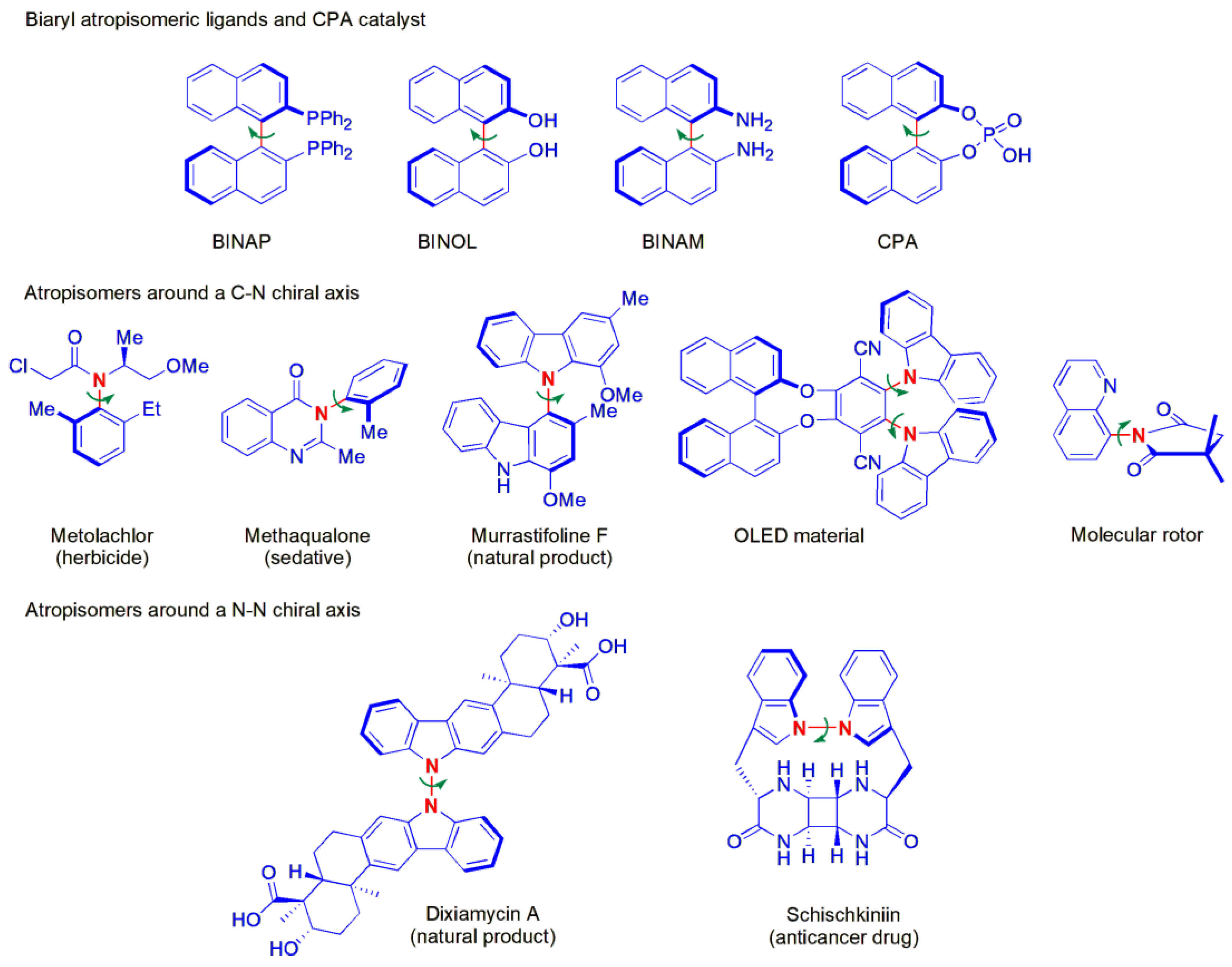
2.1. C-H Amination
Non-biaryl atropisomers synthesized from 2-naphthol by means of asymmetric organocatalysis were described for the first time by Bella, Jørgensen and co-workers in 2006 [
34]. The rotation along the chiral axis in these substances can be hindered by substituents in the
peri position. Until then, the syntheses reported involved chiral resolution to obtain pure racemates [
35], with one exception being an account of enantioselective synthesis utilizing a metal-catalyzed approach [
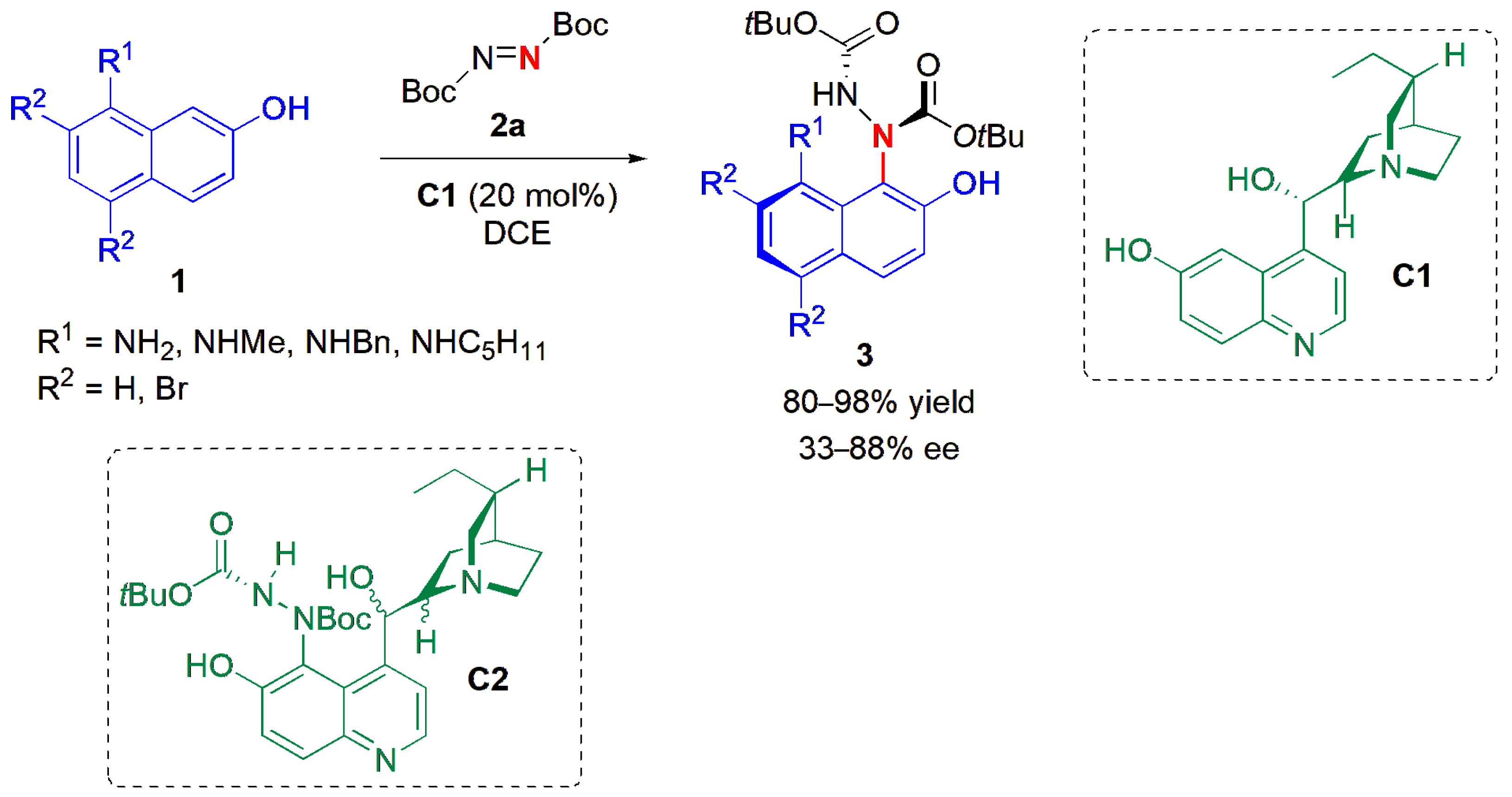
36]. The amination of a series of 8-amino-2-naphthols (
1) with di-
tert-butyl azodicarboxylate (
2a) in combination with dihydrocupreidine (
C1) as a catalyst provided products (
3) in high yields and good ees (
Figure 3). The ee of
3a (R
1 = NH
2; R
2 = R
3 = H) did not change much when the compound was kept at −20 °C, and at the room temperature, the ee dropped only 3% in 10 days. In an attempt to obtain more stereoselective catalysts, the group performed the same asymmetric Friedel–Crafts amination reaction of the cinchona alkaloid catalysts themselves, and subsequently used them, after chromatographic separation of the two diastereoisomers obtained, as catalysts to perform the reactions shown in
Figure 3. Substantial enhancements in ee were obtained, e.g., with
C2, compounds
3 were obtained with 87–98% ee.
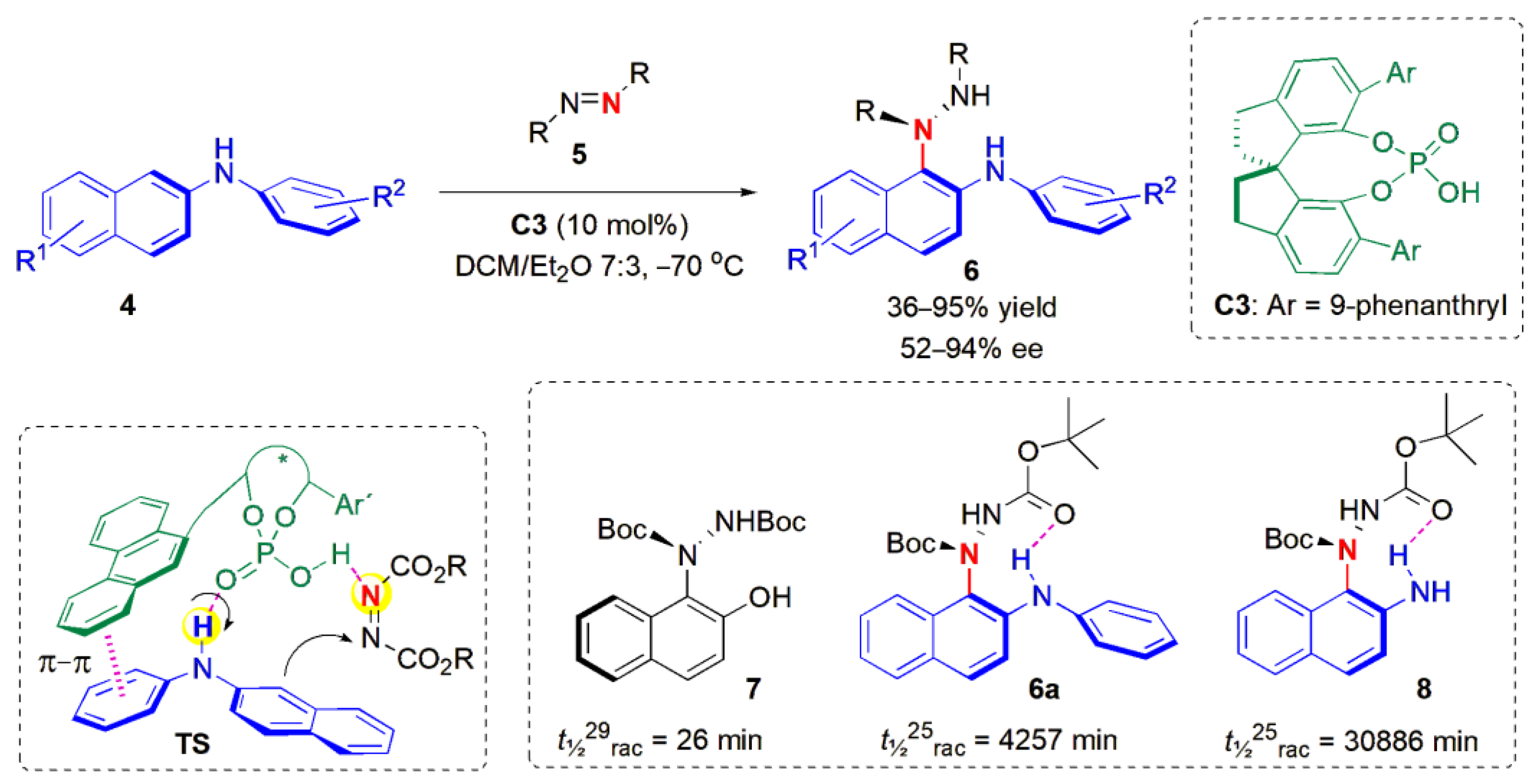
In 2019, Bai, Zhang and co-workers described the synthesis of novel nonbiaryl C-N atropisomers, naphthalene-1,2-diamines
6, obtained from
N-aryl-2-naphthylamines
4 with azodicarboxylates
5 as amino sources (
Figure 4) [
37]. Until then, chiral nonbiaryl C-N atropisomers of 2-naphthylamine derivatives had not yet been discovered. A chiral phosphoric acid catalyst, C3, was found to promote the amination reaction, and a range of products could be obtained in high yields and ee values, provided a di-
tert-butyl azodicarboxylate was used. With other dicarboxylates, the ees were substantially lower. Interestingly, when R
2 =
p-CN, or if the substrate contained a
peri NH
2 substituent, no product was obtained at all. It was assumed that the stereoselectivity could be controlled by concerted π-π interactions and dual hydrogen bonding between the chiral phosphoric acid catalyst and the two reactant molecules, as shown in A. Half-lives of racemization (in
n-hexane at 25 °C) were measured to verify the stereo-stability of the products. The ee of 6a in the solid state (90.10% ee) remained 90.04% ee after 90 h at −18 °C. Presumably, H-bonding interactions help to stabilize the products.
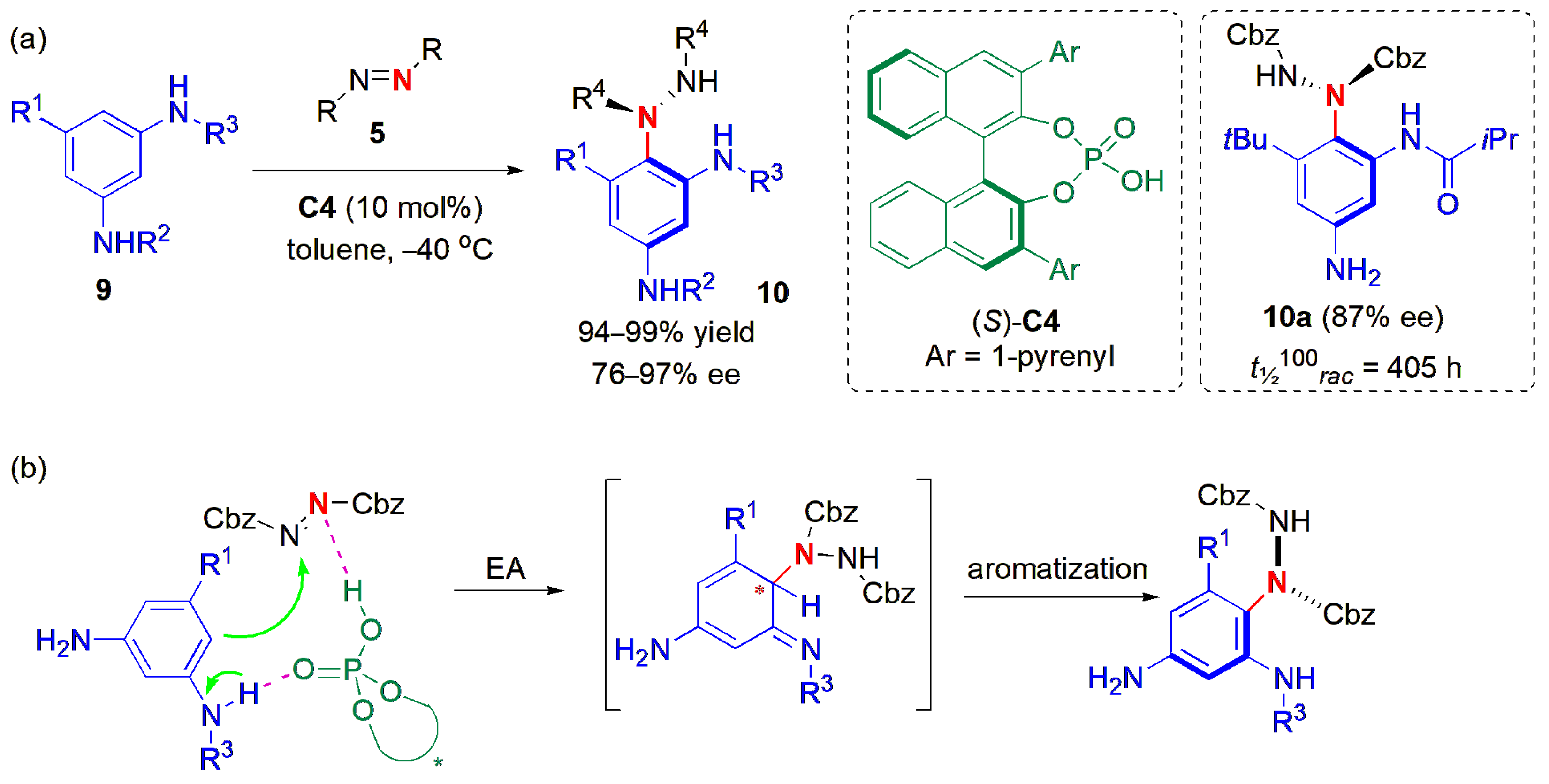
The direct aminations of 1,3-benzenediamines
9 with azodicarboxylates
5 have also been enabled by atropselective synthesis with chiral phosphoric acid catalysis, i.e., with (
S)-
C4 (
Figure 5a) [
38]. By varying the nature of the N-substituents, benzene-substituents, and azodicarboxylates, a range of C-N atropisomers
10 with high configurational stability were produced, with high yields and ees.
