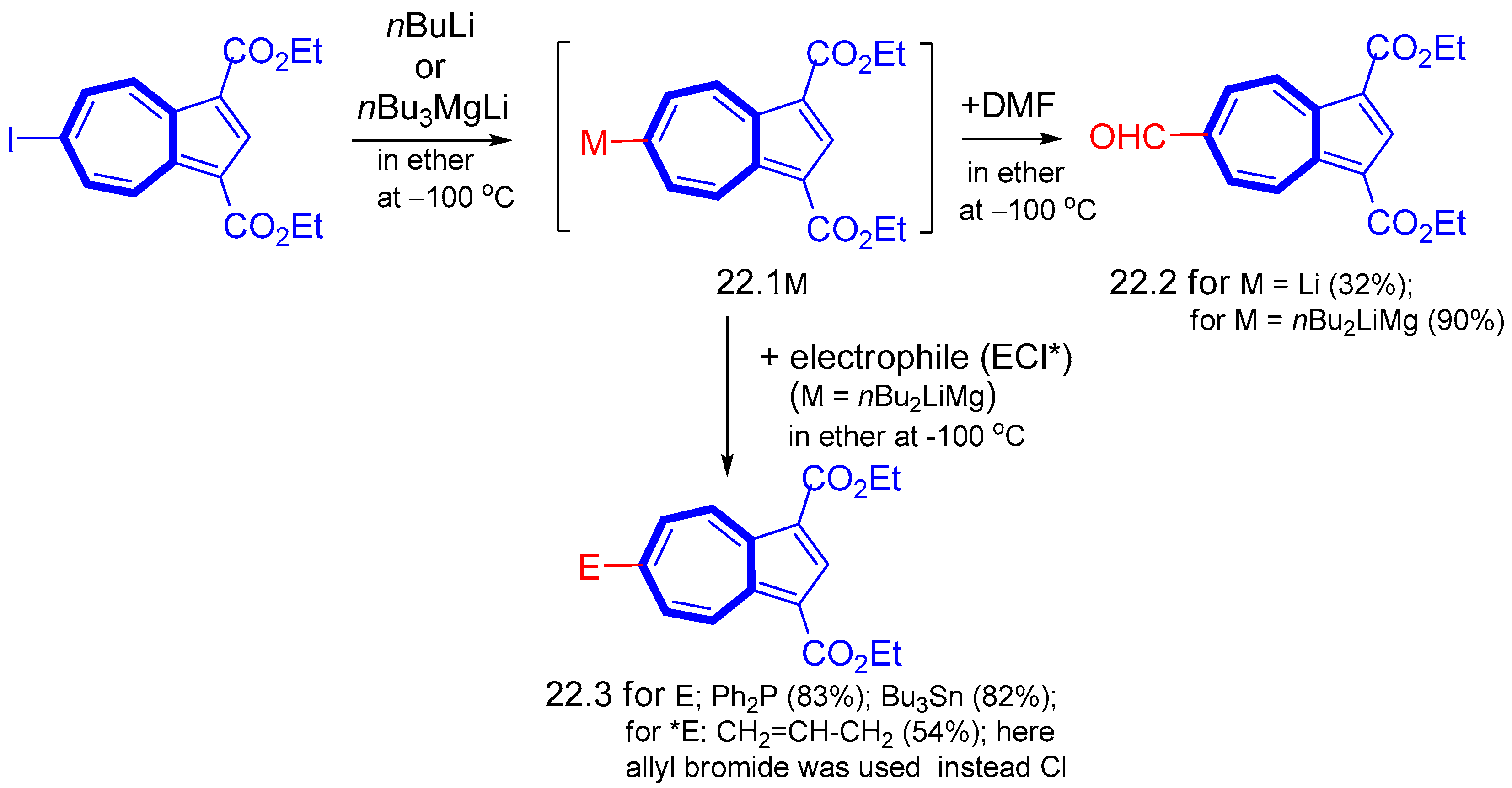{kind=link}
{kind=link}
{kind=link}
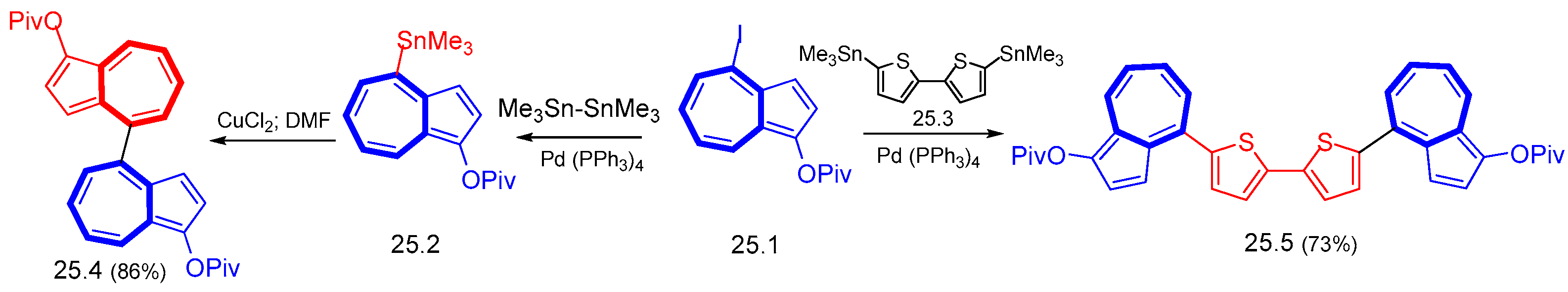{kind=link}
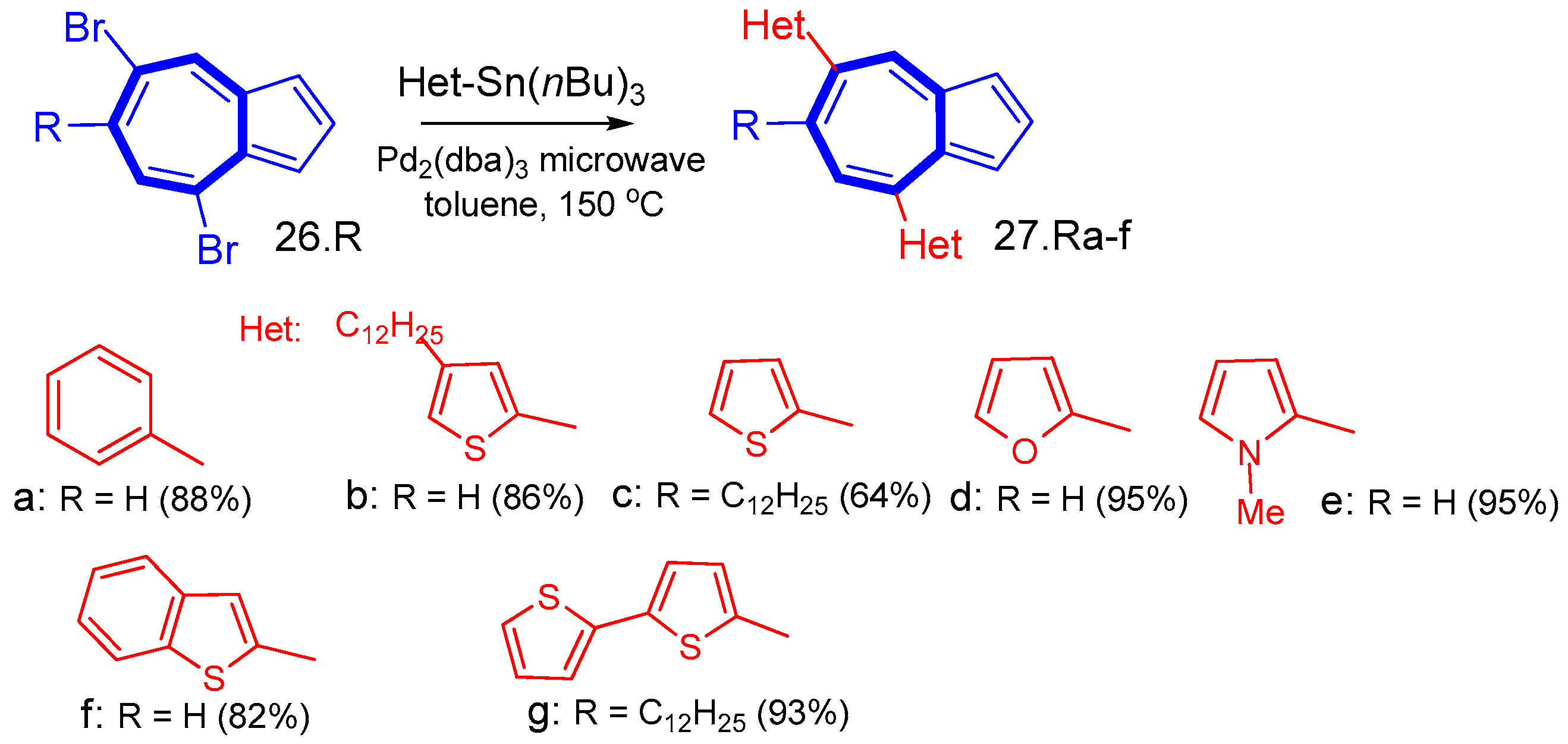{kind=link}
{kind=link}
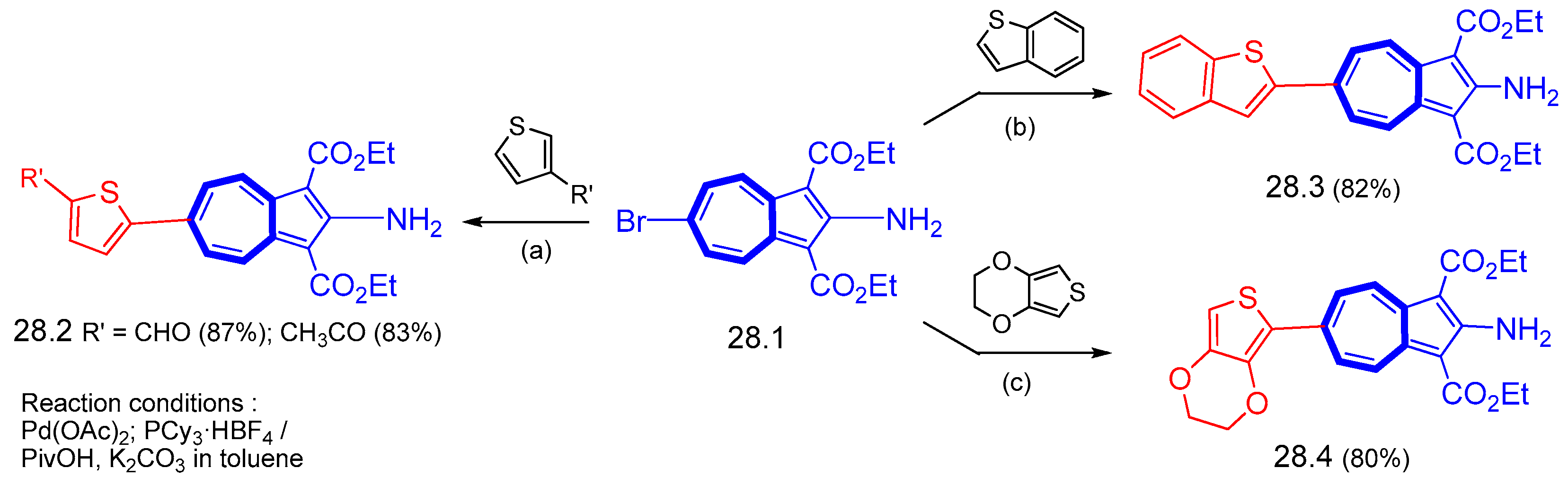{kind=link}
{kind=link}
{kind=link}
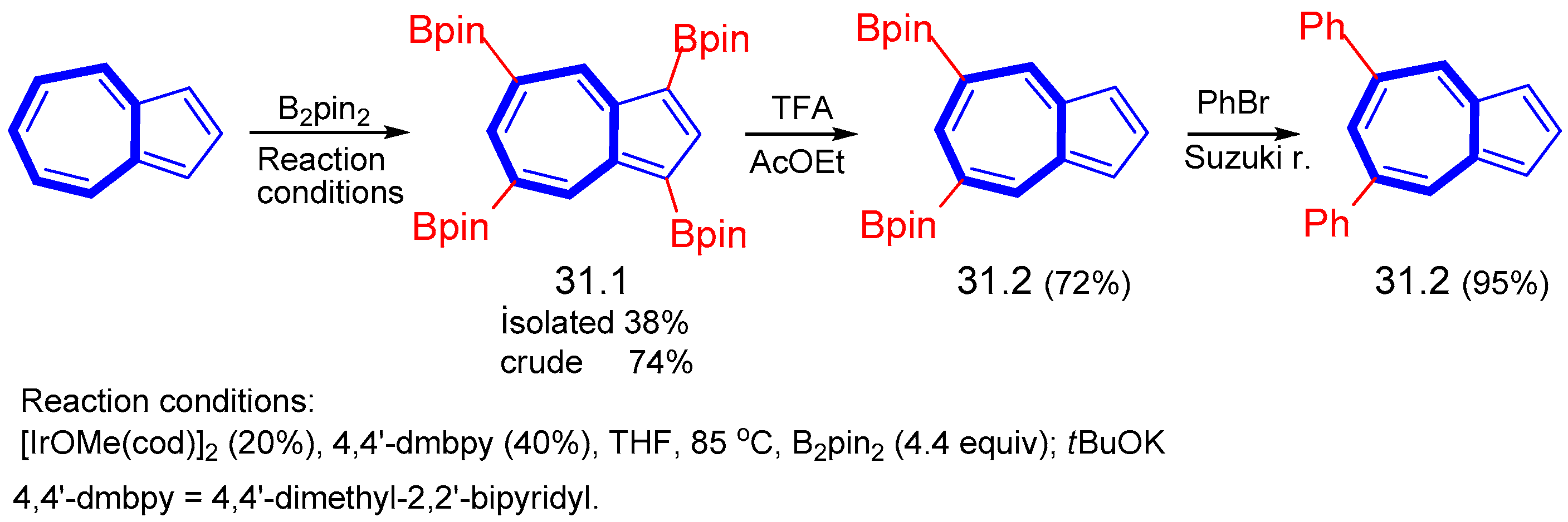{kind=link}
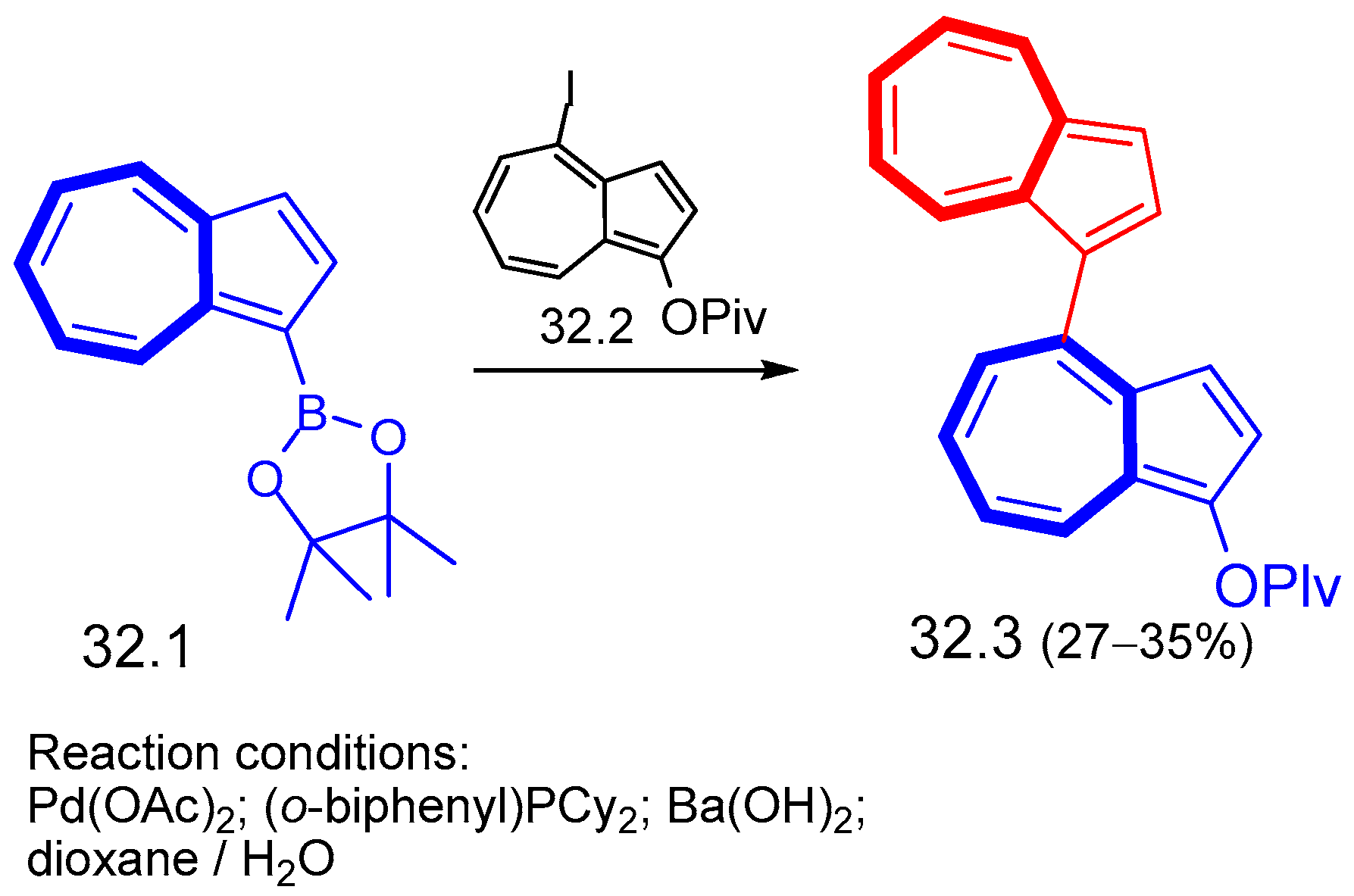{kind=link}
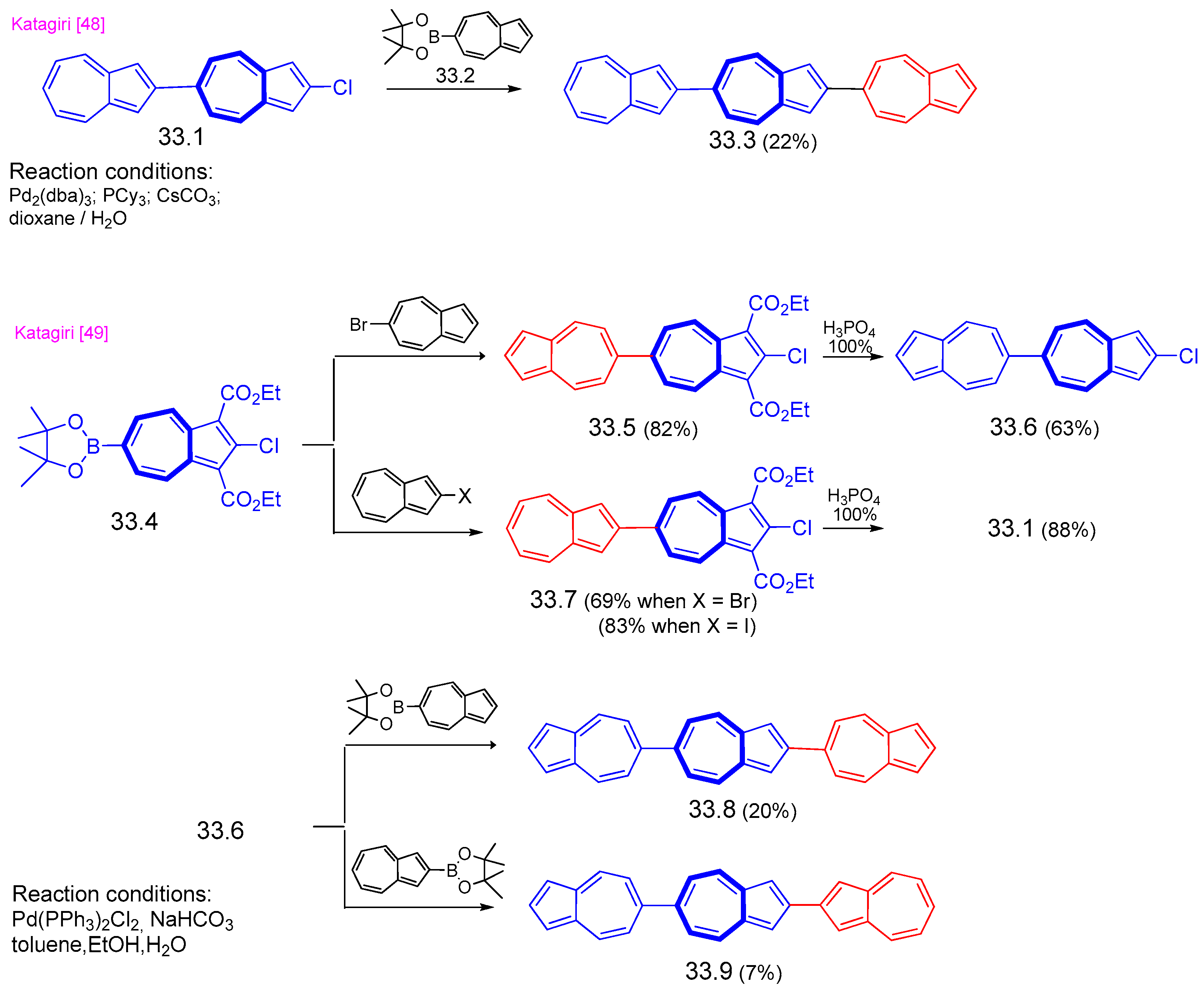{kind=link}
{kind=link}
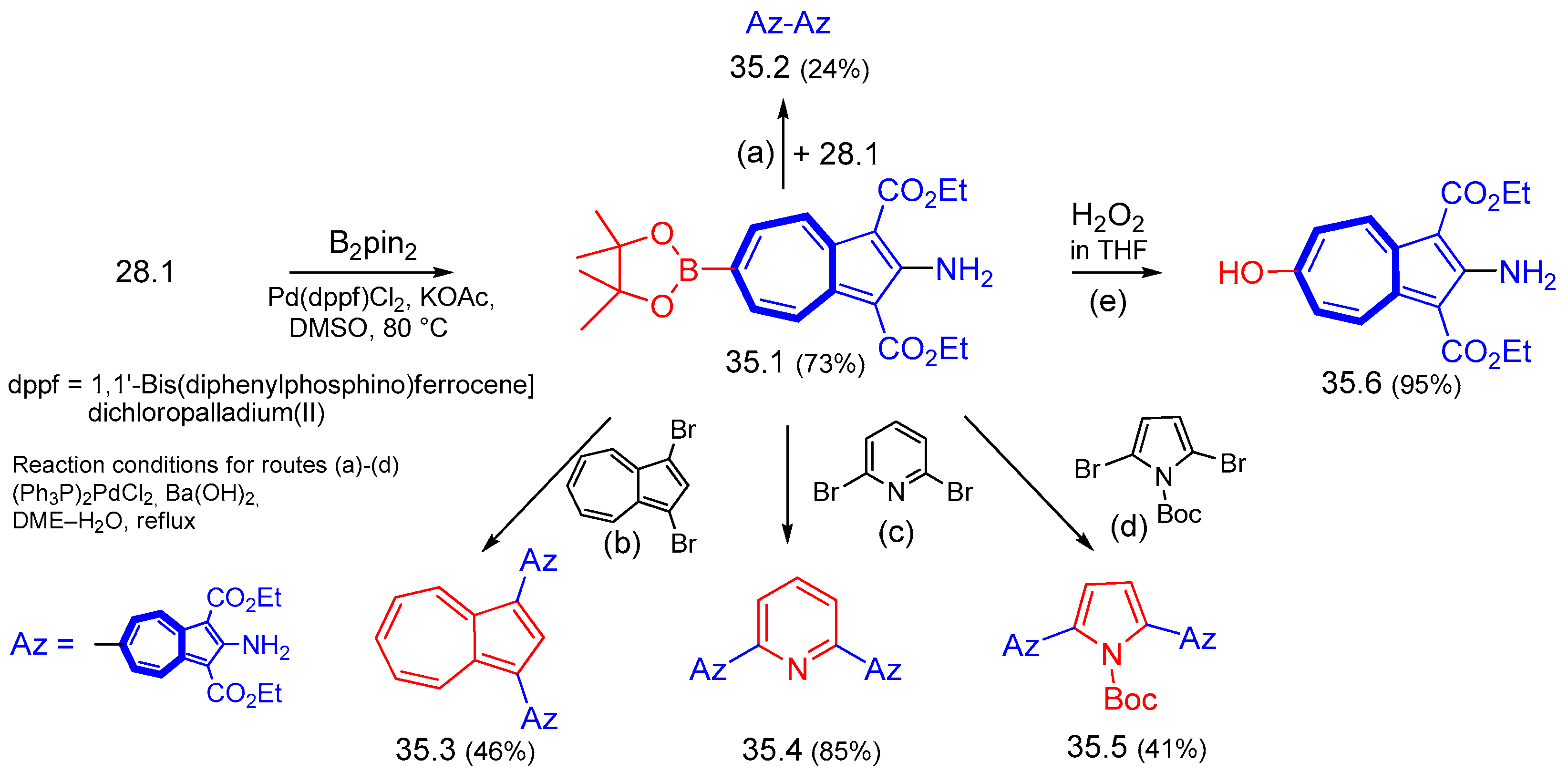{kind=link}
{kind=link}
{kind=link}
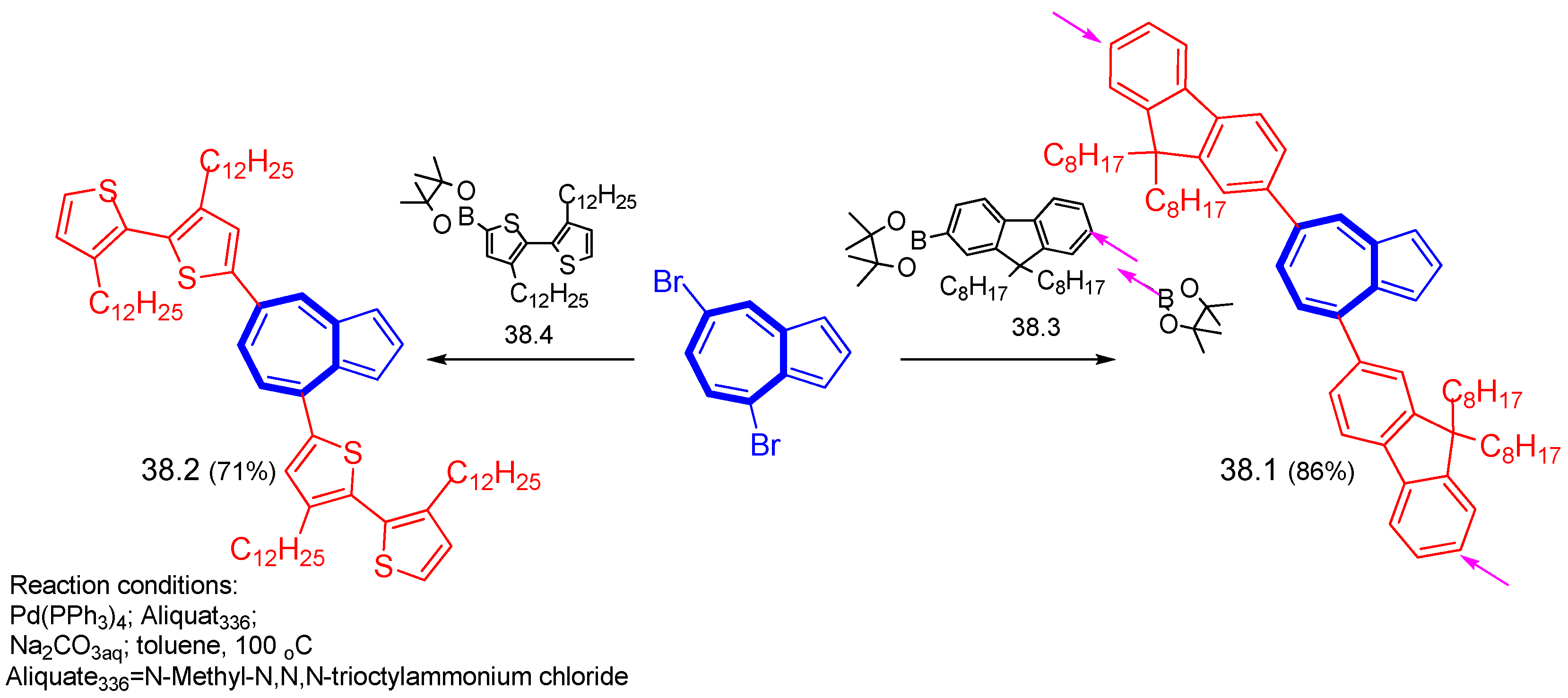{kind=link}
{kind=link}
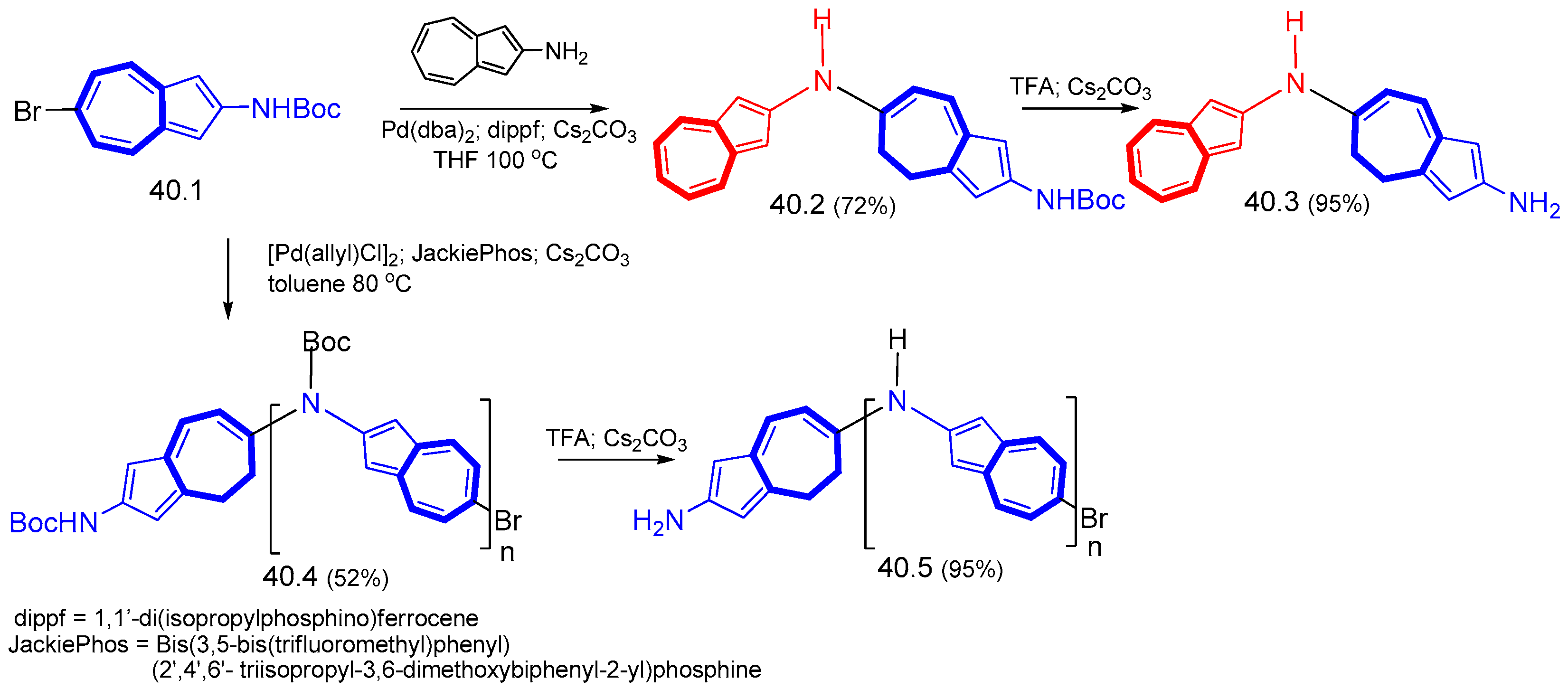{kind=link}
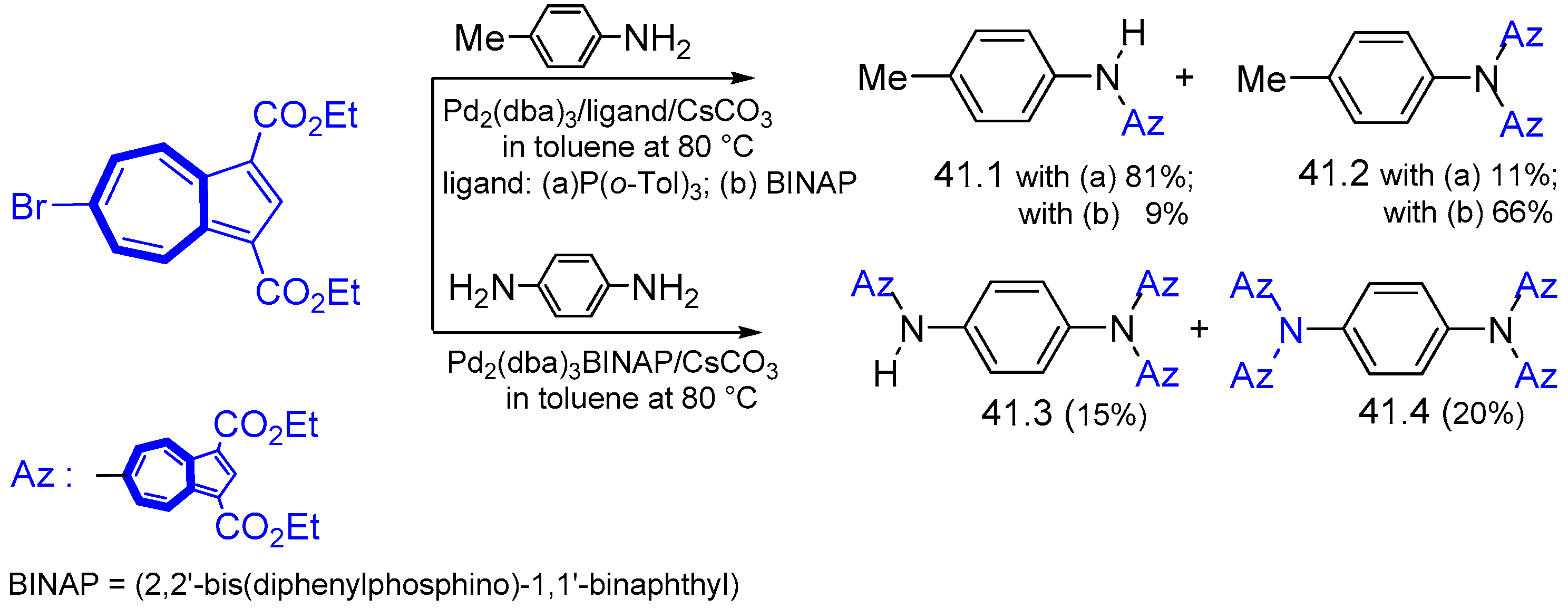{kind=link}
{kind=link}
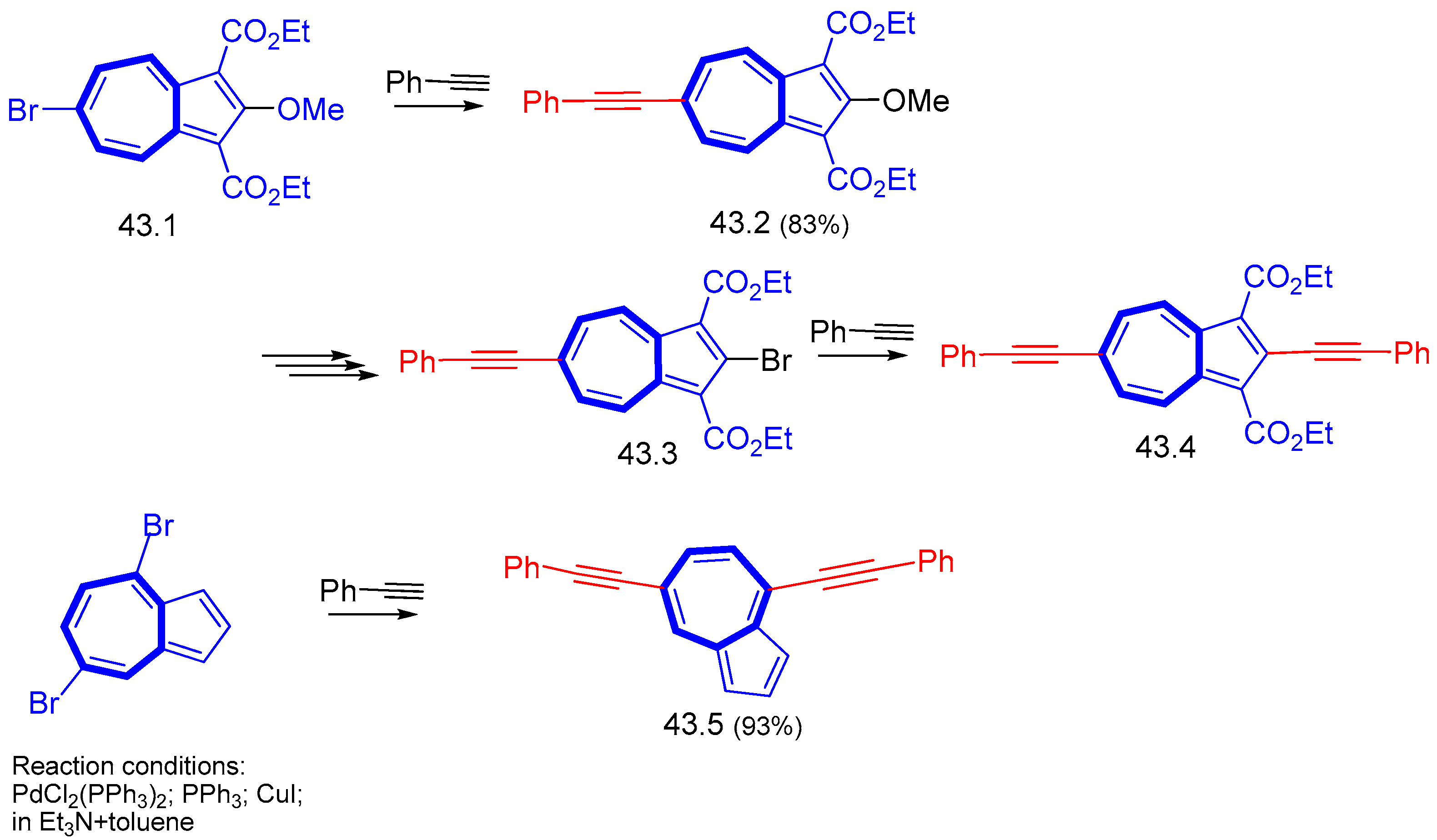{kind=link}
{kind=link}
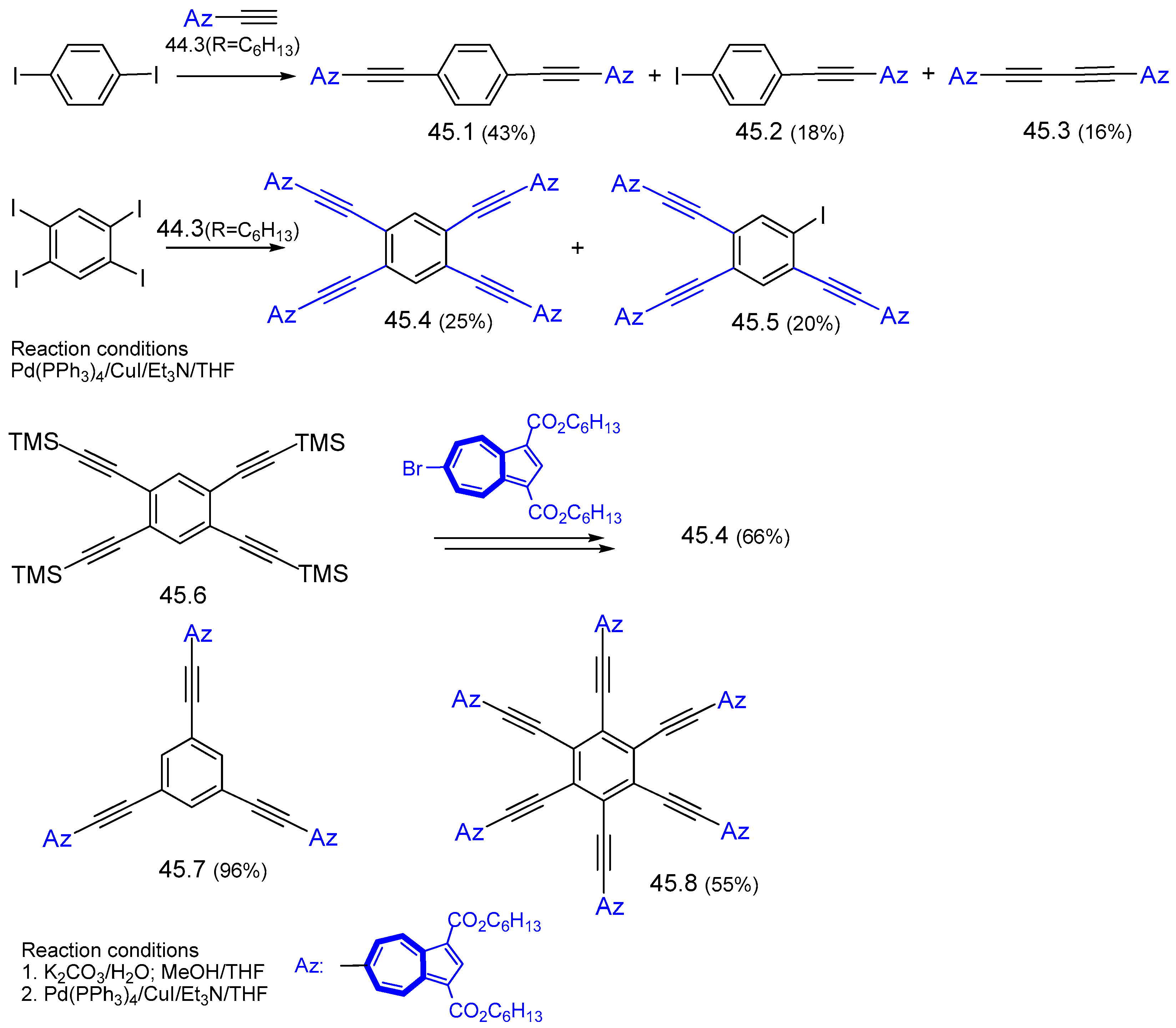{kind=link}
{kind=link}
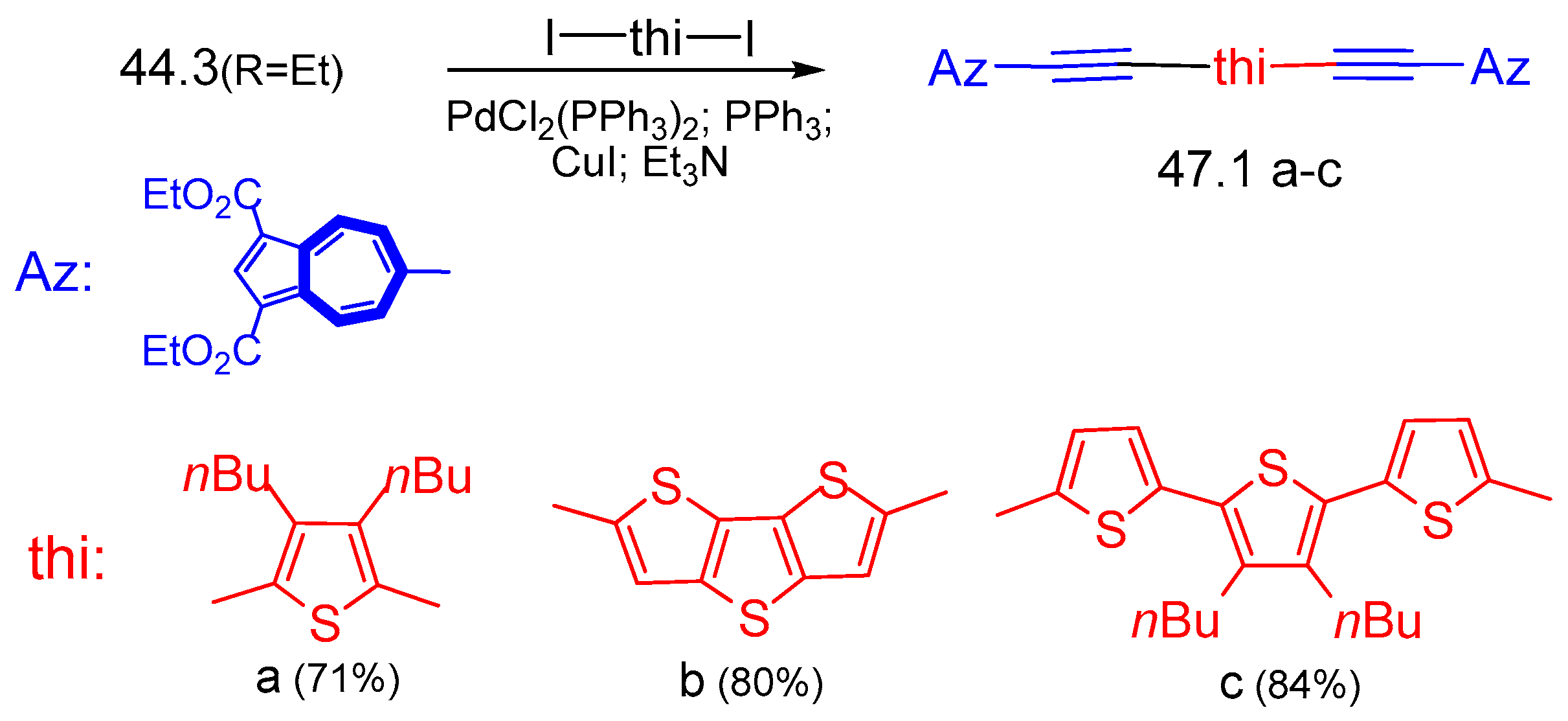{kind=link}
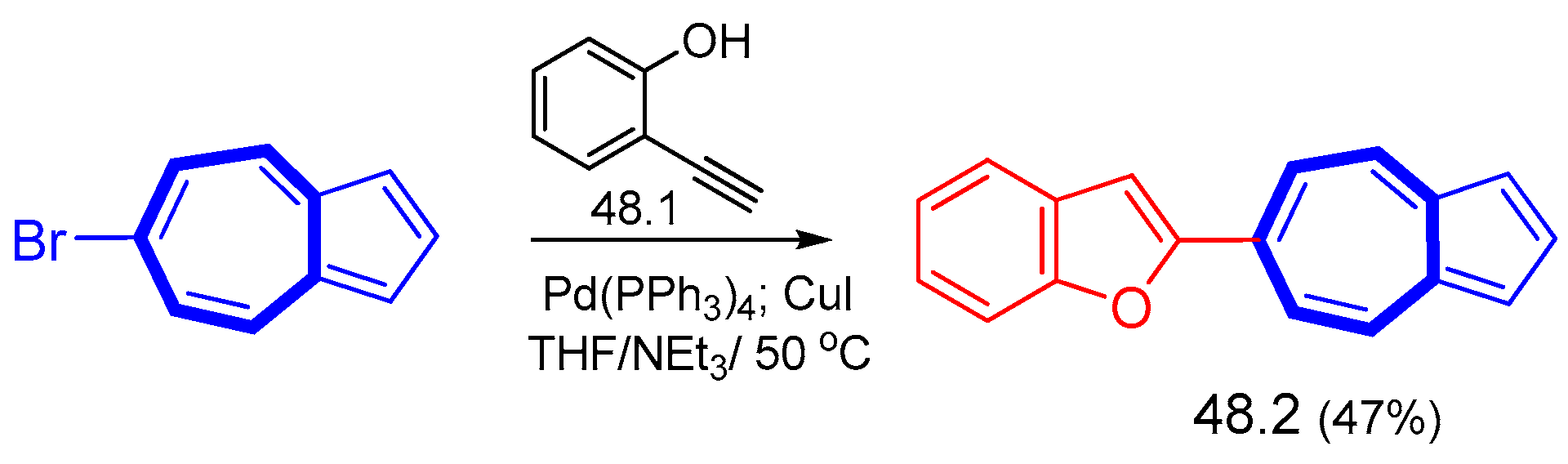{kind=link}
{kind=link}
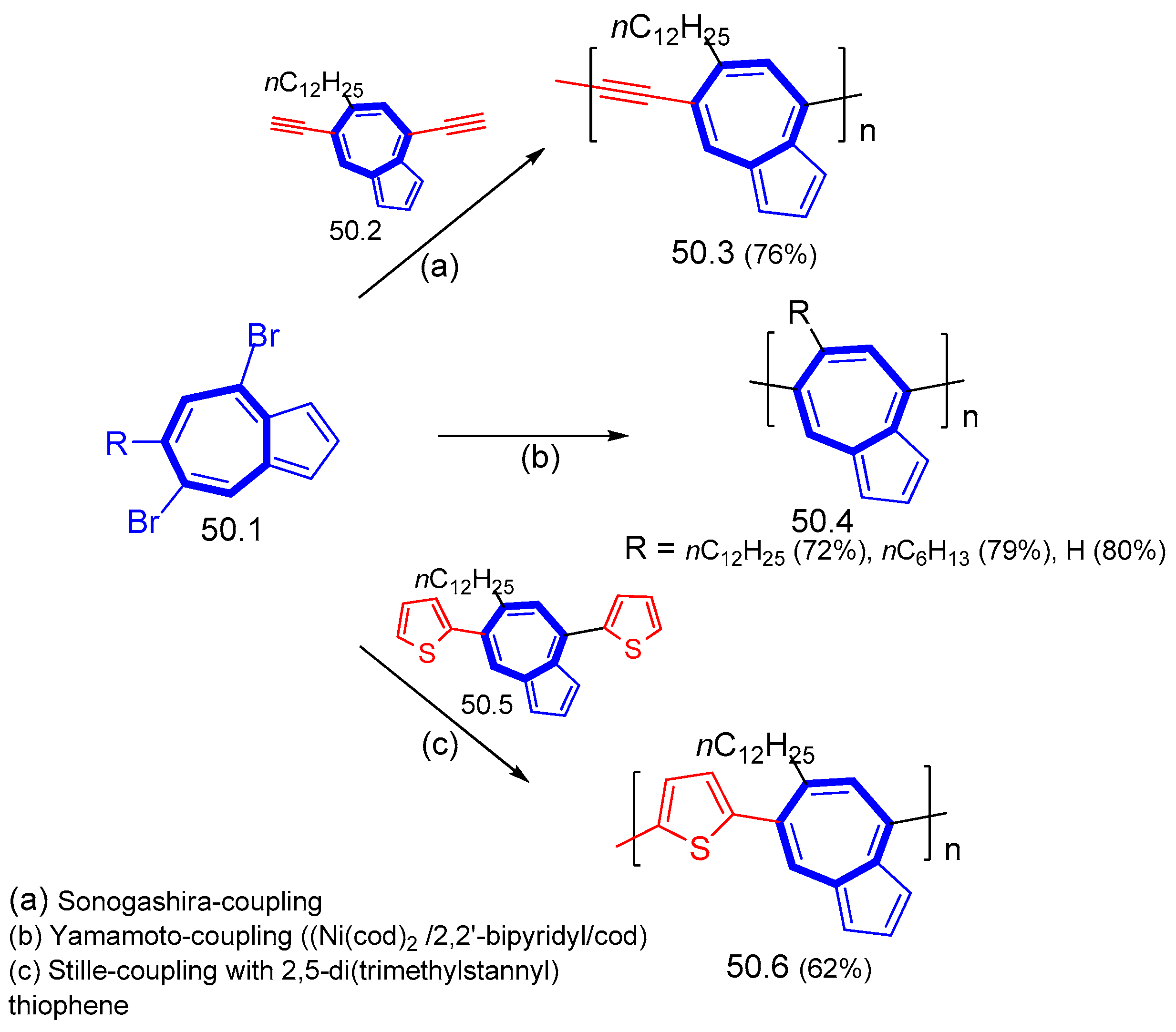{kind=link}
{kind=link}
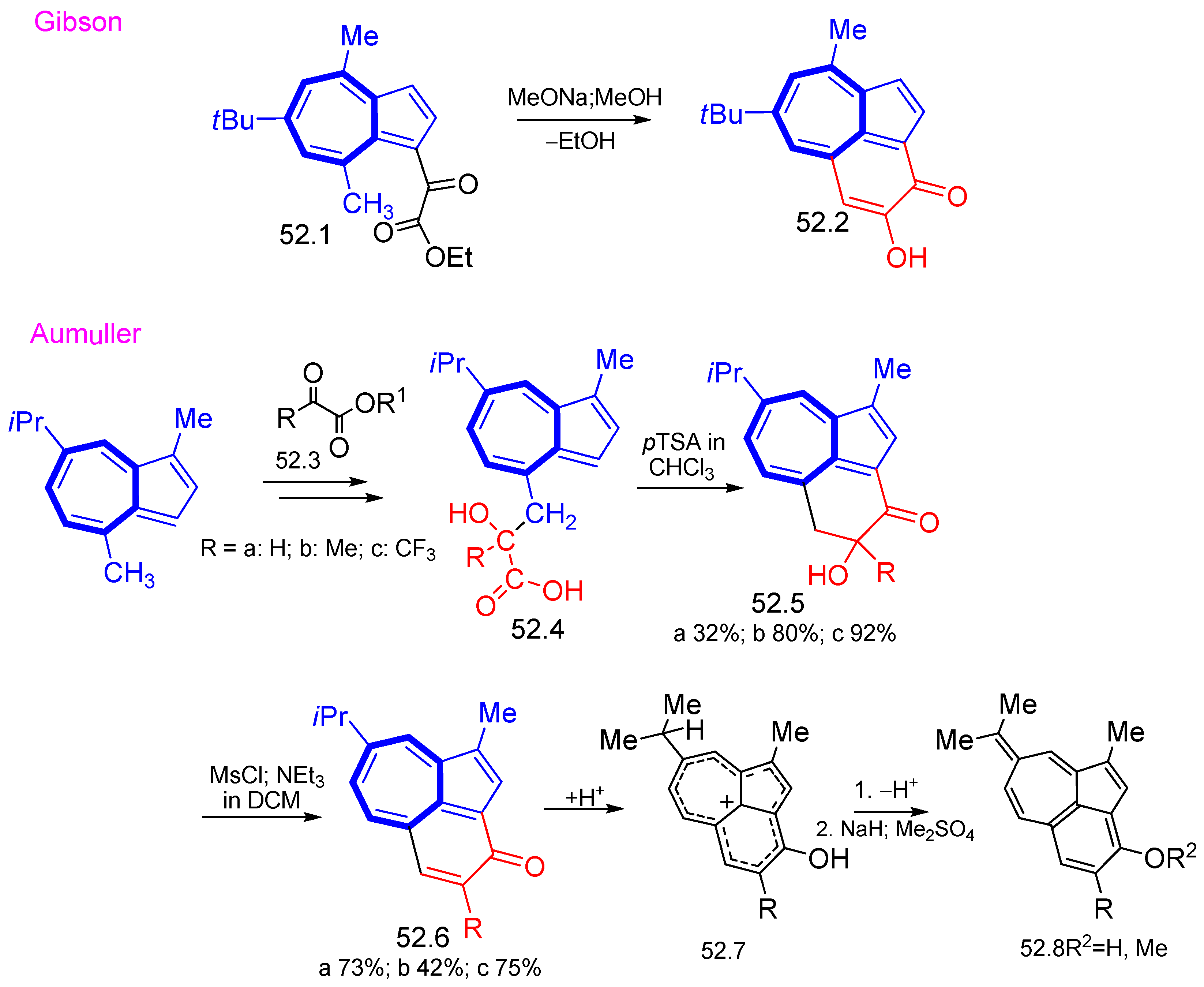{kind=link}
{kind=link}
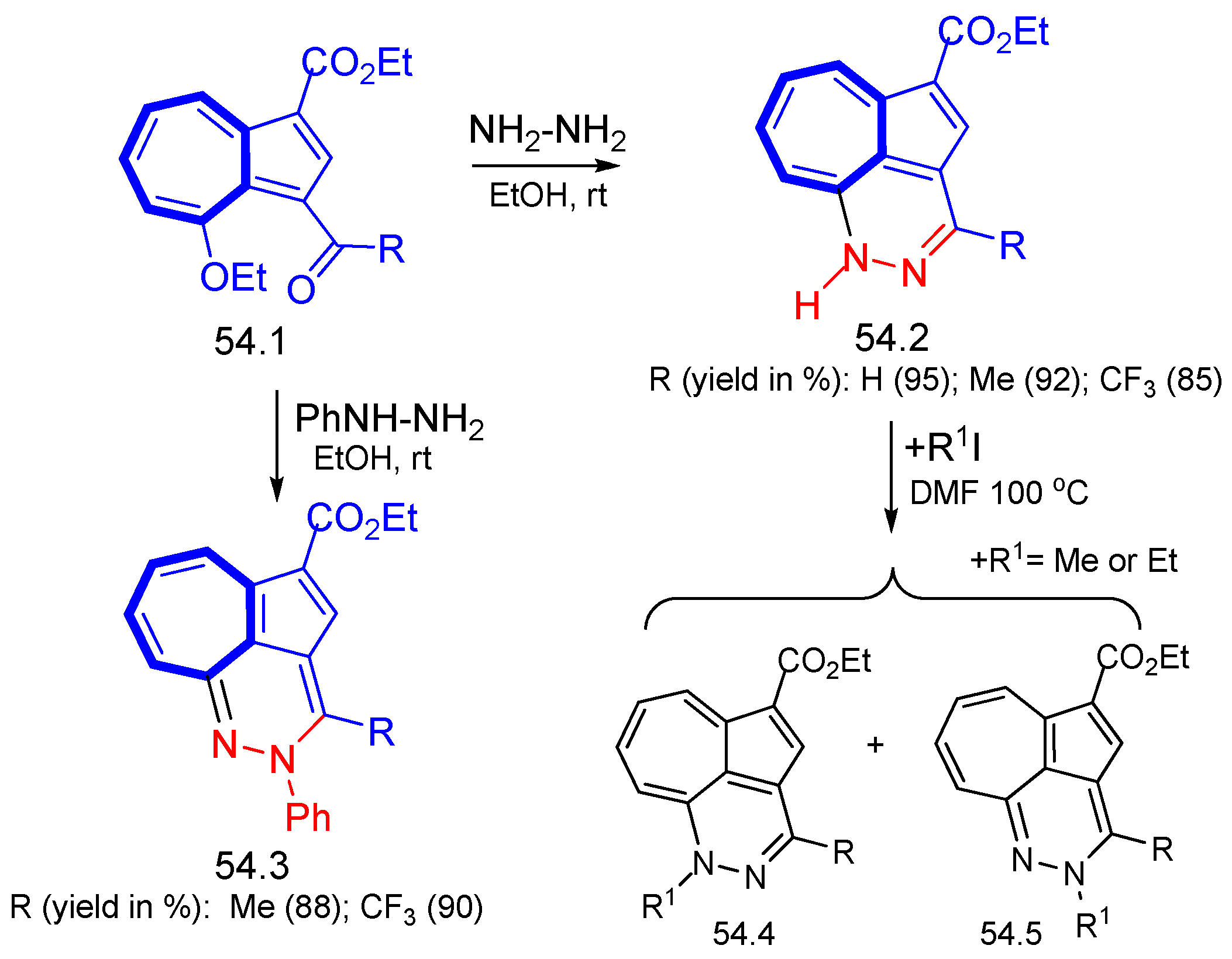{kind=link}
{kind=link}
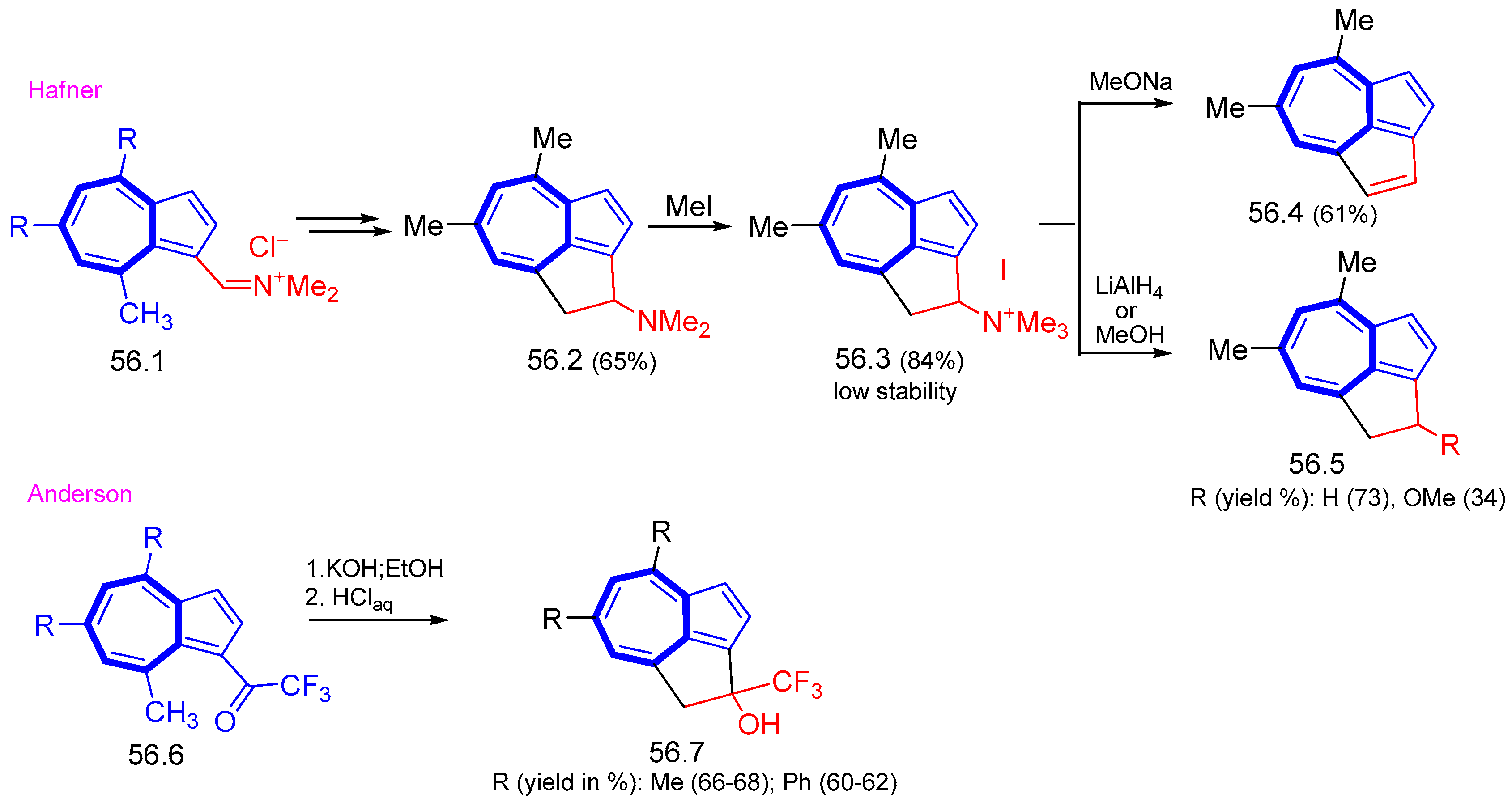{kind=link}
{kind=link}
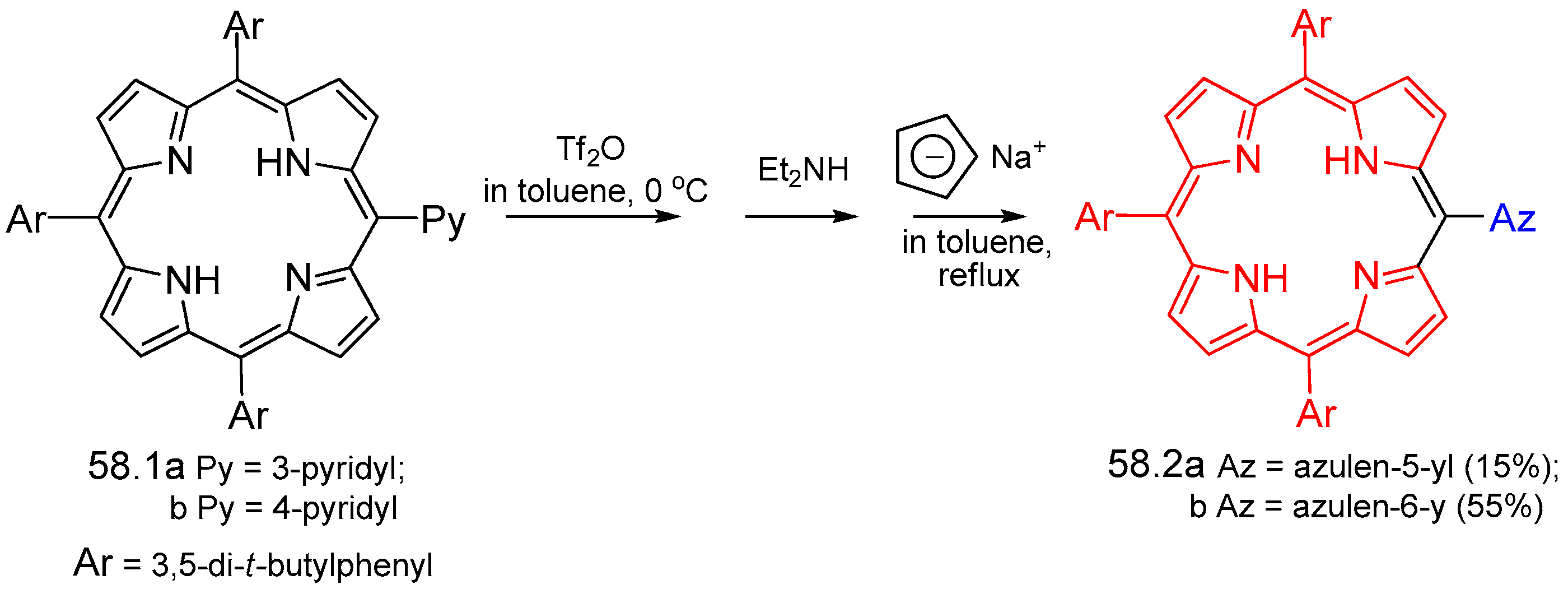{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
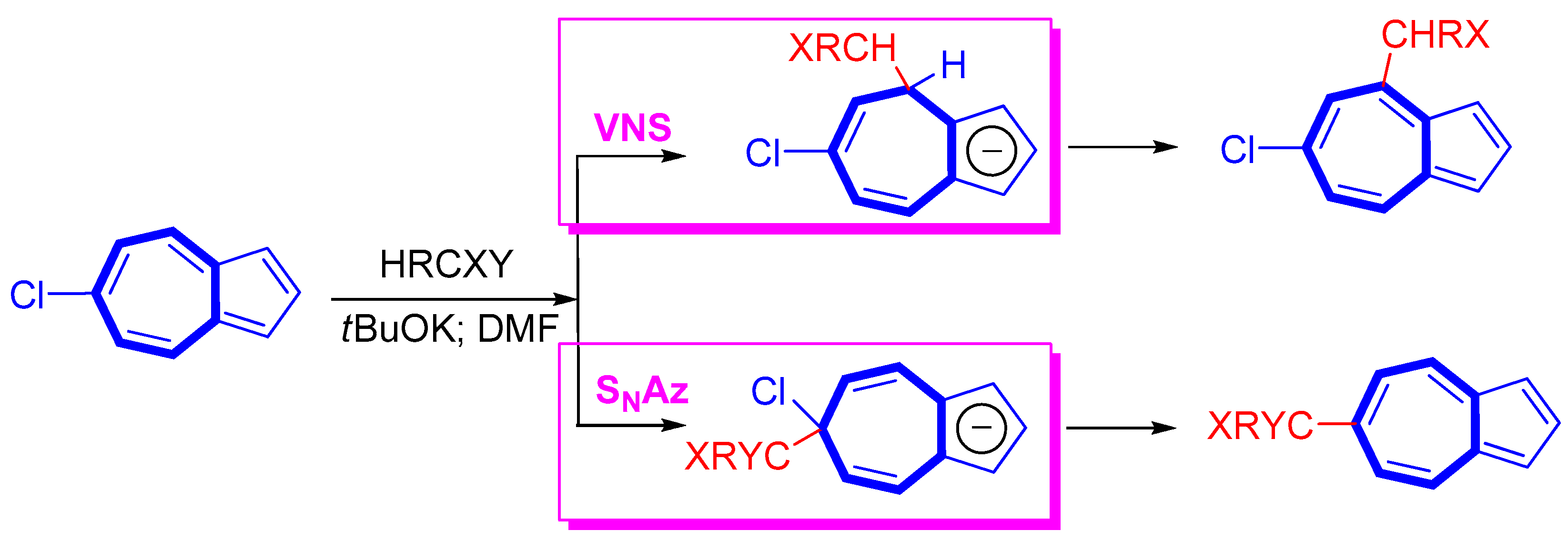
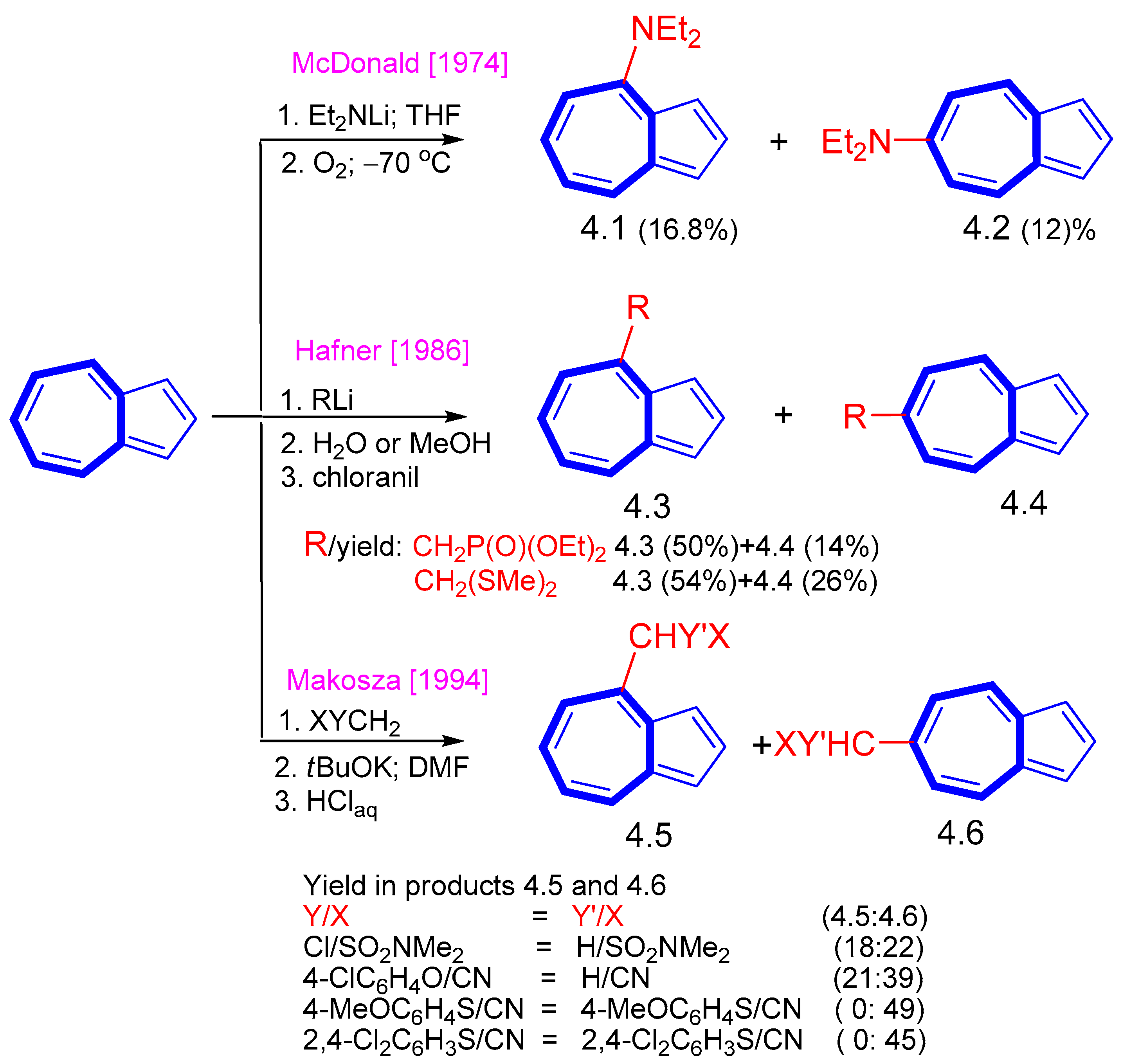
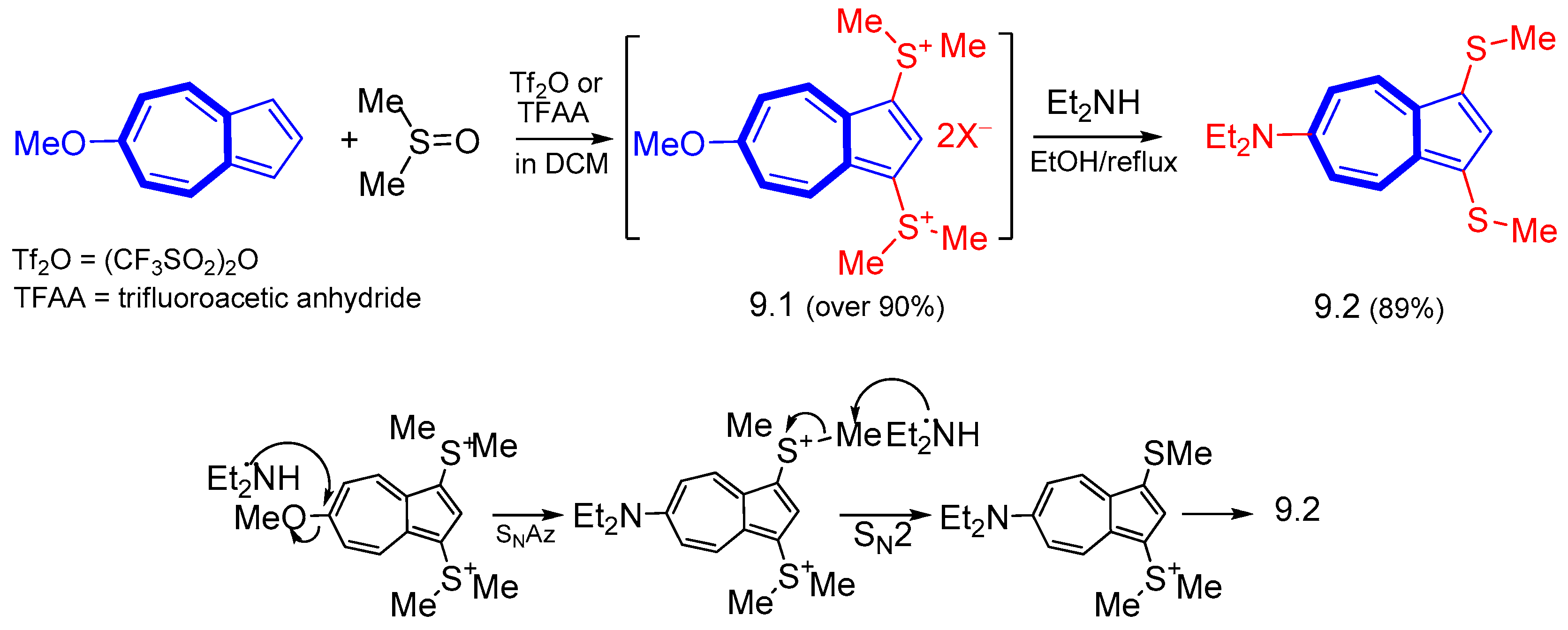
2. Nucleophilic Reactions at Azulene Moiety, SNAz
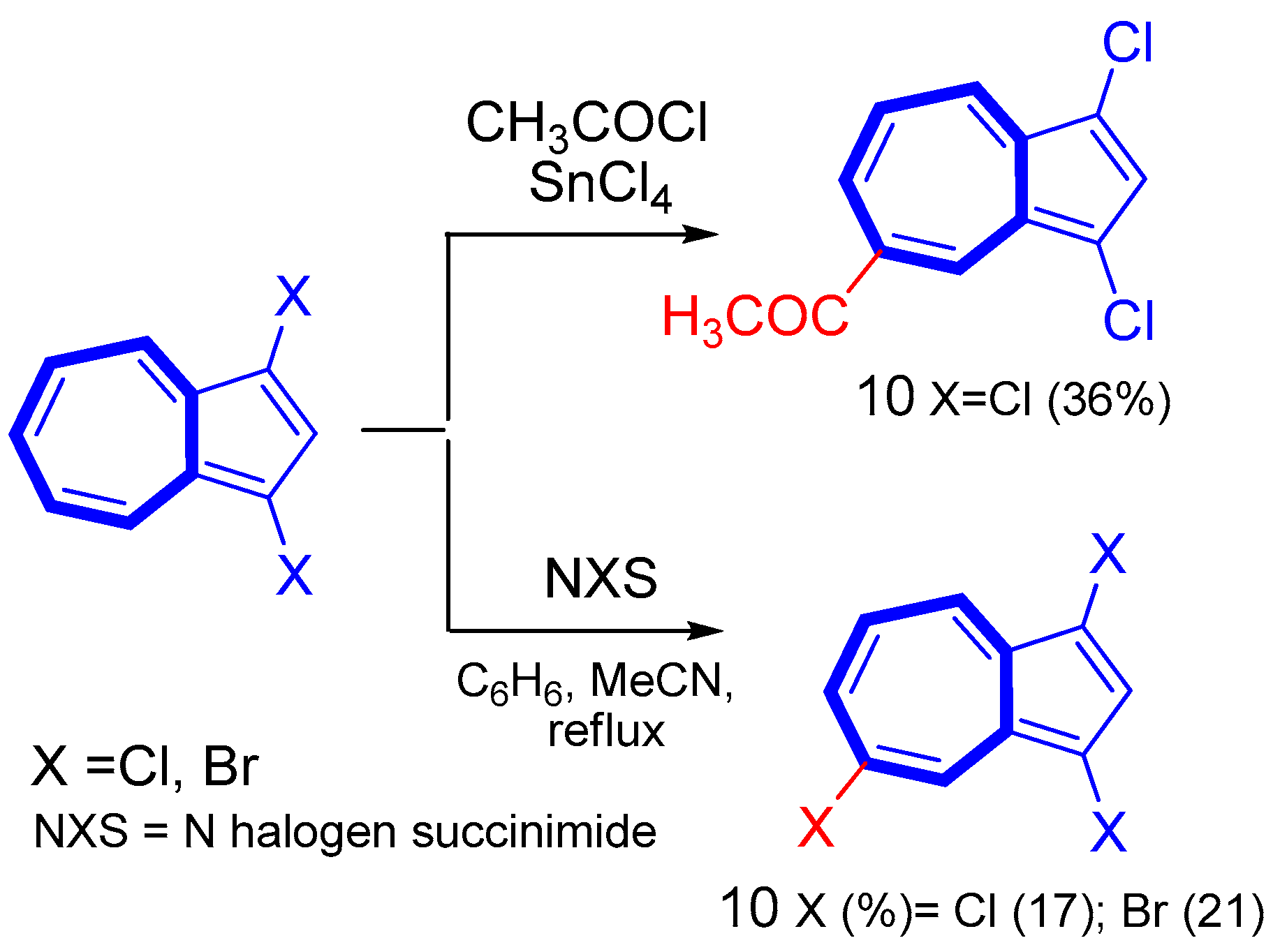
3. Electrophilic Substitutions at Azulene Moiety, SEAz
4. Nucleophilic Reactions at Azulene Substituents
5. Reaction with Organometallic Derivatives
6. Reactions Catalyzed by Transition Metal Complexes
6.1. Stille Cross-Coupling
6.2. Heck–Negishi Condensation
6.3. Syntheses of Other Thienylazulene Derivatives
6.4. Suzuki–Miyaura Cross-Condensation
6.5. Various Syntheses Catalyzed by Transition Metal Complexes
7. Azulene Derivatives Containing Ethynyl Group(s)
7.1. Azulenes Substituted with Ethynyl Groups
7.2. Polymers including Ethynyl Groups
8. Polycyclic Compounds That Include the Azulenic System
9. Porphyrinogenic Systems and Phthalocyanines Containing Azulenyl Moiety (Azulenocyanine)
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Razus, A.C. Azulene, reactivity and scientific interest inversely proportionate to ring size. Part 1. Five-membered ring. Symmetry 2023, 15, 310. [Google Scholar] [CrossRef]
- Xin, H.; Gao, X. Application of Azulene in Constructing Organic Optoelectronic Materials: New Tricks for an Old Dog. ChemPlusChem 2017, 82, 945–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razus, A.C. Azulene Moiety as Electron Reservoir in Positive Charged Systems; Short Survey. Symmetry 2021, 13, 526. [Google Scholar] [CrossRef]
- Zeller, K.-P. Houben Weyl, Methoden der Organischen Chemie; Thieme, G., Ed.; Verlag: Stuttgart, Germany; New York, NY, USA, 1985; Volume V/2c, pp. 249–405. [Google Scholar]