Electrophilic addition facilitated by dual H-bonding interactions followed by an aromatization (in which there is central axial chirality transfer) has also been reported to be involved, as shown in
Figure 5b. Contrary to the report on the amination of naphthylamines, in this case, benzyl chloroformate (Cbz)-substituted azodicarboxylates were more efficient in providing the desired stereoselectivity than Boc-substituted ones. The yields varied within the range of 94–99%, except in the case ofa NHPh-substituted benzenediamine. A large range of electron-withdrawing R
3 substituents could be used, as well as 5-substituents, and the products were obtained in high yields and ees. However, the configurational stability of products with R
1 = primary alkyl groups was relatively low compared to that of the 5-
tBu-substituted products, presumably due to the lower steric hindrance provided by these groups. The highest stabilities were observed in
N-acyl substituted products bearing a 5-
tBu and 3-NH
2 substituents (with
t1/2 ranging from 226 to 405 h at 100 °C), e.g., as for
10a.
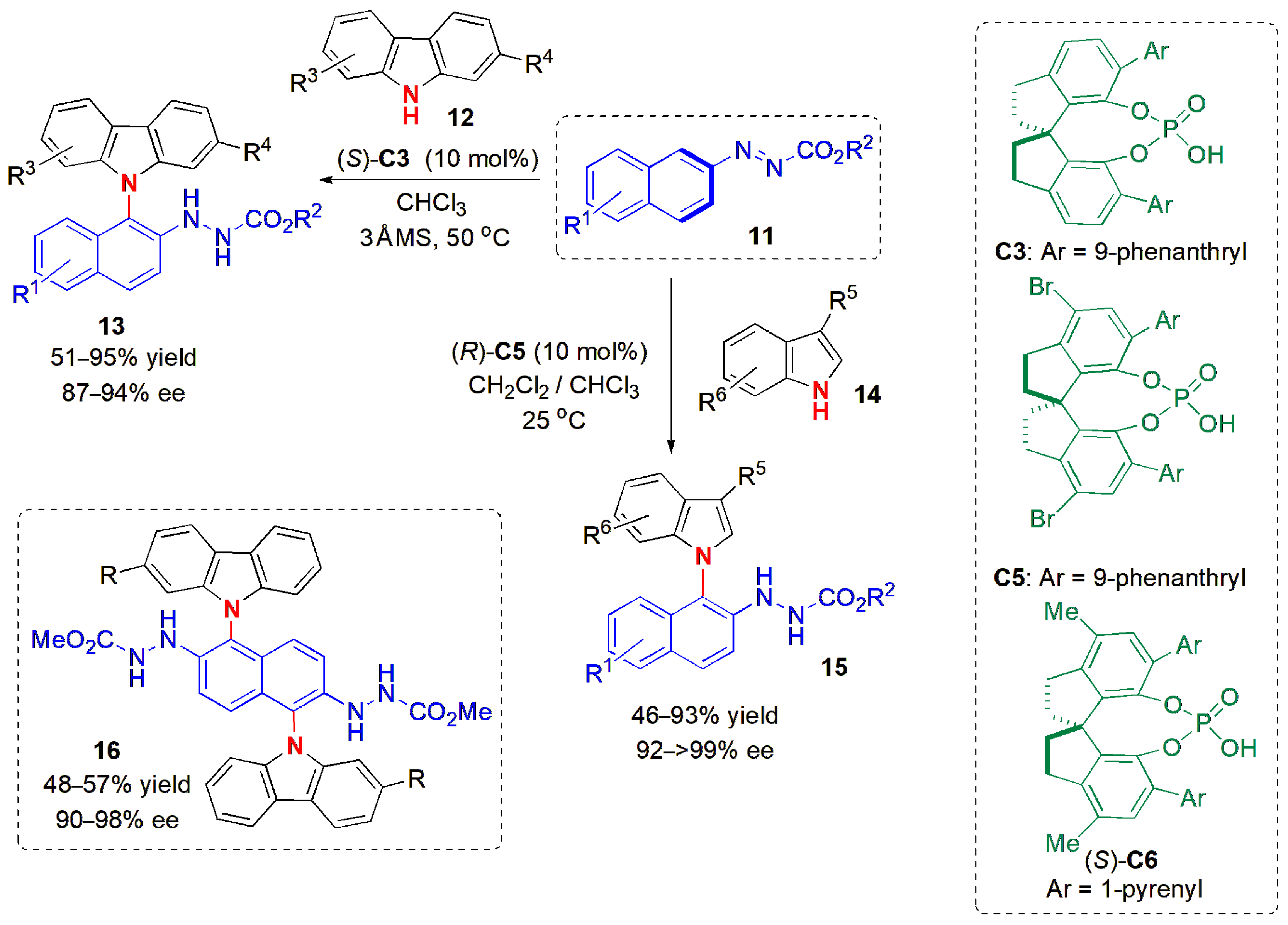
The reaction of azonaphthalines
11 with
N-arylcarbazoles
12 in the presence of chiral phosphoric acid
C3 also gave rise to axially chiral products (
13) in an atropselective manner with excellent ees (
Figure 6) [
39]. The work by Li, Tan, and co-workers was inspired by the fact that
N-arylcarbazoles are one of the most widely used host materials for OLEDs, due to the fact that they may display high triplet energies and competitive hole transport abilities. These properties often drive research towards obtaining new structurally diverse
N-arylcarbazoles, with the aim of obtaining materials with good properties. The method developed in this study was compatible with a wide range of substituents in the starting materials, with ees remaining within 87 and 94%, with electron-donating or electron-withdrawing substituents in the aromatic rings. In addition, the reaction of the azonaphthalines with indoles
14 was also explored, allowing the synthesis of various C-N axial compounds
15 with ees up to >99%, although in this case, the reaction worked better with a different chiral phosphoric acid,
C5.
Double enantioselective arene C-H amination reactions were also performed, although more forcing conditions were required in this case; reactions at 50 °C for 7 days allowed good yields of compounds 16 to be obtained, although this change did not affect the ees much. A different catalyst, C6, performed better in this case. The new products contain two chiral N-aryl axes.
2.2. N-H Functionalization
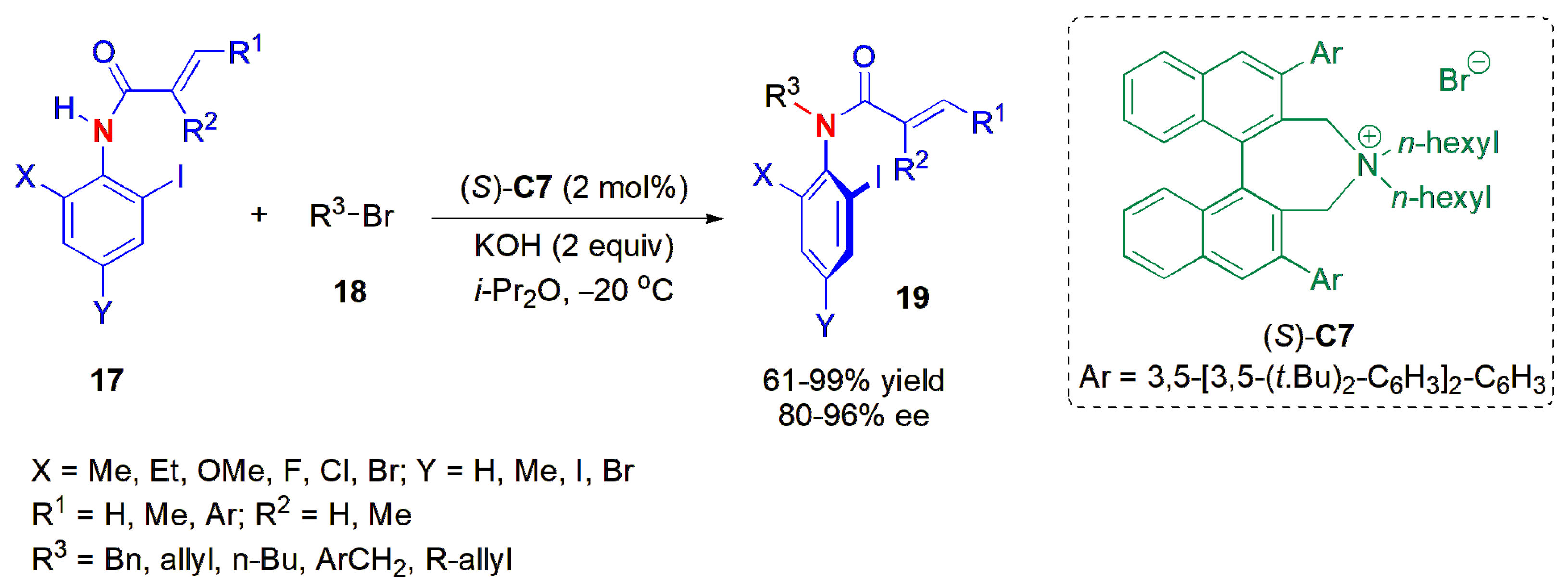
The first organocatalytic asymmetric synthesis of optically active axially chiral
o-iodoacrylanilides and
N-allyl-
o-iodoanilides from compounds
17 and bromides was described by Maruoka and co-workers in 2012 [
40].
O-iodoanilides
19 were obtained via phase-transfer catalyzed
N-alkylation with organocatalyst
C7, an organocatalyst that is itself also axially chiral (
Figure 7). The presence of bulky extended aromatic
ortho-substituents and
n-hexyl groups on the binaphthyl-modified chiral ammonium salt were crucial for high ees to be obtained, alongside a low temperature of −20 °C. The X-ray crystal structure of one product helped to indicate a mechanism of the reaction, suggesting that the catalyst can recognize the steric difference between iodide and methyl groups as
ortho-substituents on the anilide, thus favoring a halide approach, preferentially, to one side of the aromatic ring rather than the other. Very high enantiomeric excesses (ees) were obtained, as determined by chiral HPLC analysis.
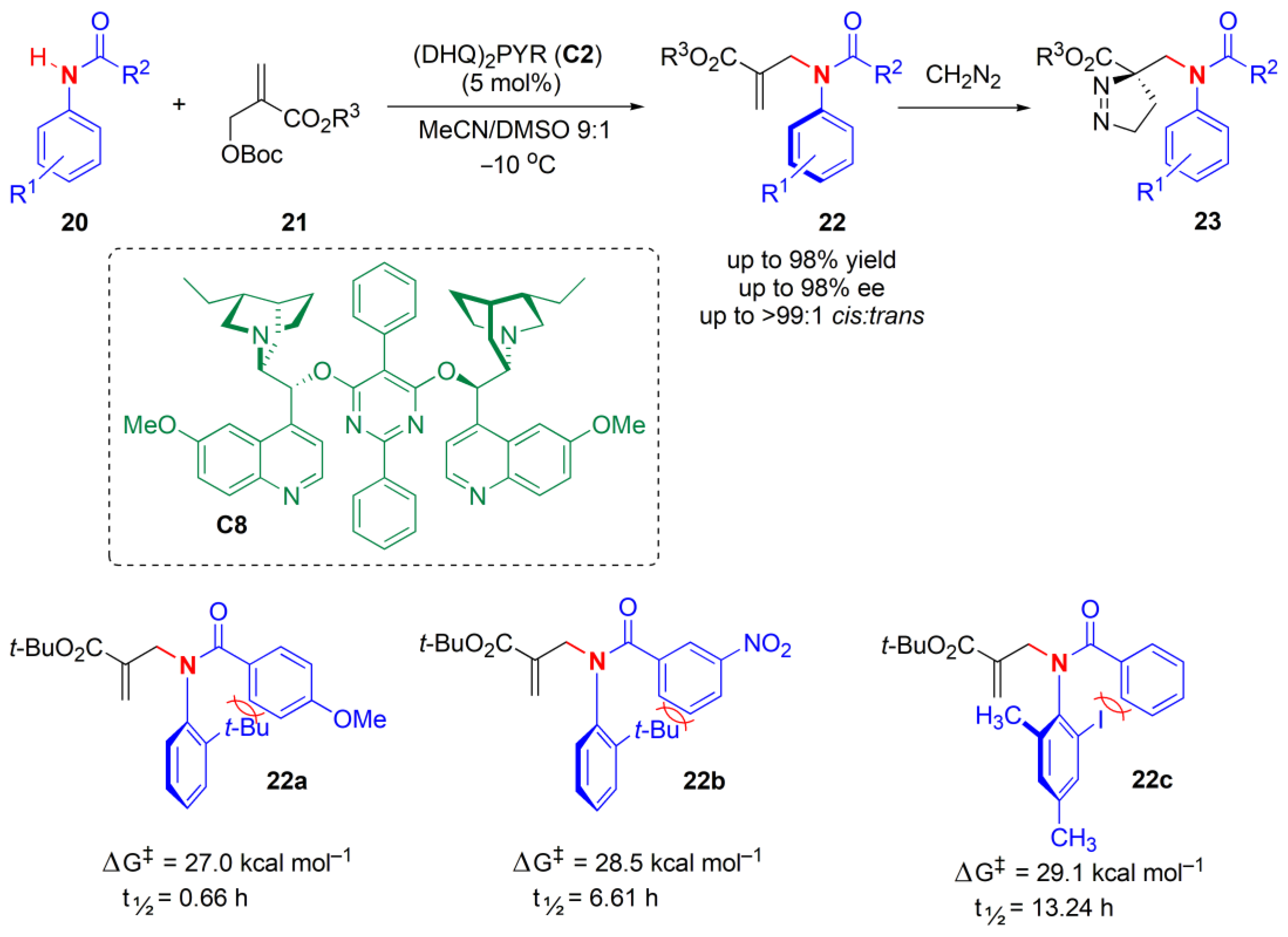
A similar N-H functionalization strategy was used by Li and co-workers to obtain axially chiral anilides, but via a Morita–Baylis–Hillman (MBH) reaction [
41]. A biscinchona alkaloid catalyst (
C8) allowed the introduction of several acyl groups, e.g., substituted phenyl, naphthyl, alkyl, enyl, styryl, and benzyl, on the amide with very good yields, moderate to excellent
cis:trans ratios, and good to excellent ees (
Figure 8). Gram-scale syntheses were also possible. However, the allylation products
22 could not be separated using column chromatography or using HPLC. Hence, they were reacted with CH
2N
2, generating a cycloaddition product
23 easily within 5 min, the isomers of which could be isolated using column chromatography to help to confirm the ee. Racemization experiments performed in isopropyl alcohol at 80 °C showed that not only the steric hindrance, but also the electronic properties and the position of substituents, had an effect on the stereochemical stability of the products
22, e.g., as for
22a–
22c.