- Hafner, K.; Bernhard, C.; Müller, R.; Zur Kenntnis der Azulene, V. Die Nucleophile Substitution des Azulens. Liebigs Ann. Chem. 1961, 650, 35–41. [Google Scholar] [CrossRef]
- McDonald, R.N.; Petty, H.E.; Wolfe, N.L.; Paukstelis, J.V. Nonbenzenoid aromatic systems. X. Formation, nuclear magnetic resonance spectral identification, and reactions of both Meisenheimer type and methyleneazulenate anions. J. Org. Chem. 1974, 39, 1877–1887. [Google Scholar] [CrossRef]
- Tilney-Bassett, J.F.; Waters, W.A. The substitution of azulene by benzyl radicals. J. Chem. Soc. 1959, 633, 3123–3139. [Google Scholar] [CrossRef]
- Makosza, M.; Kuciak, R.; Wojciechowski, K. Vicarious Nucleophilic Substitution (VNS) of Hydrogen in Azulenes. Liebigs Ann. Chem. 1994, 1994, 615–618. [Google Scholar] [CrossRef]
- Hunig, S.; Hafner, K.; Ort, B.; Muller, M. Einfuhrung funktioneller Gruppen in den siebengliedrigen Ring des Azulens. Liebigs Ann. Chem. 1986, 1986, 1222–1240. [Google Scholar] [CrossRef]
- Makosza, M.; Podraza, R. Hydroxylation and Amination of Azulenes by Vicarious Nucleophilic Substitution of Hydrogen. Eur. J. Org. Chem. 2000, 2000, 193–198. [Google Scholar] [CrossRef]
- Makosza, M. On amination and Diazotization of Azulene and Its Derivatives. Pol. J. Chem. 2001, 75, 275–281. [Google Scholar]
- Hafner, K.; Patzelt, H.; Kaiser, H. Zur Kenntnis der Azulene, XI. Nucleophile Substitution halogenierter Azulene. Liebigs Ann. Chem. 1962, 656, 24–33. [Google Scholar] [CrossRef]
- Nozoe, T.; Takase, K.; Tada, M. The Anionoid Substitution Reaction of Diethyl 6-Bromoazulene-1, 3-dicarboxylate. Bull. Chem. Soc. Jpn. 1965, 38, 247–251. [Google Scholar] [CrossRef] [Green Version]
- Morita, T.; Ise, F.; Takase, K. Synthesis of methyl 1,5- and 1,7-azulenequinone-3-carboxylates. Chem. Lett. 1982, 11, 1303–1306. [Google Scholar] [CrossRef] [Green Version]
- Shoji, T.; Sugiyama, S.; Takeuchi, M.; Ohta, A.; Sekiguchi, R.; Ito, S.; Yatsu, T.; Okujima, T.; Yasunami, M. Synthesis of 6-Amino- and 6-Arylazoazulenes via Nucleophilic Aromatic Substitution and Their Reactivity and Properties. J. Org. Chem. 2019, 84, 1257–1275. [Google Scholar] [CrossRef]
- Shoji, T.; Fujiwara, Y.; Maruyama, A.; Maruyama, M.; Ito, S.; Yasunami, M.; Yokoyama, R.; Morita, N. Synthesis of 2,6-Diaminoazulenes by the SNAr Reaction with Cyclic Amines. Heterocycles 2015, 90, 85–88. [Google Scholar] [CrossRef]
- Shoji, T.; Maruyama, A.; Maruyama, M.; Ito, S.; Okujima, T.; Higashi, J.; Toyota, K.; Morita, N. Synthesis and Properties of 6-Methoxy- and 6-Dimethylamino-1-methylthio- and 1,3-Bis(methylthio)azulenes and Triflic Anhydride-Mediated Synthesis of Their Biaryl Derivatives. Bull. Chem. Soc. Jpn. 2014, 87, 141–154. [Google Scholar] [CrossRef]
- Anderson, A., Jr.; Replogle, L. Communications-Electrophilic Substitution of 1,3-Dichloroazulene. J. Org. Chem. 1960, 25, 1275. [Google Scholar] [CrossRef]