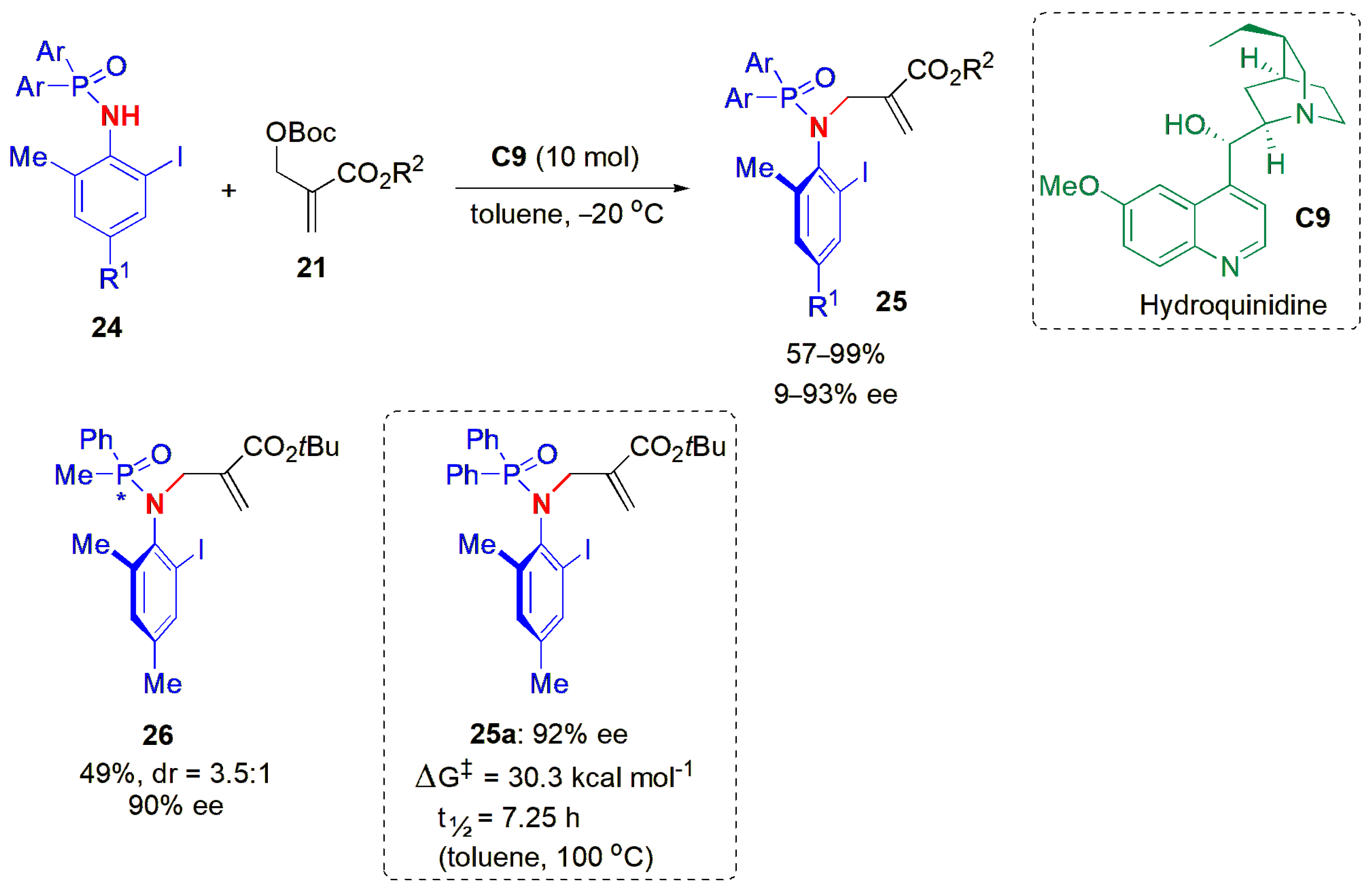
Although axially chiral anilides are a class of compounds that are emerging as biologically active scaffolds, their synthesis is complicated due to low rotation barriers. It has been noted previously that the presence of a heteroatom in the axis of rotation often causes a decrease in the rotational barrier [
21]. In 2020, Li and co-workers reported a procedure to obtain axially chiral phosphamides
25 via an atropselective
N-allylic alkylation reaction of phosphamides
24 and MBH carbonates
21 (
Figure 9) [
42]. The cinchona alkaloid hydroquinidine (
C9) was an efficient catalyst for this atroposelective strategy, and the products
25 were obtained in good yields and high ees. A good linear correlation was found to exist between the ees and the Charton values (R
2 = 0.91), parameters which are commonly used to describe the steric hindrance of substituents [
43]. From this linear free energy relationship analysis, it could be seen that substrates with large steric substituents, such as
tBu and Ad, gave better ees. However, steric hindrance was not the only factor at play, since when an
iPr group was present in the aromatic ring (at an
ortho position), a sharp decrease in ee also occurred. Hence, the nature of the substituents was also a significant factor. Higher ees were obtained when a halogen atom was present at this position, suggesting the possibility of a halogen bonding interaction between the substrate and the catalyst, which helps to improve the ees.
A dissymmetric phosphamide was also used in an attempt to obtain a product with two different types of stereogenic elements, and indeed, the desired product 26 could be obtained in 49% yield (dr = 3.5:1, 90% ee) through a kinetic resolution process. This seems to be the first example of the synthesis of compounds containing both a P-stereogenic center and C-N axial chirality.
Racemization experiments with 25a showed that the barrier to rotation was high. The potential of the newly synthesized compounds to act as chiral hypervalent iodine(III) catalysts was also demonstrated with the asymmetric oxidative dearomatization of phenol, and in Kita’s reaction, i.e., the asymmetric oxidative spirolactonization of phenol derivatives.
2.3. C-H Functionalization and Related Approaches
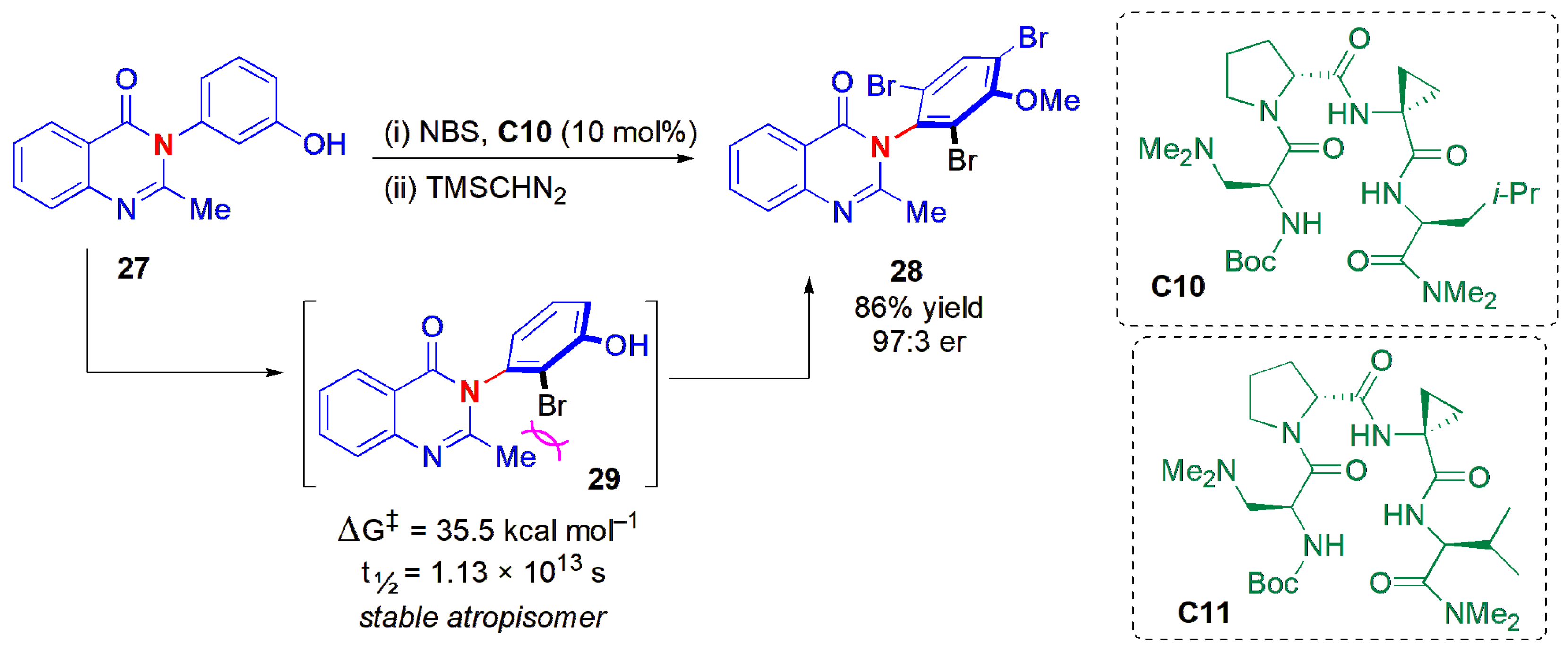
Miller and co-workers instead chose an atropselective C-H functionalization strategy to obtain axially chiral products [
44]. In the presence of a tertiary amine-containing β-turn peptide (
C10), 3-arylquinazolin-4(3
H)-ones (quinazolinones)
27, which are of pharmaceutical interest, could be brominated with high levels of stereoinduction to yield compounds
28 (
Figure 10). Screening experiments with a range of peptides suggested that the peptide β-turn secondary structure is important to achieving high ees in this reaction. The slow addition of NBS also helped to raise the ees. A broad range of substrates
27 with different substituents on the quinazolinone aromatic ring, at C-2 and on the
N-Ar ring, were compatible with the reaction conditions, with the products
28 being obtained in high yields and very high ees. These products were isolated after phenol methylation with (trimethylsilyl)diazomethane, and acetic acid addition as the reaction’s quench material. Mechanistic studies suggest that the initial bromination is achieved in the stereodetermining step, with the major monobromide intermediate, an
ortho-substituted isomer, being atropisomerically stable. Evidence for this came from an experiment with peptide
C11 (Boc-Dmaa-
d-Pro-Acpc-Val-NMe
2) as a catalyst, which provides the opposite sense of induction to
C10 when the reaction is explored at the early stages (low conversion). The major product obtained was the
ortho-monobrominated product, with a configuration opposite to that of
28, which means that the stereodetermining bromination event is the first bromination to give
29. The barrier to rotation about the chiral axis in
29 was calculated to be 35.5 kcal mol
−1 using DFT calculations, corresponding to a half-life of 1.13 × 10
13 s. Further transformations of products
28 or
29, e.g., by a dehalogenation Suzuki–Miyaura cross-coupling sequence to
ortho-arylated derivatives, and a regioselective Buchwald–Hartwig amination procedure to afford a
para-amine substituted quinazolinone, proceeded with retention of configuration.
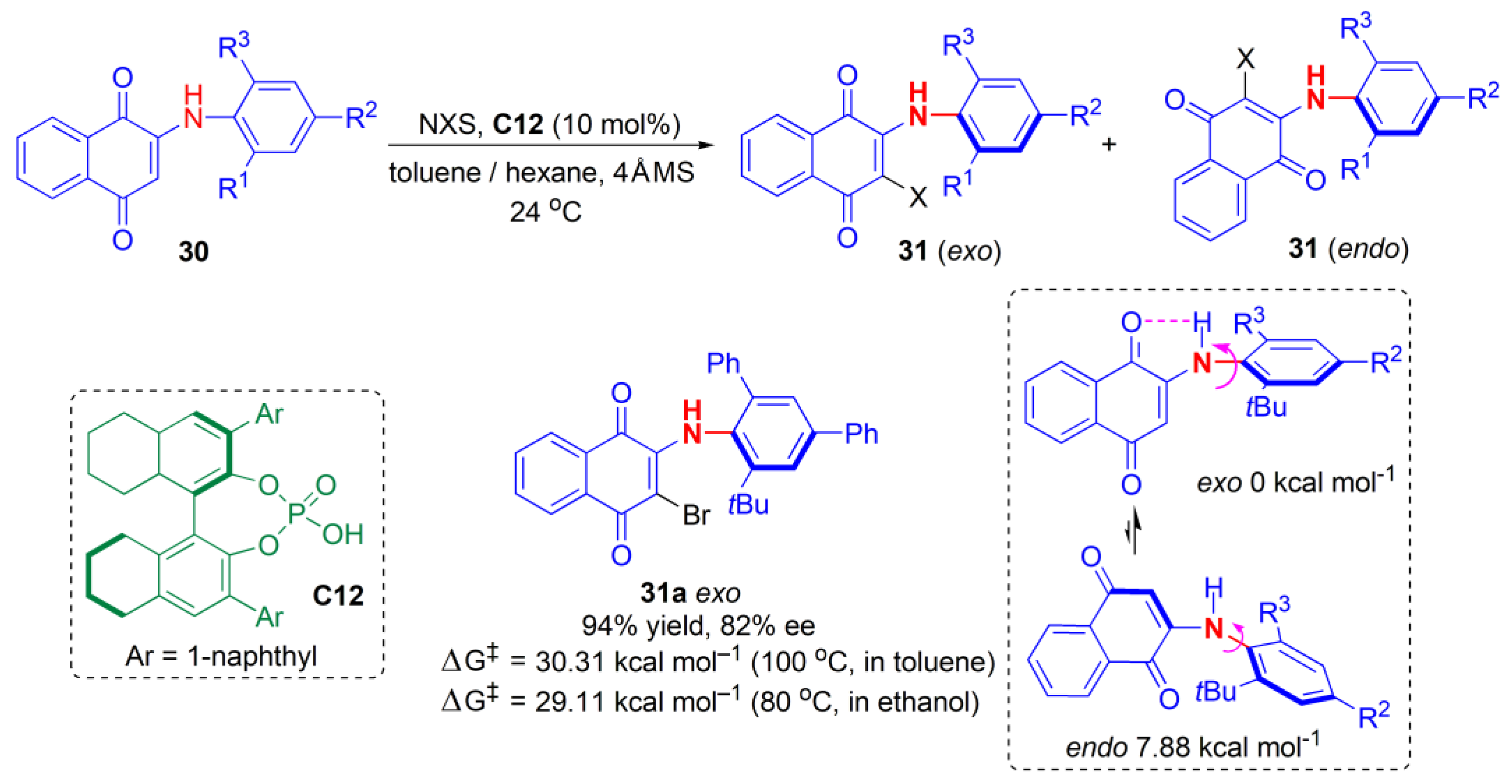
Stereochemically stable diarylamines and related substances are few in number, because their axes typically possess lower stereochemical stabilities. In 2020, Gustafson and co-workers described the CPA-catalyzed atroposelective electrophilic halogenation of
N-aryl quinoids
30, which afforded diarylamine-like scaffolds
31 in an atropselective manner for the first time (
Figure 11) [
45]. The resulting
N-aryl quinoids possess a five-membered intramolecular N-H-O hydrogen bond that stabilizes them, with barriers to racemization approaching and exceeding 30 kcal mol
−1 (
t1/2 (37 °C) > 4.5 years) in both protic and aprotic solvents, e.g., as
31a. They are thereforer considered sufficiently stereochemically stable for drug development [
45]. High yields and ees were obtained for a variety of substrates. However, the aryl groups played an important role; if they were replaced by Me, a large drop (by more than 60%) in ee took place. When the aryl groups contained
ortho-fluorine substituents, ees greater than 90% were observed. Fused ring systems, e.g., naphthyl, benzofuran, and benzothiophene, reacted to provide products in very high yields and ees up to 90%.
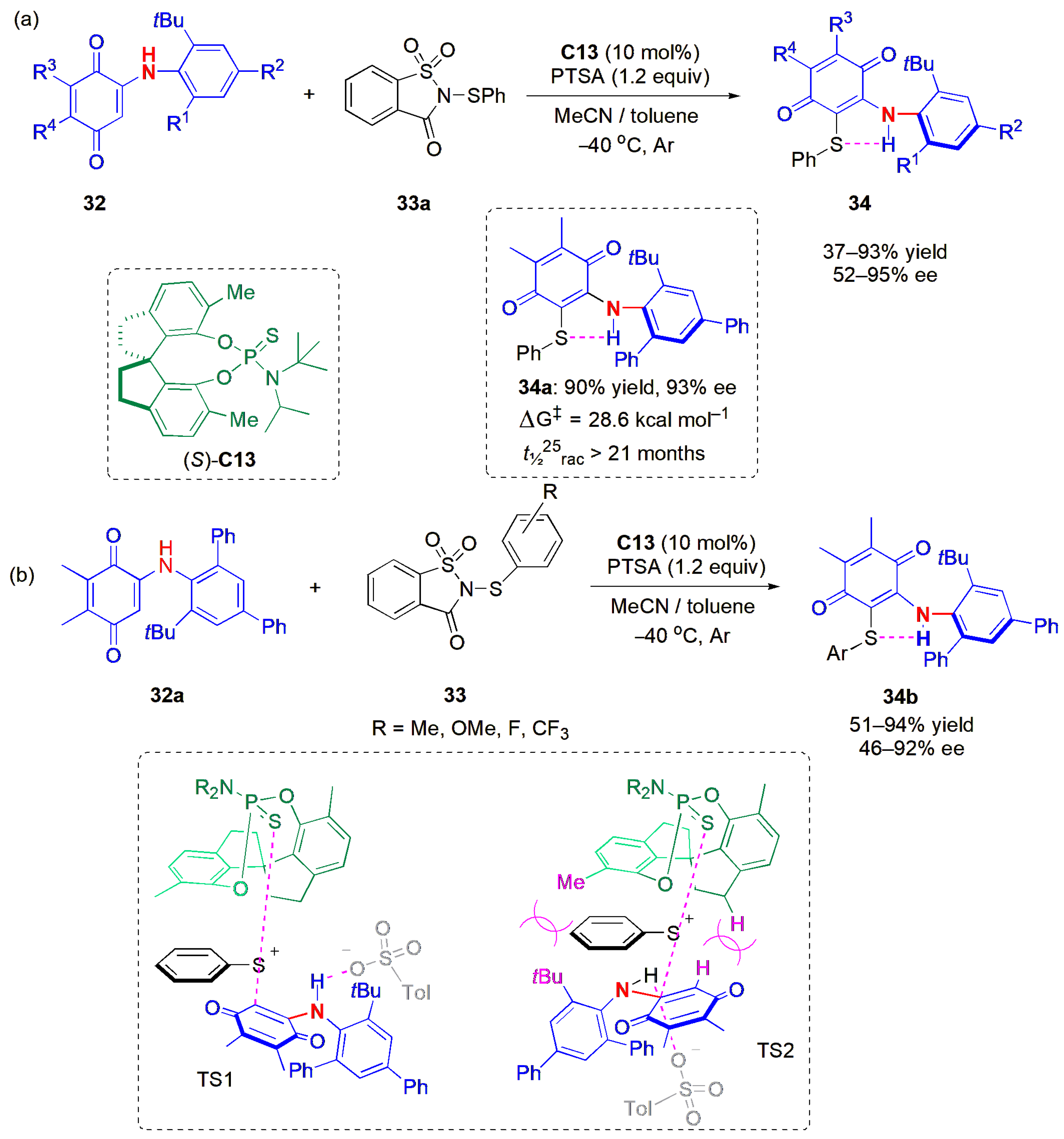
Modifications of non-aromatic rings incorporating the C-N axis of symmetry have also been used to restrict rotation and allow the formation of distinct rotamers. An atroposelective electrophilic sulfenylation of
N-aryl aminoquinone derivatives
32 was achieved for the first time in 2022 [
46]. This development brings us one step closer to control of axial chirality in diarylamines, which are useful structures in drug discovery; although they may possess two contiguous atropisomeric C-N axes, they are less stable, and their rotational barriers are not sufficiently high to prevent racemization. In order to achieve atropselectivity in the related
N-aryl aminoquinone derivatives, Xue, Chen, and co-workers introduced an hydrogen acceptor sulfide group in the quinone ring, aiming to obtain an intramolecular N-H-S bond capable of locking one of the C- N axes into a planar conformation, in a manner similar to the example above, e.g.,
34a in
Figure 12a. Sulfenylating reagent
33a in combination with the new CPA (
S)-
C13 afforded products
34 in very high yields and ees, irrespective of the nature of the substituents on the aryl ring. The quinone substituents could also be varied, but lower yields and ees were obtained in these cases. When benzofuran groups were present as substituents at the phenylamine moiety, a very low ee of 16% was also obtained.