- Anderson, A.G.; Replogle, L.L. Electrophilic Substitution of Some 1,3-Disubstituted Azulenes1,2,3. J. Org. Chem. 1963, 28, 2578–2581. [Google Scholar] [CrossRef]
- Hafner, K.; Moritz, K.L. Zur Kenntnis der Azulene, XIII. Elektrophile Substitutionen des Azulens in 2-und 5-bzw. 7-Stellung. Liebigs Ann. Chem. 1962, 656, 40–53. [Google Scholar] [CrossRef]
- Nozoe, T.; Asao, T.; Oda, M. Some Electrophilic Substitution Reactions of 2-Hydroxyazulene Derivatives. Bull. Chem. Soc. Jpn. 1974, 47, 681–686. [Google Scholar] [CrossRef] [Green Version]
- Shoji, T.; Ito, S.; Toyota, K.; Morita, N. Synthesis of 5-heteroaryl- and 5,7-bis(heteroaryl)azulenes by electrophilic substitution of 1,3-di-tert-butylazulene with triflates of N-containing heterocycles. Eur. J. Org. Chem. 2010, 80, 1059–1069. [Google Scholar] [CrossRef]
- Hafner, K.; Pelster, H.; Schneider, J. Zur Kenntnis der Azulene, VIII. 4-bzw. 8- un 6 Alkyliden-azuleniat Salze. Liebigs Ann. Chem. 1961, 650, 80–92. [Google Scholar] [CrossRef]
- Neidlein, R.; Kramer, W. Heterocyclische und carbocyclische 12-π- and 14-π-Molekelsysteme, 47. Mitteilung. Herstellung von 7,9-Dimethyl-4,5-dihydro-3H-benz [cd]-azulen-3-on and 7,9-Dimethyl-3H-benz [cd]azulen-3-on. Eine einfache Synthese eines Azulenopseudophenalenons. Helv. Chim. Acta 1982, 65, 280–285. [Google Scholar] [CrossRef]
- Wörner, M. Dissertation, Technische Hochschule Darmstadt. 1984. [Google Scholar]
- Herrmann, R.; Wagner, G.; Zollfrank, C. Ferrocene and Azulene—A Promising Combination for NLO Materials. 1995. Available online: http://www.ch.ic.ac.uk/ectoc/papers/49 (accessed on 20 March 2023).
- Herrmann, R.; Pedersen, B.; Wagner, G.; Youn, J.-H. Molecules with potential applications for non-linear optics: The combination of ferrocene and azulene. J. Organomet. Chem. 1998, 571, 261–266. [Google Scholar] [CrossRef]
- Song, J.; Hansen, H.-J. An Improved and Simplified Synthesis of 4-Styrylazulenes. Helv. Chim. Acta 1999, 82, 309–314. [Google Scholar] [CrossRef]
- Razus, A.C.; Birzan, L.; Tecuceanu, V.; Cristea, M.; Enache, C. Condensation of alkylazulenes with thiophene-2-carboxaldehyde and the corresponding azomethines. Arkivoc 2008, 11, 210–226. [Google Scholar] [CrossRef] [Green Version]
- Birzan, L.; Tecuceanu, V.; Draghici, C.C.; Hanganu, A.; Razus, A.C. Preparation of Azulenes Substituted at Seven-Membered Cycle with 2- and 3-thiophenevinyl Groups. Rev. Chim. 2020, 71, 212–224. [Google Scholar] [CrossRef]
- Ghazvini Zadeh, E.H.; Tang, S.; Woodward, A.W.; Liu, T.; Bondar, M.V.; Belfield, K.D. Chromophoric materials derived from a natural azulene: Syntheses, halochromism and one-photon and two-photon microlithography. J. Mater. Chem. C 2015, 3, 8495–8503. [Google Scholar] [CrossRef]
- Kurotobi, K.; Kim, K.S.; Noh, S.B.; Kim, D.; Osuka, A. A quadruply azulene-fused porphyrin with intense near-IR absorption and a large two-photon absorption cross section. Angew. Chem. Int. Ed. Engl. 2006, 45, 3944–3947. [Google Scholar] [CrossRef]