Different sulfenylating agents
33 were tried, within which the aryl group was varied and the products, e.g.,
34b, were obtained with moderate to good yields and moderate to high ees (
Figure 12b). Steric hindrance caused by this group, as well as its nature, affected enantioselectivity. Electron-poor substituents in the aromatic ring improved the reactivity and hence the yields, as well as the ees in relation to aryl groups bearing electron-donating groups, which not only afforded products with lower ees, but also meant that the reactions had to be performed at higher temperatures. The reaction was also tried on a
N-methyl protected substrate, but no product was obtained in this case.
The energy barrier to racemization of
34a was found to be approximately 28.6 kcal mol
−1 (
t1/225 °C
rac > 21 months) in toluene; thus,
34a is considered to be stereochemically stable [
4]. DFT calculations were also performed, and revealed the origin of the atropselectivity. It was found that the key factor is the presence of strong steric repulsions between the catalyst‘s methylene unit on the spirocyclic skeleton and the methyl and
tert-butyl substituents on the aromatic ring and the substrate‘s quinone group, respectively, which mean that TS1 is favored over TS2. This is in agreement with the fact that when the N atom is methylated, there is no reaction.
2.4. Desymmetrization
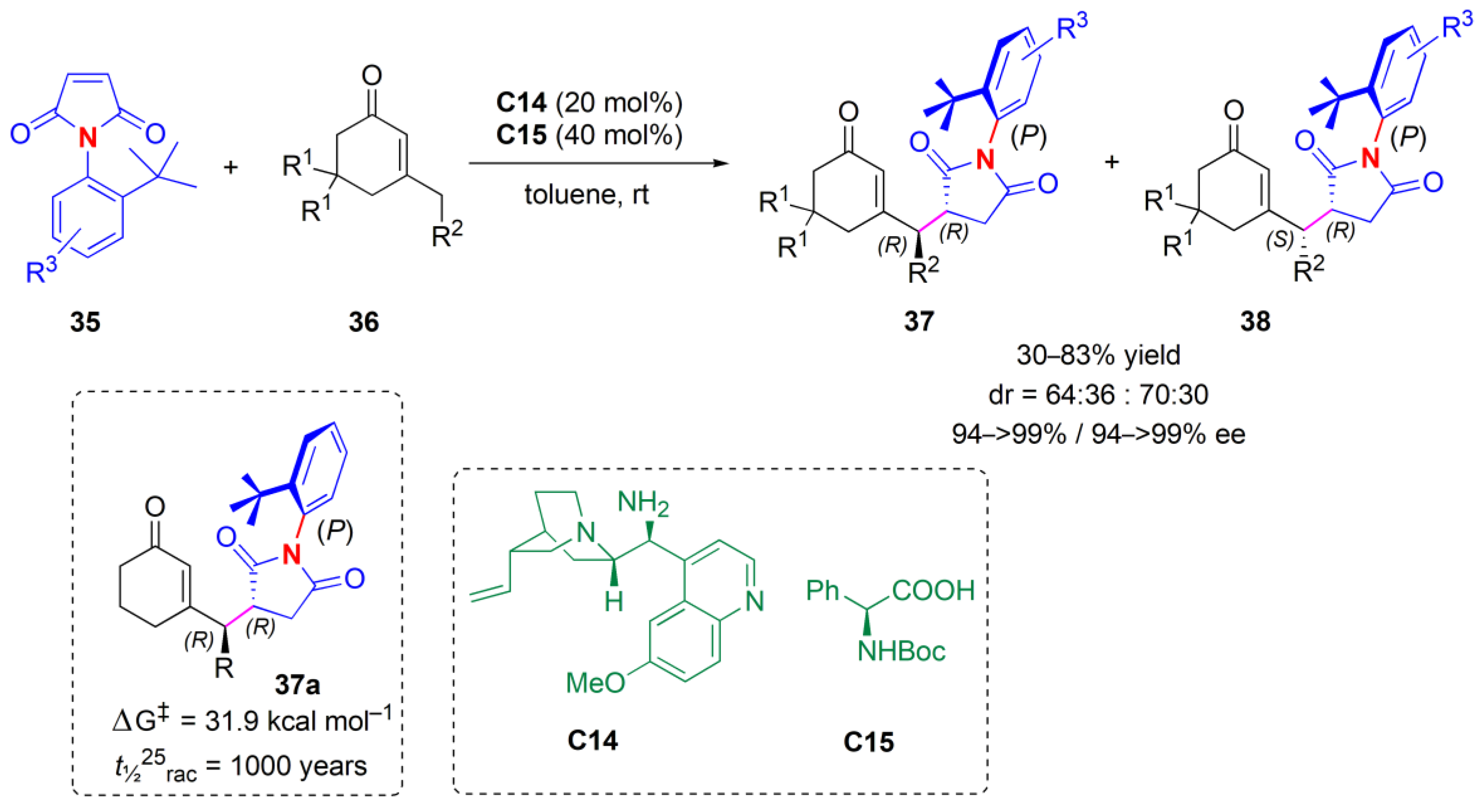
Enantioselective desymmetrization reactions provide another approach for the introduction of axial chirality.
N-(2-
t-butylphenyl)succinimides
35 were subjected to a desymmetrization reaction, through a vinylogous Michael addition of 3-substituted cyclohexenones
36 by Bencivenni and co-workers in 2014 (
Figure 13) [
47]. In the presence of cinchona alkaloid
C14, the reactions proceeded in an enantioselective fashion, with remote control of the axial chirality, to afford atropisomeric succinimides
37 and
38 with two adjacent stereocenters. The utilization of
N-Boc-
l-phenylglycine
C15 as a co-catalyst helped to raise the ee.
The ees were very high, although the diastereoselectivity was moderate. A P,R,R and a P,R,S absolute configuration were assigned to the major and minor diastereosiomers obtained, based on X-ray diffraction and EDC spectra experiments. The scope of the reaction was examined with different substituents on the N-aryl ring, and it was found that when halogen, phenyl or methoxy substituents were present, and with an amino group, the yield and ees were high. On the contrary, the presence of a second t-butyl group at position 5 of the maleimide aromatic ring resulted in only traces of products, and with ortho- iodo-, triethylsilyl- or phenyl groups, the products have chiral axes that quickly epimerize at 25 °C. Enones with various substituents were also tried; the most effective ones were methyl and isopropyl.
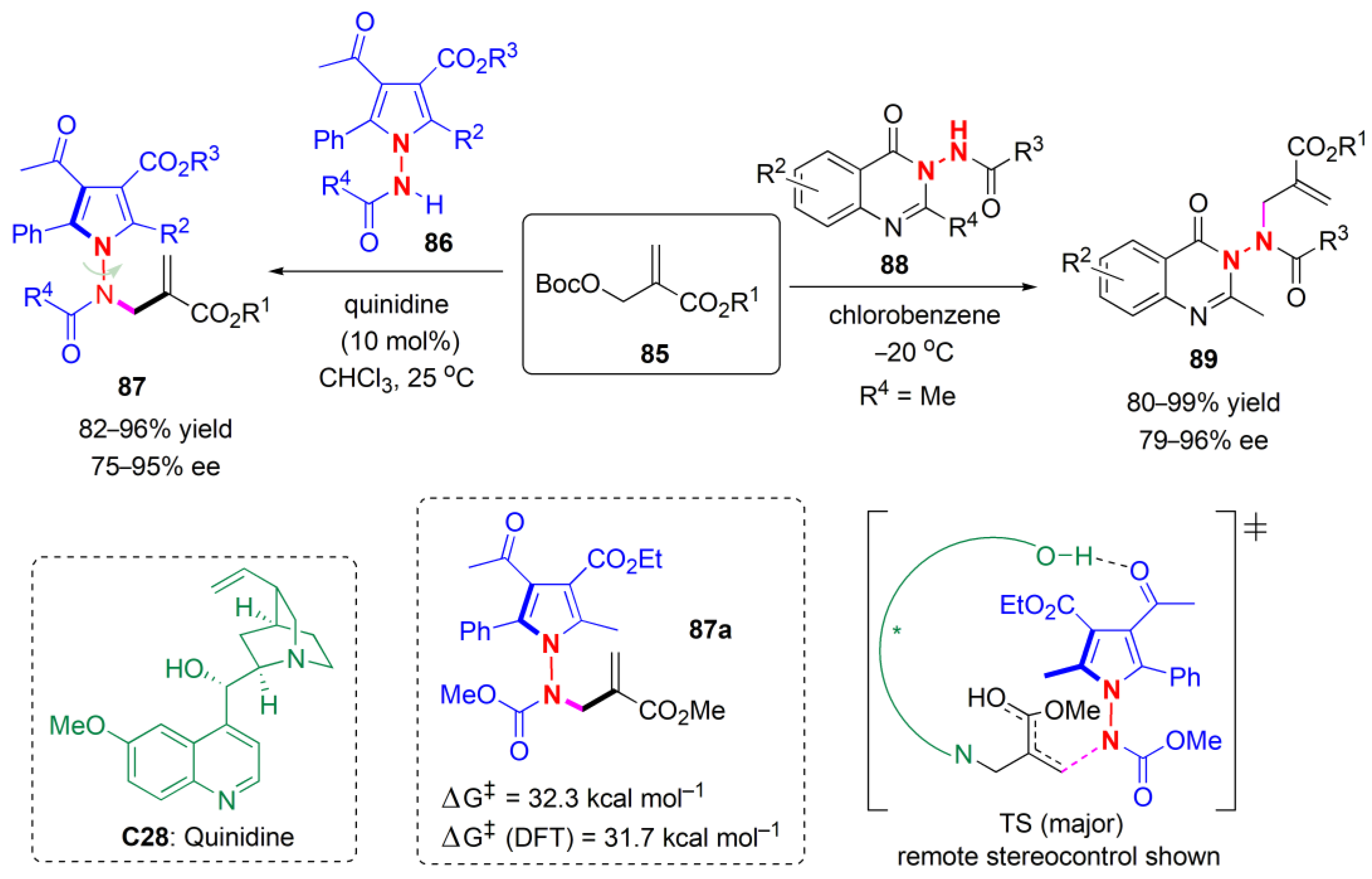
The thermal stability with respect to the epimerization of the chiral axis was also probed. When 37a was heated to 130 °C in C2D2Cl4, 10 h later, an equilibrium ratio of 62:38 and a new diastereoisomer were observed. The fact that this new compound was indeed a rotamer and not another diastereoisomer was confirmed using NMR analysis and an NOE experiment. The energy barrier to rotation was found to be ΔGepi‡ = 31.9 kcal mol−1, corresponding to a t1/225 = 1000 years.
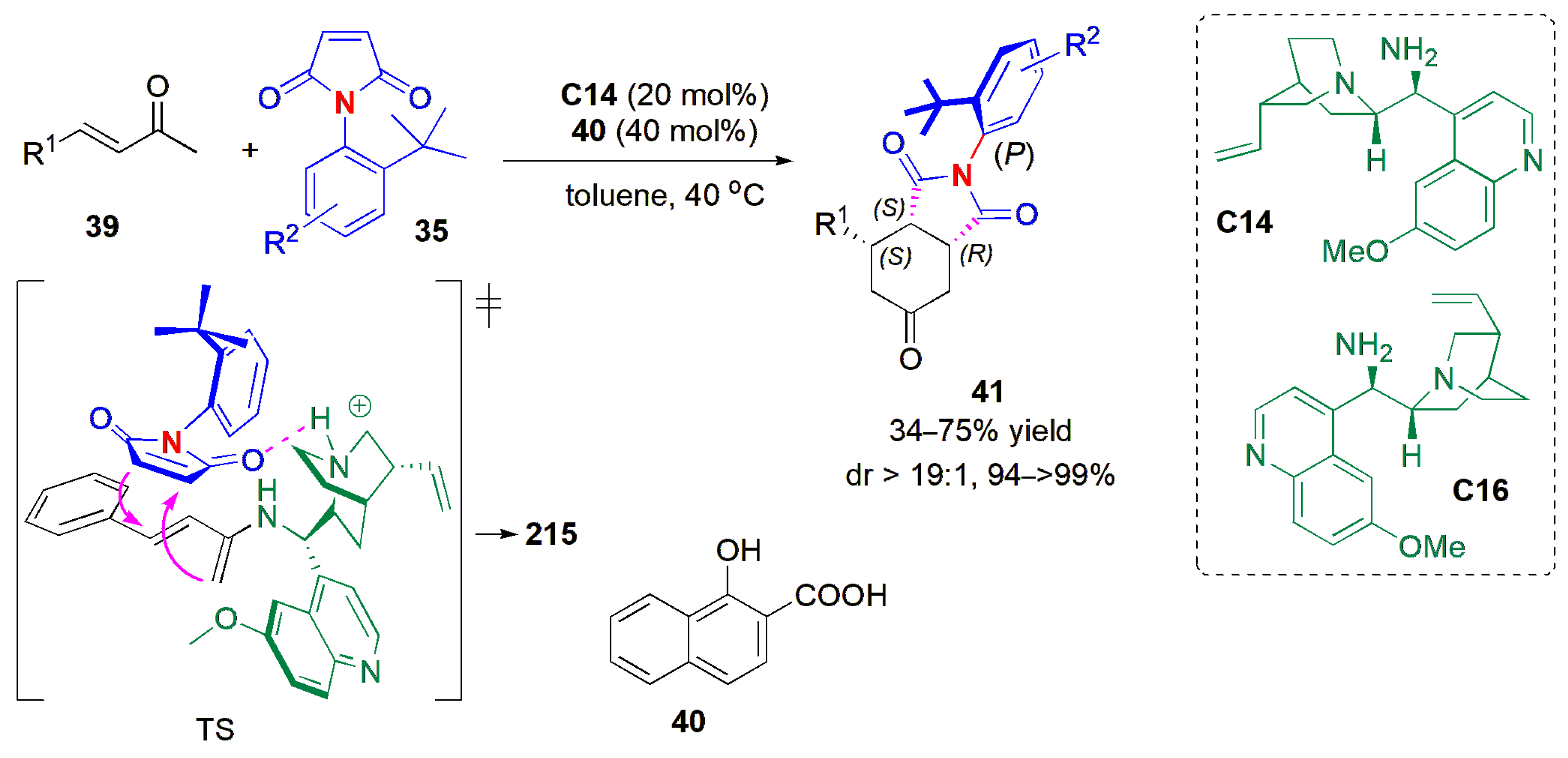
The Bencivenni group developed soon after a formal Diels–Alder reaction of enones
39 catalyzed by a primary amine
C14 to achieve an atroposelective desymmetrization of
N-arylmaleimides
35 (
Figure 14) [
48]. 1-Hydroxy-2-naphthoic acid (
40) worked well as an additive. The product succinimides
41 were obtained as single diastereoisomers (dr > 19:1 in all cases) in good yield and excellent ees with either cinchona alkaloid derivative
C14 or
C16,
C16 giving the opposite enantiomers of
C14. Electron-withdrawing and electron-donating substituents could be used, but the reactivity was suppressed completely by an o-bromo substituent in the aryl group of the enone, and also when an alkyl chain and an ester substituent were used. Similarly, when 5-
tBu and 5-NO
2 substituents were employed in the
N-Ar group, no product was obtained. The chiral axis is generated under catalyst remote control, The stereochemical outcome depends on the way that the α,β-unsaturated enamine, produced by the reaction between the catalyst and ketone, and the maleimide, approach one another. The unsaturated enamine approaches from the maleimide side that is not shielded by the
tert-butyl group in the TS. Both central and axial chirality are created in this example.