- Leino, T.O.; Baumann, M.; Yli-Kauhaluoma, J.; Baxendale, I.R.; Wallén, E. Synthesis of 1,3,6-Trisubstituted Azulenes. J. Org. Chem. 2015, 80, 11513–11520. [Google Scholar] [CrossRef]
- Williams, G.E.; Kociok-Köhn, G.; James, T.D.; Lewis, S.E. C4-aldehyde of guaiazulene: Synthesis and derivatisation. Org. Biomol. Chem. 2021, 19, 2502–2511. [Google Scholar] [CrossRef]
- Morita, T.; Abe, N.; Takase, K. Reactions of diethyl azulene-1,3-dicarboxylate derivatives and 1-azaazulene derivatives with Grignard reagents, and alkyl- and aryllithium. And reference therein. J. Chem. Soc. Perkin Trans. 1 2000, 18, 3063–3070. [Google Scholar] [CrossRef]
- Hünig, S.; Ort, B. Mehrstufige reversible Redoxsysteme, XXXVII. Biazulenyle und ω,ω′-Biazulenylpolyene: Synthesen und spektroskopische Eigenschaften. Liebigs Ann. Chem. 1984, 12, 1905–1935. [Google Scholar] [CrossRef]
- Maekawa, H.; Honda, J.; Akaba, R. Regioselective coupling reaction of azulene with α,β-unsaturated ketones by Mg-promoted reduction. Tetrahedron Lett. 2012, 53, 6519–6522. [Google Scholar] [CrossRef]
- Ito, S.; Kubo, T.; Morita, N.; Matsui, Y.; Watanabe, T.; Ohta, A.; Fujimori, K.; Murafuji, T.; Sugihara, Y.; Tajiri, A. Preparation of azulenyllithium and magnesium reagents utilizing halogen-metal exchange reaction of several iodoazulenes with organolithium or magnesium ate complex. Tetrahedron Lett. 2004, 45, 2891–2894. [Google Scholar] [CrossRef]
- Okujima, T.; Ito, S.; Morita, N. Preparation and Stille cross-coupling reaction of the first organotin reagents of azulenes. An efficient Pd(0)-catalyzed synthesis of 6-aryl- and biazulenes. Tetrahedron Lett. 2002, 43, 1261–1264. [Google Scholar] [CrossRef]
- Azizian, H.; Eaborn, C.; Pidcock, A. Synthesis of organotrialkylstannanes. The reaction between organic halides and hexaalkyldistannanes in the presence of palladium complexes. J. Organomet. Chem. 1981, 215, 49–58. [Google Scholar] [CrossRef]
- Ito, S.; Okujima, T.; Morita, N. Preparation and Stille cross-coupling reaction of the first organotin reagents of azulenes. Easy access to poly(azulen-6-yl)benzene derivatives. J. Chem. Soc. Perkin Trans. 1 2002, 16, 1896–1905. [Google Scholar] [CrossRef]
- Crombie, A.L.; Kane, J.L., Jr.; Shea, K.M.; Danheiser, R.L. Ring Expansion-Annulation Strategy for the Synthesis of Substituted Azulenes and Oligoazulenes. 2. Synthesis of Azulenyl Halides, Sulfonates, and Azulenylmetal Compounds and Their Application in Transition-Metal-Mediated Coupling Reactions. J. Org. Chem. 2004, 69, 8652–8667. [Google Scholar] [CrossRef]
- Amir, E.; Amir, R.J.; Campos, L.M.; Hawker, C.J. Stimuli-Responsive Azulene-Based Conjugated Oligomers with Polyaniline-like Properties. J. Am. Chem. Soc. 2011, 133, 10046–10049. [Google Scholar] [CrossRef]
- Amir, E.; Murai, M.; Amir, R.J.; Cowart, J.S.; Chabinyc, M.L.; Hawker, C.J. Conjugated oligomers incorporating azulene building blocks—Seven- vs. five-membered ring connectivity. Chem. Sci. 2014, 5, 4483–4489. [Google Scholar] [CrossRef]
- Shoji, T.; Maruyama, A.; Araki, T.; Ito, S.; Okujima, T. Synthesis of 2- and 6-thienylazulenes by palladium-catalyzed direct arylation of 2- and 6-haloazulenes with thiophene derivatives. Org. Biomol. Chem. 2015, 13, 10191–10197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujinaga, M.; Murafuji, T.; Kurotobi, K.; Sugihara, Y. Polyborylation of azulenes. Tetrahedron 2009, 65, 7115–7121. [Google Scholar] [CrossRef]
- Scott, L.; Nishimura, H.; Eliseeva, M.; Wakamiya, A. 1,3,5,7-Tetra(Bpin)azulene by Exhaustive Direct Borylation of Azulene and 5,7-Di(Bpin)azulene by Selective Subsequent Deborylation. Synlett 2015, 26, 1578–1580. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Ogawa, K.; Nakayama, K.; Ohba, Y.; Katagiri, H. Terazulene: A High-Performance n-Type Organic Field-Effect Transistor Based on Molecular Orbital Distribution Control. J. Am. Chem. Soc. 2013, 135, 19095–19098. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Takubo, M.; Ogawa, K.; Nakayama, K.; Koganezawa, T.; Katagiri, H. Terazulene Isomers: Polarity Change of OFETs through Molecular Orbital Distribution Contrast. J. Am. Chem. Soc. 2016, 138, 11335–11343. [Google Scholar] [CrossRef]
- Xin, H.; Ge, C.; Yang, X.; Gao, H.; Yang, X.; Gao, X. Biazulene diimides: A new building block for organic electronic materials. Chem. Sci. 2016, 7, 6701–6705. [Google Scholar] [CrossRef] [Green Version]
- Kurotobi, K.; Tabata, H.; Miyauchi, M.; Murafuji, T.; Sugihara, Y. Coupling Reaction of Azulenyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolanes with Haloazulenes. Synthesis 2002, 2002, 1013–1016. [Google Scholar] [CrossRef]
- Murfin, L.C.; Weber, M.; Park, S.J.; Kim, W.T.; Lopez-Alled, C.M.; McMullin, C.L.; Pradaux-Caggiano, F.; Lyall, C.L.; Kociok-Köhn, G.; Wenk, J.; et al. Azulene-Derived Fluorescent Probe for Bioimaging: Detection of Reactive Oxygen and Nitrogen Species by Two-Photon Microscopy. J. Am. Chem. Soc. 2019, 141, 19389–19396. [Google Scholar] [CrossRef] [Green Version]
- Xin, H.; Ge, C.; Jiao, X.; Yang, X.; Rundel, K.; McNeill, C.R.; Gao, X. Incorporation of 2,6-Connected Azulene Units into the Backbone of Conjugated Polymers: Towards High-Performance Organic Optoelectronic Materials. Angew. Chem. Int. Ed. 2017, 57, 1322–1326. [Google Scholar] [CrossRef]
- Kitai, J.; Kobayashi, T.; Uchida, W.; Hatakeyama, M.; Yokojima, S.; Nakamura, S.; Uchida, K. Photochromism of a Diarylethene Having an Azulene Ring. J. Org. Chem. 2012, 77, 3270–3276. [Google Scholar] [CrossRef]
- Tsurui, K.; Murai, M.; Ku, S.-Y.; Hawker, C.J.; Robb, M.J. Modulating the Properties of Azulene-Containing Polymers through Controlled Incorporation of Regioisomers. Adv. Funct. Mater. 2014, 24, 7338–7347. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Ge, C.; Hou, B.; Xin, H.; Gao, X. Incorporation of 1,3-Free-2,6-Connected Azulene Units into the Backbone of Conjugated Polymers: Improving Proton Responsiveness and Electrical Conductivity. ACS Macro Lett. 2019, 8, 1360–1364. [Google Scholar] [CrossRef]