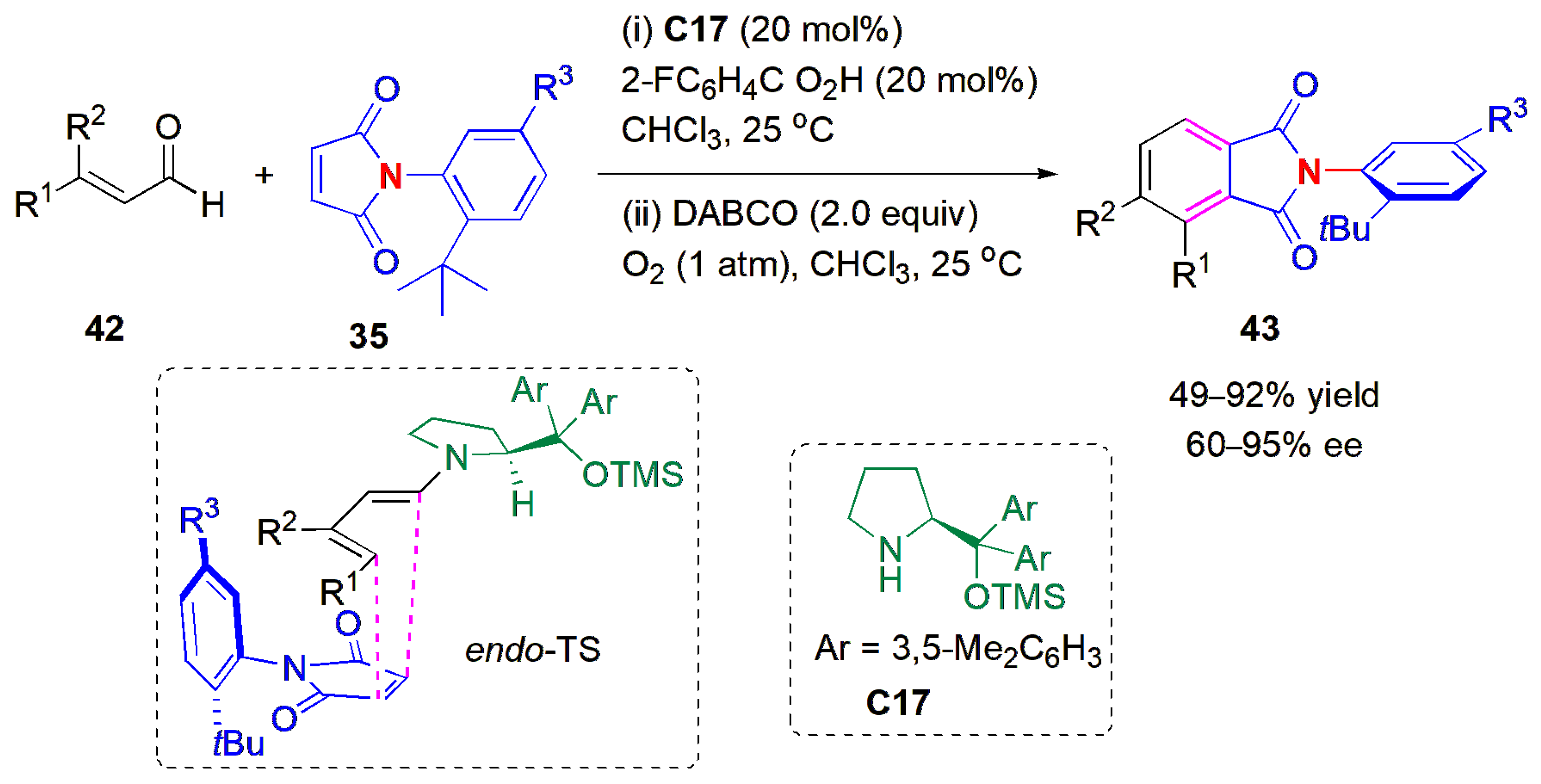
When instead of α,β-unsaturated ketones, the corresponding enals
42 are used to react with maleimides
35 in the presence of a secondary amine,
C17, N-substituted phthalimides
43 are produced in an atropselective desymmetrization reaction (
Figure 15). This de novo arene construction, proceeding through an oxidative [4 + 2] cycloaddition, was described by Mondal and Mukherjee in 2022 [
49]. The dienamine catalysis obtained allows the formation of products in very high ees, using remote stereocontrol via the
endo TS. Only axial chirality is produced in this example. The highest yields were obtained when R
3 = NO
2 and R
2 was an electron-donating group, and the lowest ee was achieved with a 2-Me substituent, when R
3 = NO
2. When R
3 = H, only an 11% yield was obtained, even after 11 days. Thus, the nitro group has a large effect on the reactivity.
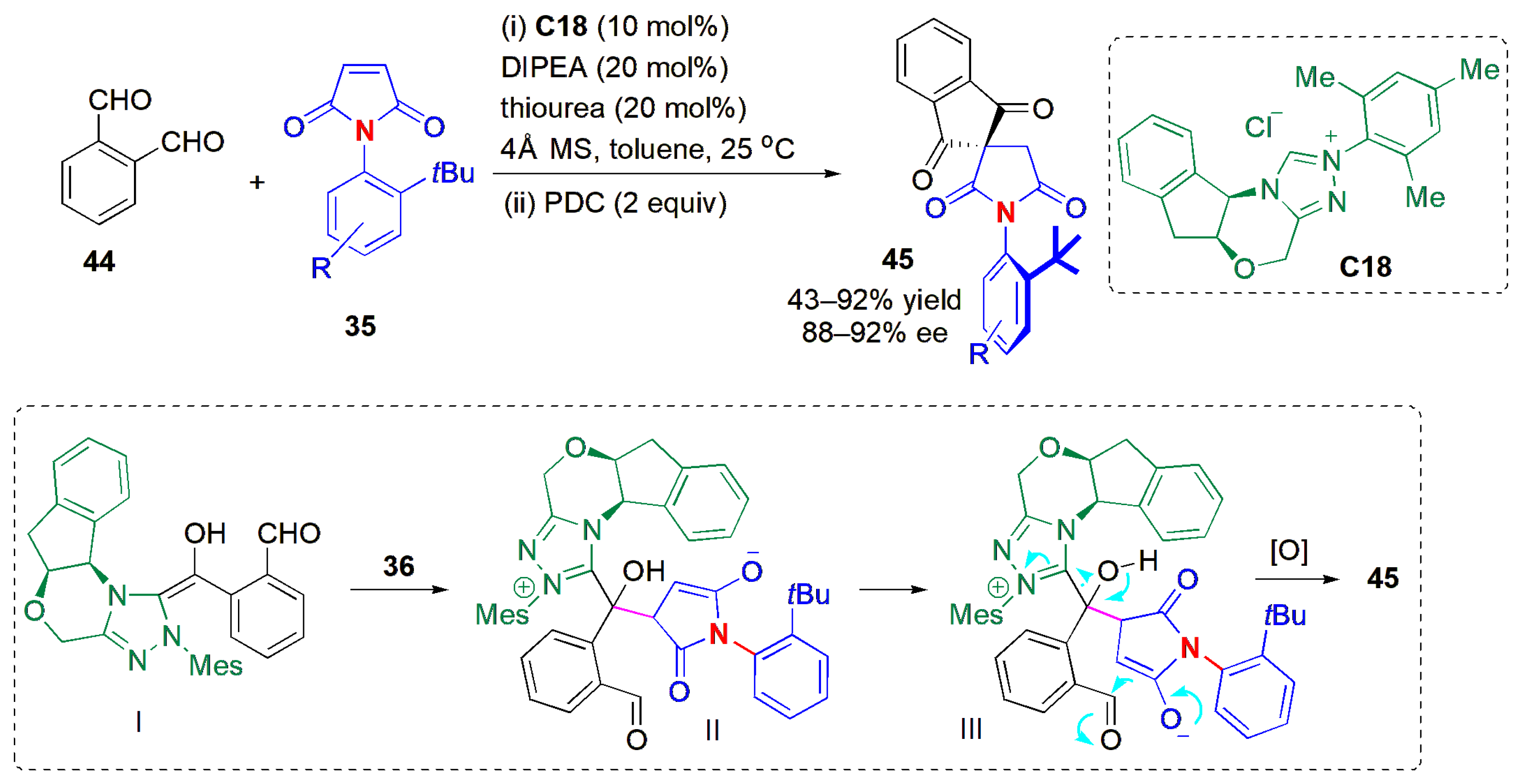
Another example of the use of a desymmetrization strategy to obtain axial chirality was described by Jindal, Mukherjee, Biju and co-workers in 2021 [
50]. In this case,
N-aryl maleimides
35 were subjected to an intermolecular Stetter reaction/oxidation sequence (
Figure 16). Although no reaction could be observed with 4-chlorobenzaldehyde, phthalaldehyde
44 afforded good yields of products
45 with high ees when reacted in the presence of the chiral N-heterocyclic carbene (NHC)
C18 and Hünig’s base, followed by in situ oxidation of the resulting product with pyridinium dichromate (PDC). Overall, the products are formed via a tandem intermolecular Stetter reaction followed by an intramolecular aldol reaction, with subsequent oxidation. Product stability was inspected by heating
45a (R = H) in toluene. Up to 90 °C, the ee was preserved, but at 110 °C, the ee dropped to 84%. With a further increase in temperature, the ee dropped even more, being almost racemic at 150 °C, suggesting that there was unrestricted rotation of the C-N bond at this temperature. The ΔG
rot‡ for the C-N bond in
45a was calculated to be 32.4 kcal mol
−1, using DFT studies. In this study, the
ortho-tert-butyl group present in the
N-aryl substituents played an important role in restricting the rotation about the C-N axis. When replaced with a dimethyl phenyl group, the ee of the reaction product was still high (43% yield, 86% ee); however, if this group was replaced with an isopropyl group, the ee dropped to 60%.
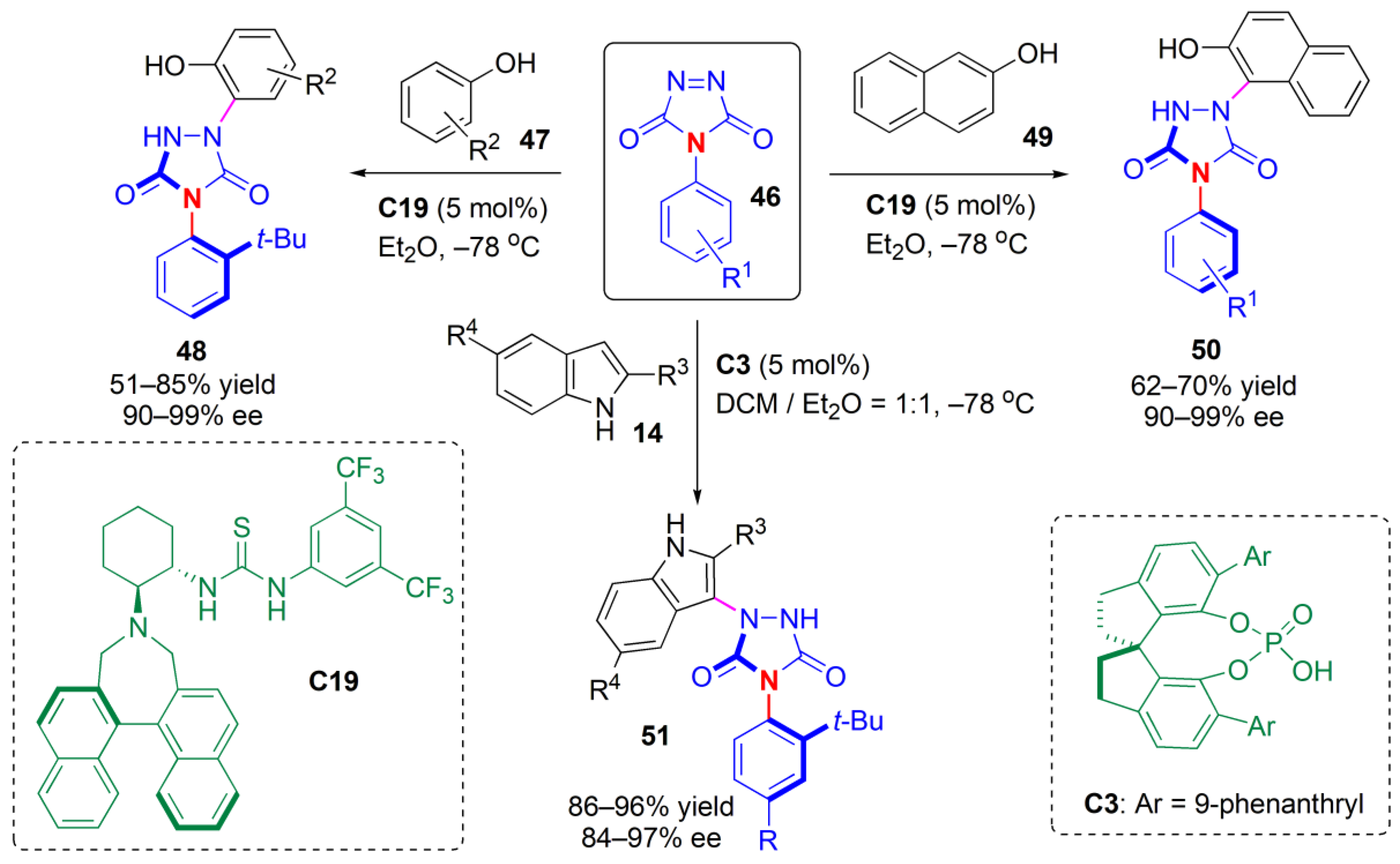
An organocatalytic tyrosine click-like reaction allowed the synthesis of axially chiral urazoles [
51]. Zhang et al. found that by using a bifunctional thiourea-based catalyst (
C19), efficient discrimination of the two reactive sites in the triazoledione ring of substrates
46 could be achieved upon reaction with phenols
47 or naphthols
48, and the stereochemical information of the catalyst could be transferred into axial chirality at a remote position, far from the reactive site, in the urazoles (
Figure 17). The reactions proceeded readily at a low temperature (−78 °C), and high yields and ees of products
48 or
50, respectively, were obtained for a variety of substrates bearing electron-withdrawing or electron-donating substituents at different positions in the aromatic rings. To probe the configurational stability of the products, a solution of product
45a (R
1 = 2-
tBu) in toluene or MeCN was heated at 80 °C for 12 h. Chiral HPLC analysis showed that there was no difference in the ee values of
13a, confirming its stability. Similar reactions were performed with indoles
14 as desymmetrization reagents, but a different catalyst was required. In this case, chiral phosphoric acid (CPA)
C3 was an efficient catalyst, and the corresponding products
51 were obtained with very high yields and ees. Gram-scale reactions could be performed successfully, with no loss of enantioselectivity.