- Fu, H.; Zhao, C.; Cheng, J.; Zhou, S.; Peng, P.; Hao, J.; Liu, Z.; Gao, X.; Jia, C.; Guo, X. Dipole-improved gating of azulene-based single-molecule transistors. J. Mater. Chem. C 2022, 10, 7803–7809. [Google Scholar] [CrossRef]
- Huang, J.; Huang, S.; Zhao, Y.; Feng, B.; Jiang, K.; Sun, S.; Ke, C.; Kymakis, E.; Zhuang, X. Azulene-Based Molecules, Polymers, and Frameworks for Optoelectronic and Energy Applications. Small Methods 2020, 4, 2000628. [Google Scholar] [CrossRef]
- Hou, I.C.-Y.; Shetti, V.; Huang, S.-L.; Liu, K.-L.; Chao, C.-Y.; Lin, S.-C.; Lin, Y.J.; Chen, L.Y.; Luh, T.-Y. Poly[2(6)-aminoazulene]: Synthesis, photophysical properties, and proton conductivity. Org. Chem. Front. 2017, 4, 773–778. [Google Scholar] [CrossRef]
- Ito, S.; Kubo, T.; Morita, N.; Ikoma, T.; Tero-Kubota, S.; Kawakami, J.; Tajiri, A. Azulene-substituted aromatic amines. Synthesis and amphoteric redox behavior of N,N-Di(6-azulenyl)-p-toluidine and N,N,N′,N′-tetra(6- azulenyl)-p-phenylenediamine and their derivatives. J. Org. Chem. 2005, 70, 2285–2293. [Google Scholar] [CrossRef]
- Murai, M.; Takami, K.; Takeshima, H.; Takai, K. Iridium-Catalyzed Dehydrogenative Silylation of Azulenes Based on Regioselective C–H Bond Activation. Org. Lett. 2015, 17, 1798–1801. [Google Scholar] [CrossRef]
- Koch, M.; Blacque, O.; Venkatesan, K. Impact of 2,6-connectivity in azulene: Optical properties and stimuli responsive behavior. J. Mater. Chem. C 2013, 1, 7400–7408. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Inabe, H.; Okujima, T.; Morita, N.; Watanabe, M.; Imafuku, K. Diels–Alder approach to the synthesis of azulene-substituted benzene derivatives. Synthesis and redox behavior of 1,2-di(6-azulenyl)tetraphenylbenzenes. Tetrahedron Lett. 2000, 41, 8343–8347. [Google Scholar] [CrossRef]
- Ito, S.; Inabe, H.; Okujima, T.; Morita, N.; Watanabe, M.; Harada, N.; Imafuku, K. Synthesis and Redox Behavior of Azulene-Substituted Benzene Derivatives and (η5-Cyclopentadienyl)[tetra- and di(6-azulenyl)cyclobutadiene]cobalt Complexes. J. Org. Chem. 2001, 66, 7090–7101. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Inabe, H.; Morita, N.; Ohta, K.; Kitamura, T.; Imafuku, K. Synthesis of poly(6-azulenylethynyl)benzene derivatives as a multielectron redox system with liquid crystalline behavior. J. Am. Chem. Soc. 2003, 125, 1669–1680. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Iida, T.; Kawakami, J.; Okujima, T.; Morita, N. Towards the Preparation of Electrochromic Materials with Strong Absorption in the Near-Infrared Region: Synthesis and Redox Behavior of Azulene-Substituted Enediyne Scaffolds Connected by a 9,10-Anthracenediyl Spacer. Eur. J. Org. Chem. 2009, 5355–5364. [Google Scholar] [CrossRef]
- Okujima, T.; Toda, A.; Miyashita, Y.; Nonoshita, A.; Yamada, H.; Ono, N.; Uno, H. Synthesis and properties of azulene-substituted thiophenes, terthiophenes and dithienothiophenes. Heterocycles 2012, 86, 637–648. [Google Scholar] [CrossRef]
- Shoji, T.; Ariga, Y.; Yamazaki, A.; Uda, M.; Nagasawa, T.; Ito, S. Synthesis, Photophysical and Electrochemical Properties of 1-, 2-, and 6-(2-Benzofuryl)azulenes. Bull. Chem. Soc. Jpn. 2021, 94, 1000–1009. [Google Scholar] [CrossRef]
- Erdogan, M. Facile One-pot Synthesis of a Novel Propargyl-Azulene Hybrid Derivative. J. Inst. Sci. Technol. 2020, 10, 2747–2758. [Google Scholar] [CrossRef]
- Zeng, H.N.; Png, Z.M.; Xu, J. Azulene in Polymers and Their Properties. Chem. Asian J. 2020, 15, 1904–1915. [Google Scholar] [CrossRef]
- Murai, M.; Amir, E.; Amir, R.J.; Hawker, C.J. Azulene-based conjugated polymers: Unique seven-membered ring connectivity leading to stimuli-responsiveness. Chem. Sci. 2012, 3, 2721. [Google Scholar] [CrossRef]
- Hafner, K.; Schaum, H. Cycloheptatrieno-Indene. Angew. Chem. Int. Ed. 1963, 2, 95–96. [Google Scholar] [CrossRef]
- Hafner, K. Structure and Aromatic Character of Non-benzenoid Cyclically Conjugated Systems. Angew. Chem. Int. Ed. 1964, 3, 165–173. [Google Scholar] [CrossRef]
- Gibson, W.K.; Leaver, D.; Roff, J.E.; Cumming, C.W. Synthesis of cycl [3,3,2]azinones and benz[cd]azulenones. Chem. Commun. 1967, 214. [Google Scholar] [CrossRef]
- Aumüller, I.B.; Yli-Kauhaluoma, J. Synthesis and Tautomerization of Benzo[cd]azulen-3-ones. Org. Lett. 2011, 13, 1670–1673. [Google Scholar] [CrossRef]
- Shoji, T.; Yamazaki, A.; Katoh, R.; Shimamura, K.; Sakai, R.; Yasunami, M.; Okujima, T.; Ito, S. Synthesis, Reactivity, and Properties of Benz[a]azulenes via the [8 + 2] Cycloaddition of 2H-Cyclohepta[b]furan-2-ones with Selenesulfonates Leading to (E)-2,3-Disulfonylpropene Derivatives. J. Org. Chem. 2022, 87, 5827–5845. [Google Scholar] [CrossRef]
- Nozoe, T.; Wakabayashi, H.; Shindo, K.; Kurihara, T.; Ishikawa, S.; Kageyama, M. A Highly Convenient, One-Pot Synthesis of 3-Bromo-1,5- and -1,7-Azulenequinones by Polybromination of Azulene. Chem. Lett. 1995, 24, 25–26. [Google Scholar] [CrossRef]
- Sigrist, R.; Hansen, H.J. Benzo[a]azulenediones and 10,10′-Bibenzo[a]azulene. Helv. Chim. Acta 2014, 97, 1165–1175. [Google Scholar] [CrossRef]
- Shoji, T.; Yamazaki, A.; Sakata, N.; Sekiguchi, R.; Ito, S.; Yasunami, M. Benz[a]azulenequinones: Reaction of Benz[a]azulenes with Pyridinium Hydrobromide Perbromide. Molbank 2022, 2022, M1467. [Google Scholar] [CrossRef]
- Devendar, B.; Wu, C.-P.; Chen, C.-Y.; Chen, H.-C.; Chang, C.-H.; Ku, C.-K.; Tsai, C.-Y.; Ku, C.-Y. Synthesis, characterization and applications of densely functionalized pyridazines and fulvene-type compounds containing azulene moiety. Tetrahedron 2013, 69, 4953–4963. [Google Scholar] [CrossRef]