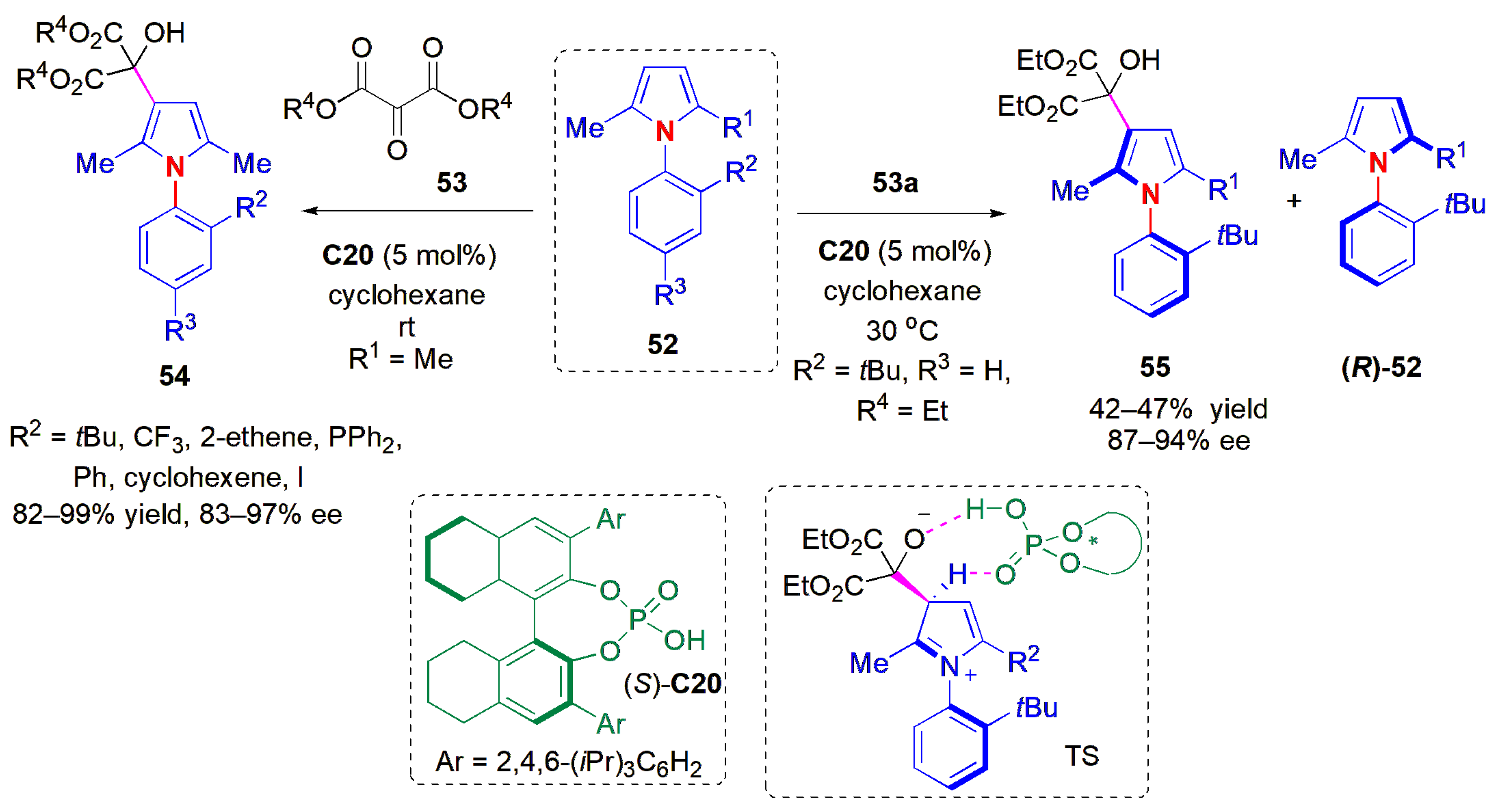
Inspired by the studies in the desymmetrization of maleimides, Tan and co-workers studied a way to desymmetrize arylpyrroles
52 and reported the results in 2019 [
52]. Axially chiral arylpyrroles are core structures of a wide range of natural products and pharmaceutical agents [
53,
54]. Chiral arylpyrroles are also present in chiral ligands; some are chiral catalysts and even chiral resolving agents. Until this report, chiral resolution or chiral chromatography was used to obtain axially chiral arylpyrroles in an optically pure state. In this work, enantioenriched axially chiral arylpyrroles were obtained either by means of organocatalytic atroposelective desymmetrization, or by kinetic resolution in the case of nonsymmetrical pyrroles (
Figure 18). Depending on the remote control of chiral catalyst, the arylpyrroles were obtained in high yields and excellent ees under mild reaction conditions. As an electrophilic reagent, diethyl ketomalonate
53a was selected, and CPA (
S)-
C20 was found to be the best catalyst. In this case, not only did the
tert-butyl group serve as an efficient
ortho substituent on the
N-aryl ring to hinder rotation, but high ees were also obtained with a range of other functional groups (
Figure 18). The nature of a 4-substituent did not matter either. Similar results were also obtained with other malonates, e.g.,
iPr,
tBu). More critical to the atropselectivity (and also to the yield) was the nature of the substituents at the 2-position in the pyrrole ring. When a change was introduced in the pyrrole ring via replacement of the substituent at C-2 with another different from methyl, the system was no longer symmetric. The products could be obtained using the same catalyst with high selectivity, irrespective of the nature of the substituent (electron-withdrawing or electron-donating), via kinetic resolution, with a good to high selectivity factor (
S = 32–69). The absolute configuration of one product,
55a (R
1 = Me, R
2 =
tBu, R
3 = H, R
4 =
iPr) was determined using X-ray crystallographic analysis to be (
aS), and those of the other products were assigned by analogy.
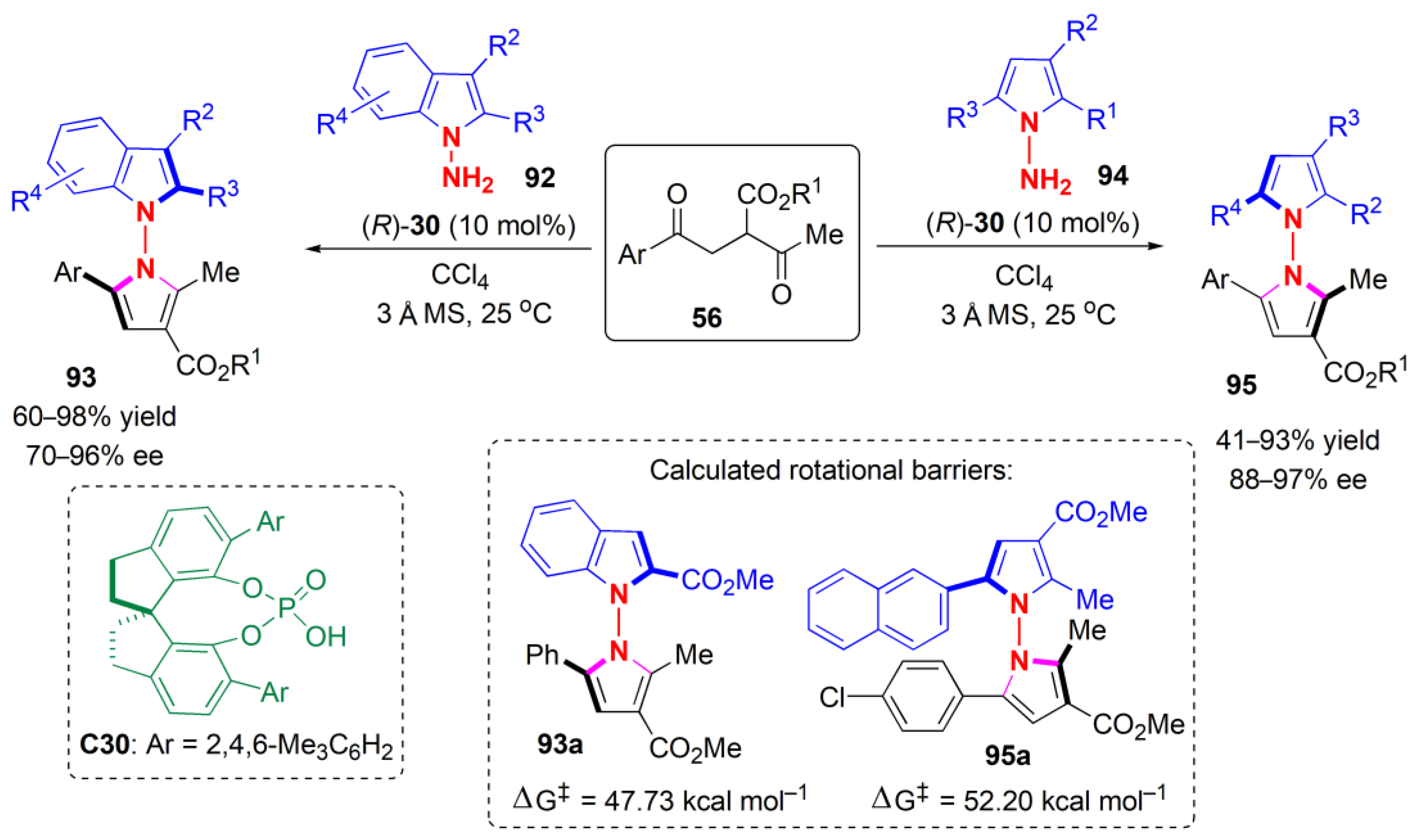
The configurational stability of the products was investigated by heating 55b in different solvents (iPrOH, DCE, and toluene) at up to 150 °C for 24 h. Deteriorations of stereochemical integrity were negligible, even when the substrate began to decompose. It was assumed that the key interactions controlling the atropselectivity were hydrogen bonding between ketomalonate and the CPA to form a chiral pocket for the induction of chirality, and H-bonding with the second carbonyl group of the ketomalonate so as to fix the whole system in a rigid configuration, as in TS1. The diphenylphosphine derivatives were applied as ligands to palladium in enantioselective allylic substitution reactions, thereby affording products with high ees.
2.5. Annulation
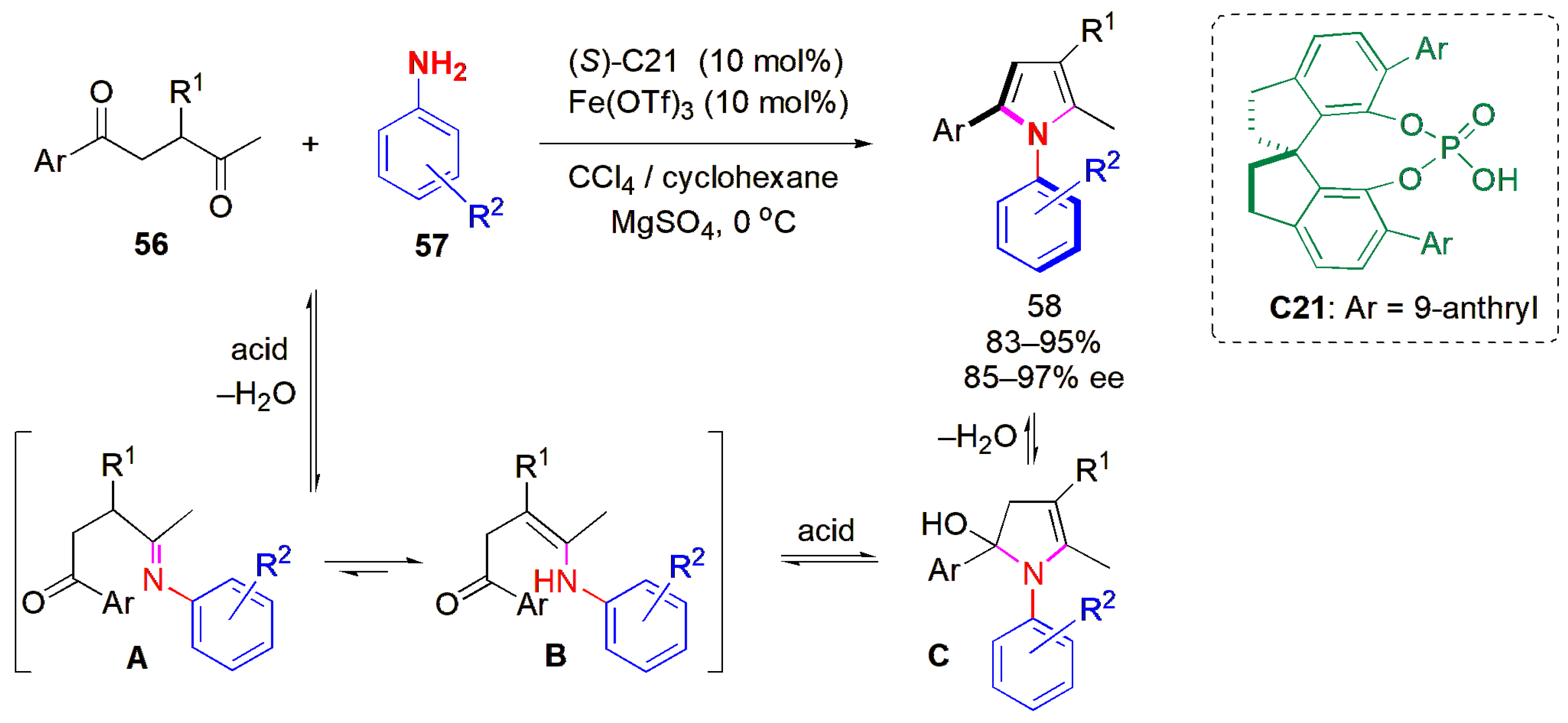
The last general approach for the construction of C-N axially chiral molecules is atropselective annulation, i.e., de novo ring construction, in which at least one ring is built up. In this context, enantiomerically pure aryl pyrroles were synthesized for the first time, by Tan and co-workers, using an atropselective method based on a catalytic asymmetric Paal–Knorr reaction, in 2017 [
55]. A functionalized 1,4-diketone
56 was reacted with an aryl amine
57 in the presence a combined-acid catalytic system consisting of a Lewis acid, ferric triflate, and chiral phosphoric acid
C21 (
Figure 19). The products were obtained in high yields and ees. The presence of electron-withdrawing groups on the aromatic substituent of the diketone favored yields and ees. The
ortho group on the
N-aryl ring was not only restricted to the
tert-butyl group; the bromo, iodo, and phenyl groups at this position were well tolerated too, and the products could be obtained with high ees.
Control experiments to trap the intermediate allowed the isolation of species B, obtained from condensation product intermediate A, which confirmed that the reaction proceeds via an enamine intermediate. This is followed by acid-catalyzed dehydrative cyclization, which is a deviation from the usual Paal–Knorr reaction mechanism.
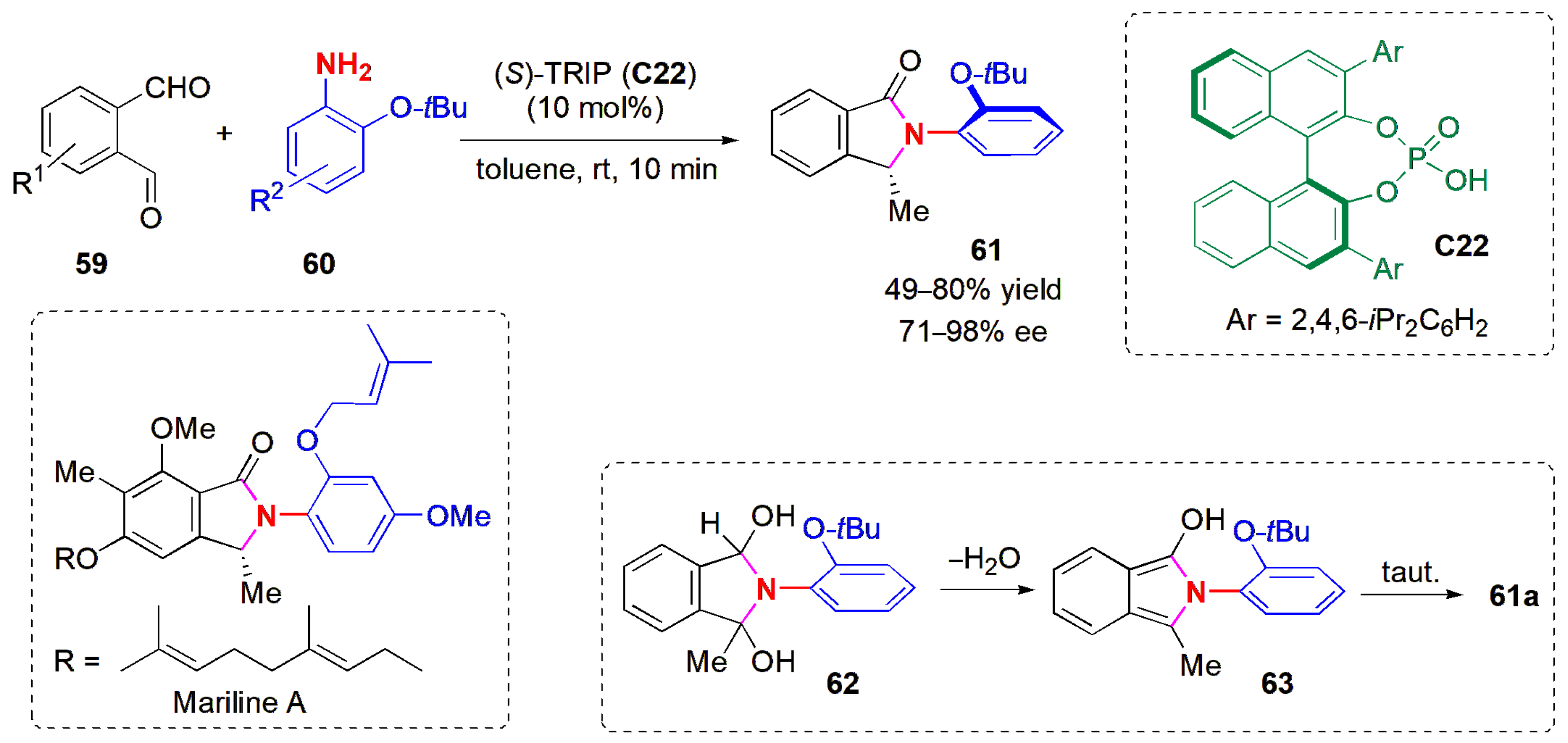
In the same year, Seidel and co-workers developed a catalytic enantioselective synthesis of isoindolinones
61 through the condensation of 2-acylbenzaldehydes
59 and anilines
60 (
Figure 20) [
56]. The reaction was catalyzed by a very low loading of CPA
C22 [(
S)-TRIP] (1 mol%), and it was complete within 10 min. Products
61 with several substituent patterns were obtained in up to 98% ee. Anilines bearing an
ortho-t-butyl group formed atropisomeric products through the simultaneous generation of axial and point chirality from two achiral substrates. The method was applied to the first catalytic enantioselective synthesis of the natural product mariline A. The highest yields were obtained with electron-rich benzaldehydes.