- Kiriazis, A.; Aumüller, I.B.; Yli-Kauhaluoma, J. Synthesis of 4-aminoguaiazulene and its δ-lactam derivatives. Tetrahedron Lett. 2011, 52, 1151–1153. [Google Scholar] [CrossRef]
- Hafner, K.; Schneider, J. Darstellung und Eigenschaften von Derivaten des Pentalens und Heptalens. Liebigs Ann. Chem. 1959, 624, 37–47. [Google Scholar] [CrossRef]
- Anderson, A.G.; Anderson, R.G.; Hollander, G.T. The Reaction of Some 1-Trihaloacetyl-8-methylazulenes with Base. J. Org. Chem. 1965, 30, 131–138. [Google Scholar] [CrossRef]
- Pigulski, B.; Shoyama, K.; Wurthner, F. NIR-Absorbing π-Extended Azulene: Non-Alternant Isomer of Terrylene Bisimide. Angew. Chem. Int. Ed. 2020, 59, 15908–15912. [Google Scholar] [CrossRef] [PubMed]
- Shoyama, K.; Mahl, M.; Seifert, S.; Würthner, F. A General Synthetic Route to Polycyclic Aromatic Dicarboximides by Palladium-Catalyzed Annulation Reaction. J. Org. Chem. 2018, 83, 5339–5346. [Google Scholar] [CrossRef] [PubMed]
- Kurotobi, K.; Osuka, A. Synthesis of meso-Azulenylporphyrins. Org. Lett. 2005, 7, 1055–1058. [Google Scholar] [CrossRef] [PubMed]
- Lash, T.D. Out of the Blue! Azuliporphyrins and Related Carbaporphyrinoid Systems. Acc. Chem. Res. 2016, 49, 471–482. [Google Scholar] [CrossRef]
- Sprutta, N.; Swiderska, M.; Latos-Grazùynski, L. Dithiadiazuliporphyrin: Facile Generation of Carbaporphyrinoid Cation Radical and Dication. J. Am. Chem. Soc. 2005, 127, 13108–13109. [Google Scholar] [CrossRef]
- Sprutta, N.; Siczek, M.; Latos-Grazùynski, L.; Pawlicki, M.; Szterenberg, L.; Lis, T. Dioxadiazuliporphyrin: A Near-IR Redox Switchable Chromophore. J. Org. Chem. 2007, 72, 9501–9509. [Google Scholar] [CrossRef]
- Lindsey, J.S.; Schreiman, I.C.; Hsu, H.C.; Kearney, P.C.; Marguerettaz, A.M. Rothemund and Adler-Longo reactions revisited: Synthesis of tetraphenylporphyrins under equilibrium conditions. J. Org. Chem. 1987, 52, 827–836. [Google Scholar] [CrossRef]
- Muranaka, A.; Yonehara, M.; Uchiyama, M. Azulenocyanine: A New Family of Phthalocyanines with Intense Near-IR Absorption. J. Am. Chem. Soc. 2010, 132, 7844–7845. [Google Scholar] [CrossRef]
- de Diesbach, H.; von der Weid, E. Quelques sels complexes des o-dinitriles avec le cuivre et la pyridine. Helv. Chim. Acta 1927, 10, 886–888. [Google Scholar] [CrossRef]




























































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Razus, A.C. Azulene, Reactivity, and Scientific Interest Inversely Proportional to Ring Size; Part 2: The Seven-Membered Ring. Symmetry 2023, 15, 1391. https://doi.org/10.3390/sym15071391
Razus AC. Azulene, Reactivity, and Scientific Interest Inversely Proportional to Ring Size; Part 2: The Seven-Membered Ring. Symmetry. 2023; 15(7):1391. https://doi.org/10.3390/sym15071391
Chicago/Turabian StyleRazus, Alexandru C. 2023. "Azulene, Reactivity, and Scientific Interest Inversely Proportional to Ring Size; Part 2: The Seven-Membered Ring" Symmetry 15, no. 7: 1391. https://doi.org/10.3390/sym15071391