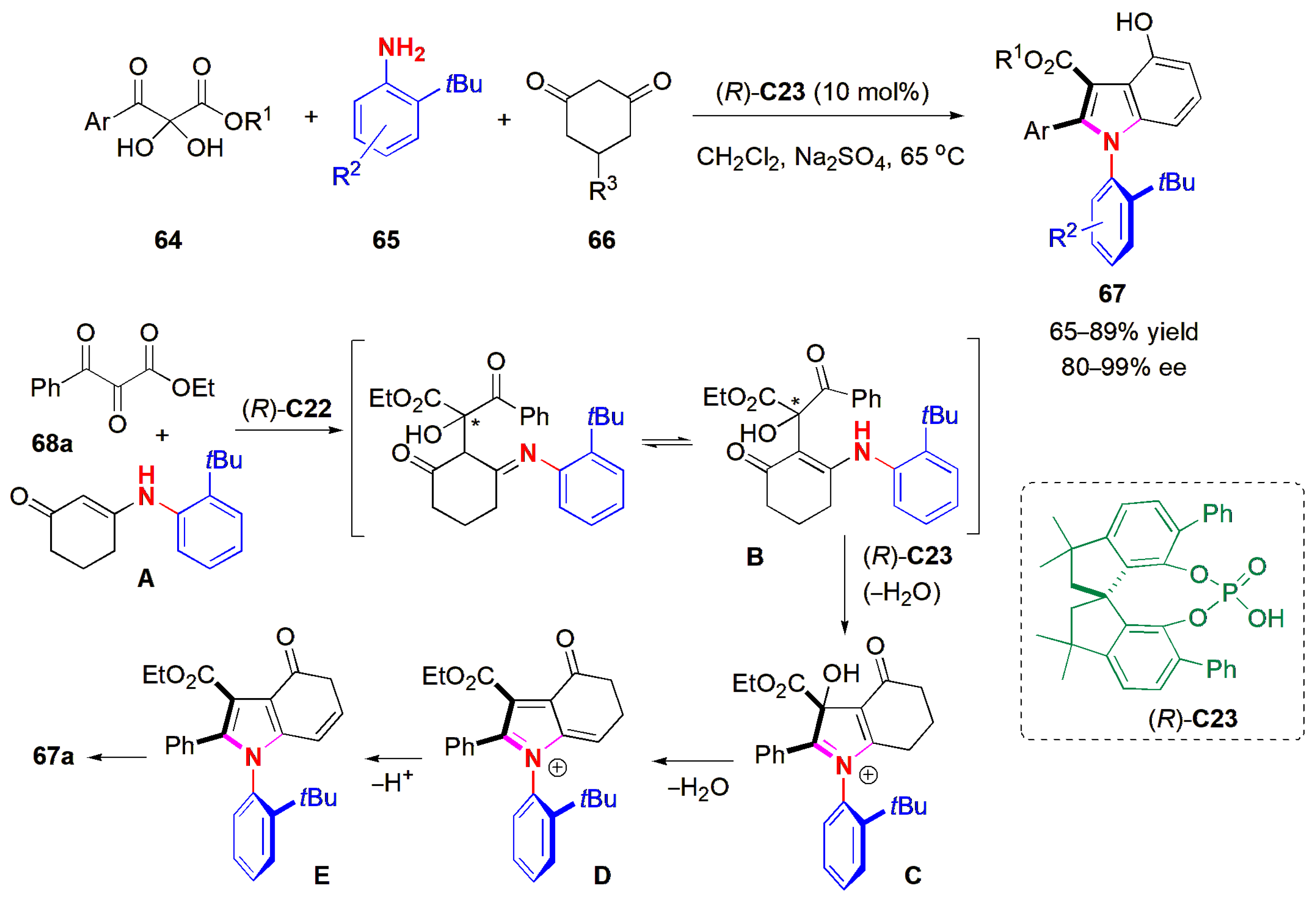
An organocatalytic atroposelective phosphoric acid-catalyzed three-component cascade reaction was utilized by Lin and co-workers to obtain axially chiral
N-arylindoles [
57]. 2,3-Diketoesters
64, aromatic amines
65, and 1,3-cyclohexanediones
66 reacted to provide a wide range of products
67 in high yields and very high ees (
Figure 21). The authors proposed that the reaction proceeds via an intermediate enamine (
A), which undergoes chiral acid-catalyzed aldol condensation with the dehydrated 2,3-diketoester to produce
B; it also undergoes dehydrative cyclization to generate
C, dehydration via 1,4-elimination to produce
D, and loss of acid to yield
E, which tautomerizes to yield the final product. This multicomponent reaction was based on a previous report by the Doyle group, whose reaction was performed without a chiral catalyst and yielded racemic products [
58].
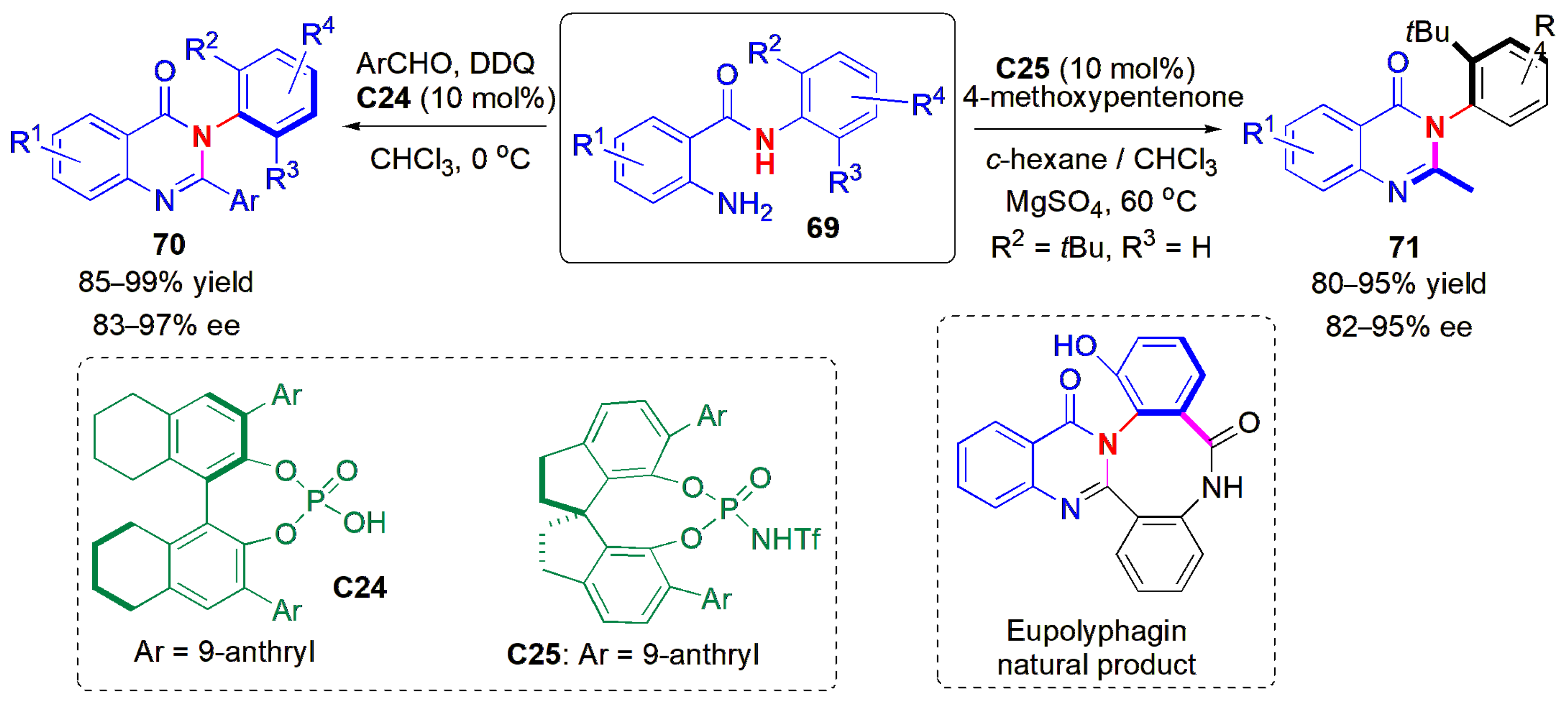
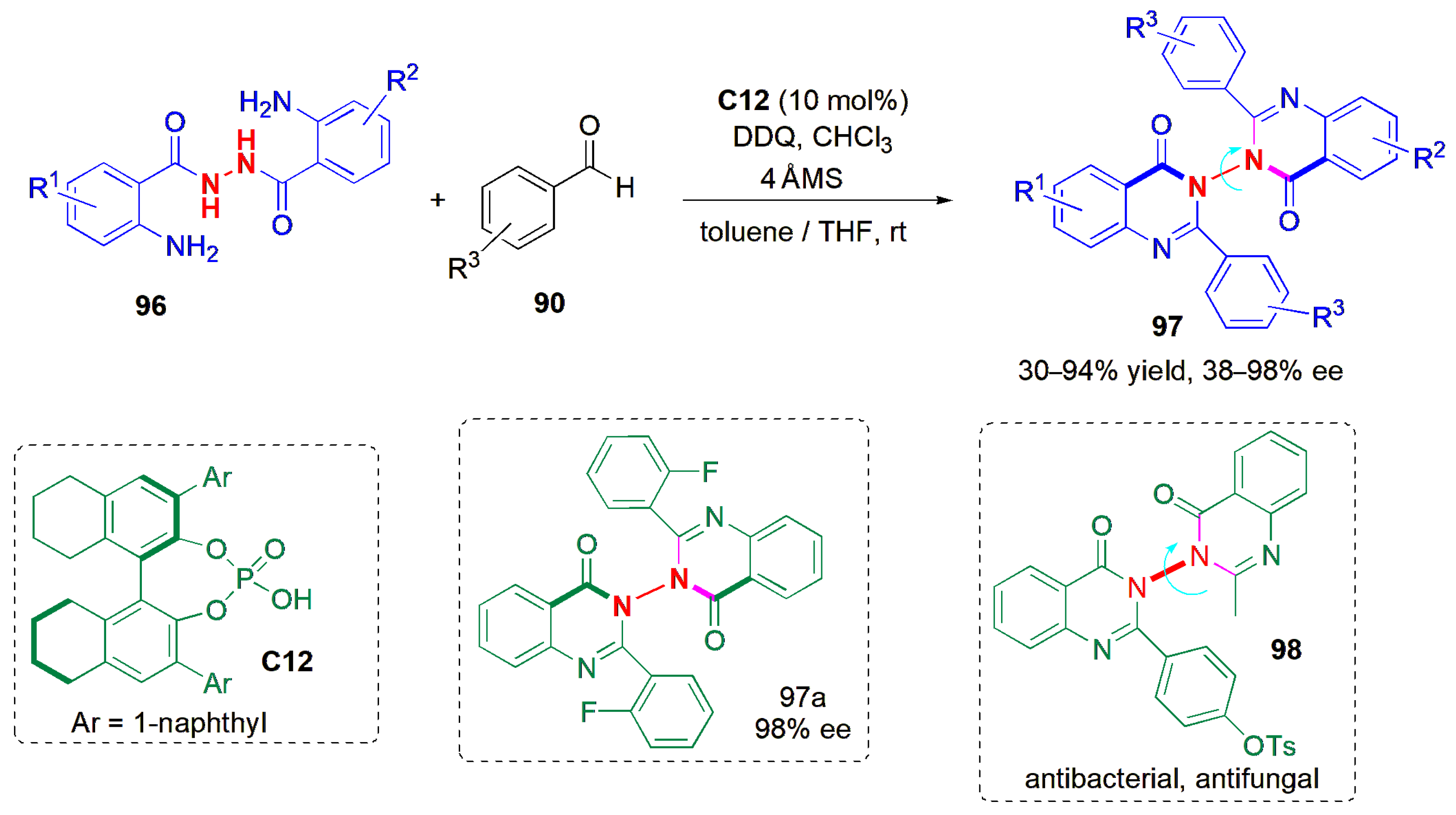
The synthesis of six-membered rings showing atropisomerism has also been described using atropselective methods involving de novo ring construction. One such method is the catalytic asymmetric construction of arylquinazolinones
70 from
N-aryl anthranilamides (
69) and benzaldehydes, as reported by Tan and co-workers in 2017 (
Figure 22) [
59]. CPA
C24 worked well in the presence of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), despite the high steric hindrance imposed by the reactant molecules. Substrates bearing both electron-withdrawing and electron-donating substituents were well tolerated. At the same time, the atroposelective synthesis of alkyl-substituted arylquinazolinones
71 was also described, and achieved via a Brønsted acid-catalyzed carbon–carbon bond cleavage strategy. In this case, the desired product could not be obtained upon reaction with ketoesters, but when 4-methoxypentenone was used as reaction partner with a more acidic catalyst
C25 in the presence of MgSO
4, several axially chiral methyl-substituted arylquinazolinones could be obtained in high yields and ees. Other diketones gave poorer results, which is a limitation of the method. It was also shown that the protocols could be applied to the asymmetric total synthesis of eupolyphagin, a natural product bearing a cyclic arylquinazolinone skeleton.
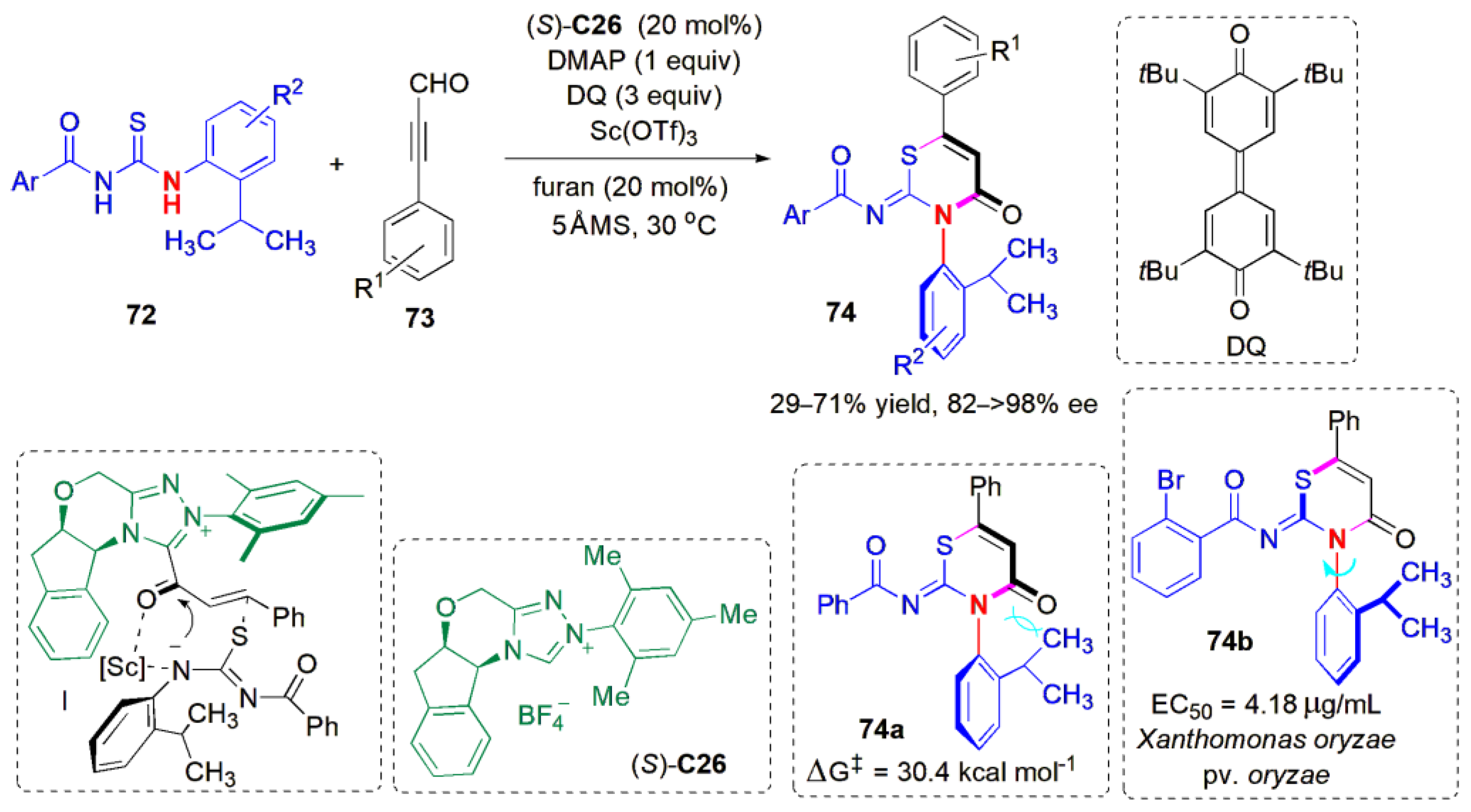
De novo ring construction was also adopted by Jin and co-workers to obtain thiazine derivatives with C-N axial chirality in 2021 [
60]. An organocatalytic atropselective cycloaddition reaction between thioureas
72 and ynals
73 was developed with an NHC [(
S)-
C26] as a catalyst. A wide range of chiral thiazine derivatives
74 could be obtained with good yields and excellent ees (
Figure 23). In this reaction, the use of an additive, scandium triflate, helped to raise the yields, which dropped considerably (to below 40%) when the benzoyl group in the thiourea was replaced by
tBuCO, or when the isopropyl
ortho-substituent in
22 was replaced by a
tert-butyl group. It was assumed that the reaction proceeded via the formation of an intermediate acetylenic acylazolium intermediate from the reaction of the catalyst with
21, which reacts to form a new form C(sp
2)-S bond, producing intermediate
I, followed by catalyst-controlled face-selective intramolecular lactam formation. The barrier to rotation determined by heating
74a (98% ee) at 100 °C for 24 h in mesitylene was 29.5 kcal mol
−1 (ΔG
‡), which agreed with the result calculated through density functional theory (DFT) (ΔG
‡ = 30.4 kcal mol
−1). The thiazine ring is an important heterocyclic motif that is present in many medicines and agricultural chemicals; it is the structural element that is usually responsible for their bioactivity, e.g., the antibiotic cephalosporins such as cefradine, omonasteine, used in the treatment of respiratory diseases [
61], and the pesticide buprofezin [
62].
The authors also probed the biological activity of the new compounds. Indeed, they found that there was antibacterial activity against
Xanthomonas oryzae pv. oryzae (Xoo), which causes leaf blight in rice plants [
63]. Among bacteria-related plant diseases, rice bacterial leaf blight is still one of the most difficult diseases to control, and may cause significant damage [
64]. In addition to resistant varieties, agrochemicals such as bismerthiazol and zinc thiazole are also used. However, they are not completely satisfactory, and the pursuit of more efficient antibiotics is still an active area of research. The axially chiral thiazines
74 have shown promise as control agents, the most effective being (
S)-
74b, with an EC
50 value of 4.18 μg/mL, which is superior to the two agrochemicals mentioned above.
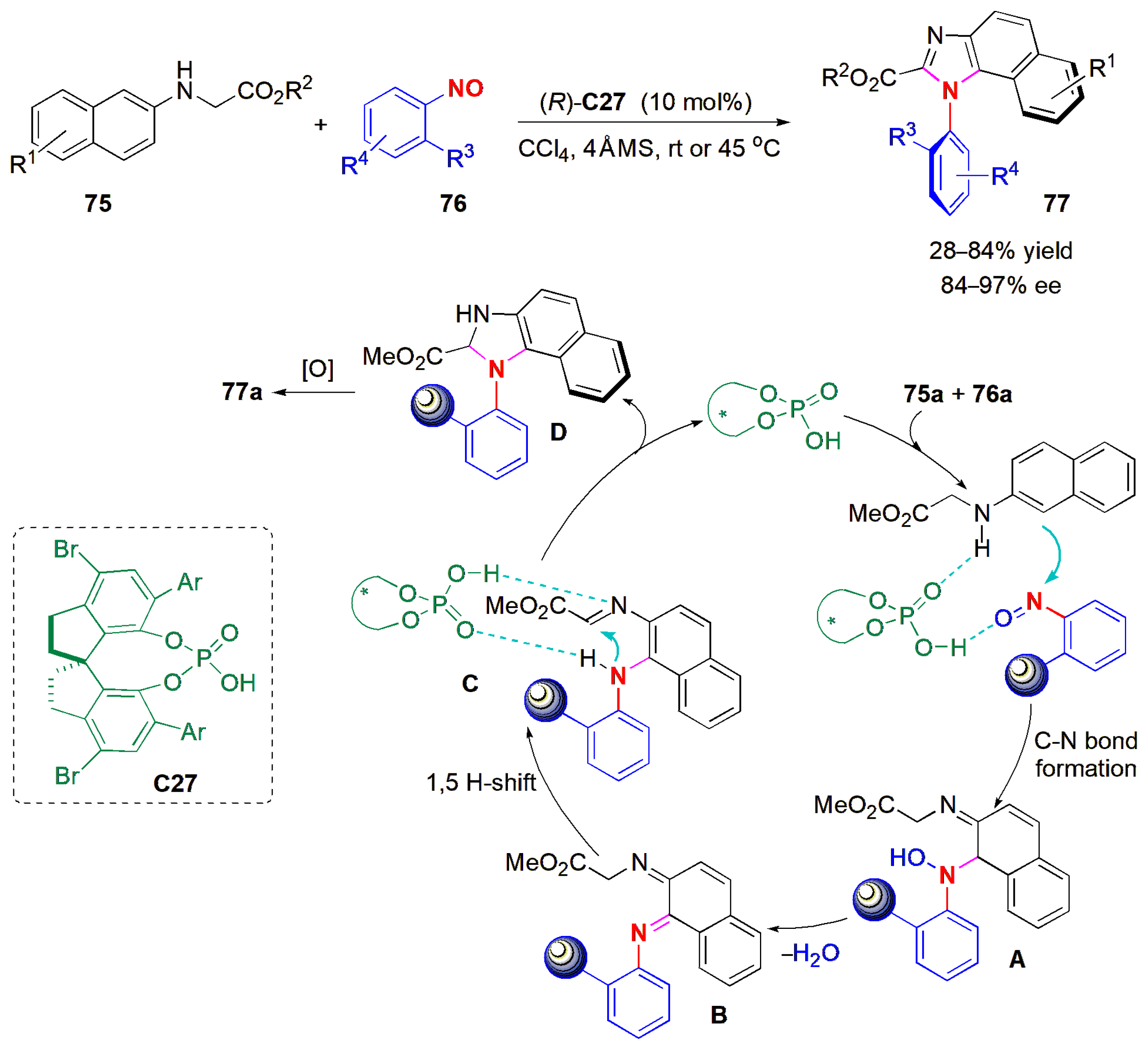
In a recent example of synthesis of enantioenriched heterobiaryl atropisomers, an imidazole ring was constructed to establish a new C-N axis of chirality (
Figure 24) [
65]. Hence, naphthylamines (glycine derivatives)
75 and nitrosobenzenes
76 were reacted by means of a domino approach to synthesize axially chiral
N-arylbenzimidazoles
77, with the catalysis provided by a chiral phosphoric acid (
C27), with excellent chemo- and regioselectivity, as well as high levels of enantiocontrol. Different alkyl and cycloalkyl substituents on the nitrobenzene ring could be used successfully, but electron-withdrawing groups at this position (Cl, CF
3) caused a considerable drop in yield; however, the ees were not affected much. Several glycine derivatives were compatible with the reaction conditions. The configurational stability of the products was studied with one example,
77a (R
1 = OMe, R
2 = Me, R
3 =
iPr, 96% ee) by heating at 120 °C for 24 h. There was no loss in ee, but there was partial decomposition of the substance during this period. A plausible reaction mechanism is shown in
Figure 24. Initially, there is the chemo- and regioselective nucleophilic addition of 2-naphthylamine
75a to
76a, enabled by dual CPA activation, to produce
A. Dehydration of
A leads to diimine
B.
B may be converted into
C via a [
1,
5] hydrogen shift or through successive reduction/oxidation steps (not shown). CPA-catalyzed intramolecular enantioselective addition of the amine to the imine provides the stereoenriched annulated intermediate
D, the stereoselectivity determination step in the domino reaction. Finally, oxidative aromatization of
D produces the desired product
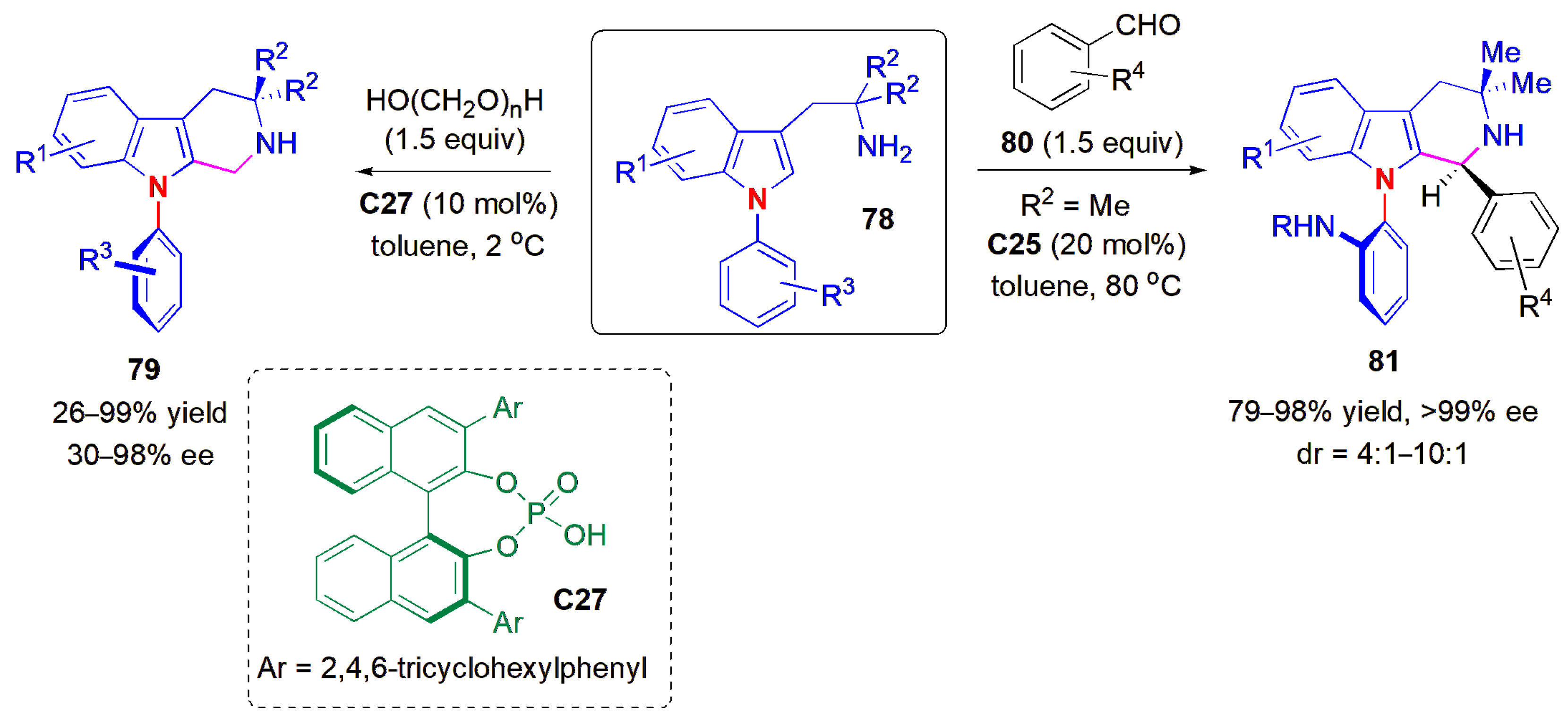
77a.An atropselective Pictet–Spengler reaction of
N-arylindoles
78 was reported in 2021 [
66]. Kwon and co-workers obtained
N-aryl-tetrahydro-β-carbolines
79 with C-N bond axial chirality via dynamic kinetic resolution using a chiral phosphoric acid (
C27) as catalyst (
Figure 25). The organocatalytic version of the Pictet–Spengler reaction, an acid-catalyzed intramolecular Friedel–Crafts-type reaction involving iminium ions, was developed by List and co-workers in 2006 [
67]. It is often applied to the synthesis of complex natural products, including β-carboline alkaloids [
68], and although several asymmetric versions have been reported since 2006, the report by Kwon and co-workers was the first example of an atropselective synthesis of this class of compounds [
64]. A wide range of substituents were compatible with the reaction conditions. The highest ees were obtained when benzaldehydes
80 were used as reaction partners instead of paraformaldehyde (all >99% ee). In these cases, both axial and point stereogenicity could be controlled. The ees were quite susceptible to the nature and number of the substituents on the
N-aryl ring, with NHBn showing the highest ees, and NBn
2 the lowest (56% ee). The authors concluded that in the first case, the hydrogen bond donor substituent forms a secondary interaction with the phosphoryl oxygen of the catalyst, which helps to increase the stereoselectivity. The presence of a
meta-substituent on this ring caused a further drop in ee. It was proposed that the reaction proceeded via the formation of an imine, itself resulting from the condensation of paraformaldehyde with the substrate, and dynamic kinetic resolution. The absolute configuration was determined via X-ray crystallography of the 4-bromobenzamide derivative of
81a (R
1 = H, R
2 = Me, R
3 = NHBn).
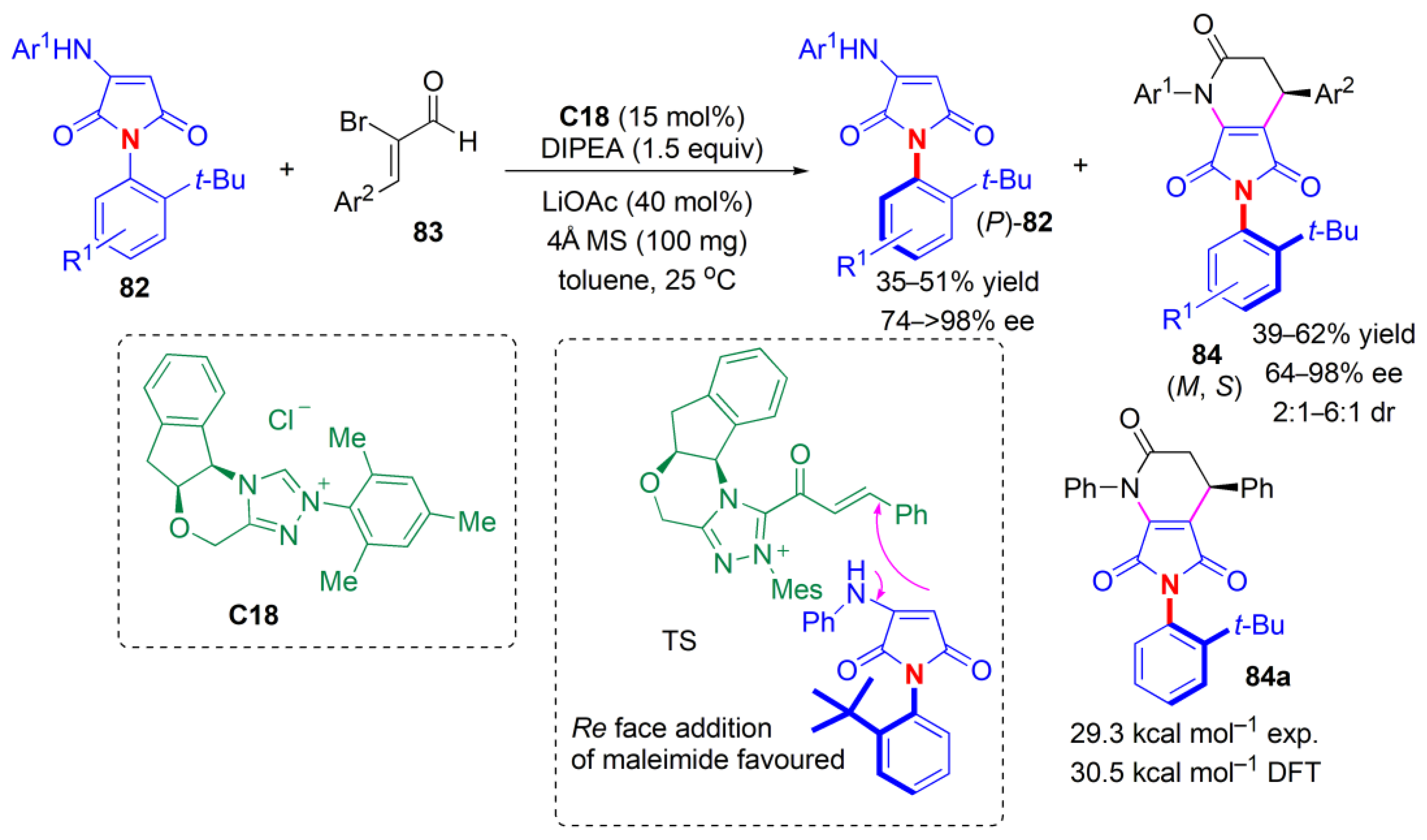
In 2022, Biju and co-workers described a procedure to obtain C–N axially chiral
N-arylaminomaleimides via kinetic resolution [
69]. Hence, in the reaction between maleimides
82 and 2-bromoenals
83, performed in the presence of chiral NHC
C18, [3 + 3] annulation with one of the enantiomers of maleimide takes place, and fused-dihydropyridinones (bearing axial/central chirality) up to 6:1 dr, >98% ee are obtained (
Figure 26). The opposite enantiomer stays unreacted and is recovered with up to >98% ee. Remote chirality control is observed. The sense of induction is determined by a
Re face addition of maleimide to the
α,
β-unsaturated acylazolium.
Si face addition (addition from the top) is hindered by both the
t-Bu group and the aminoindanol moiety of the carbine. There was also remote chirality induction governed by the bulky
t-Bu group of the maleimide substrate and the aminoindanol moiety of the NHC catalyst.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





























