
Advanced Materials Based on Azulenyl-Phenyloxazolone

 , , ,
, , ,
Abstract
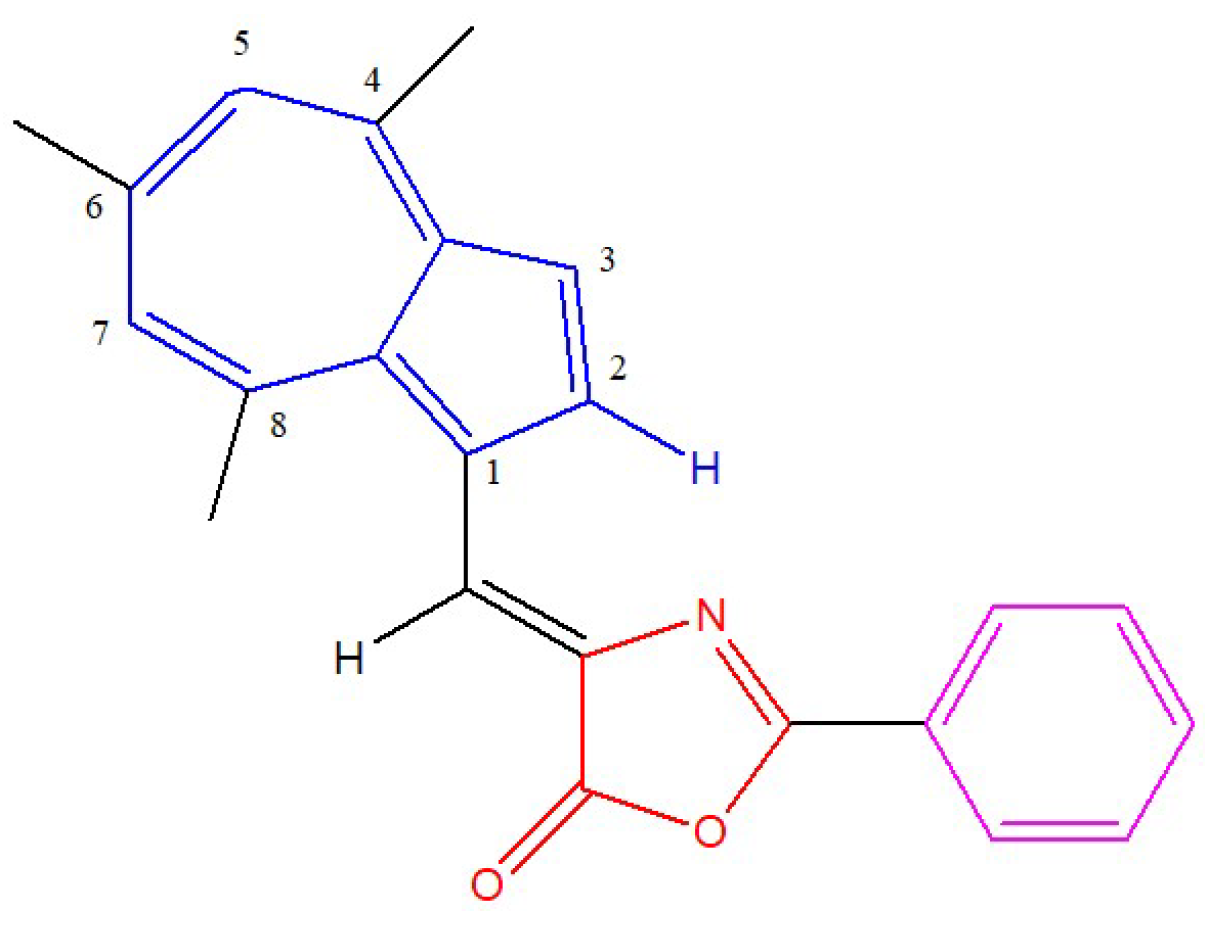
:1. Introduction
2. Materials and Methods
3. Results
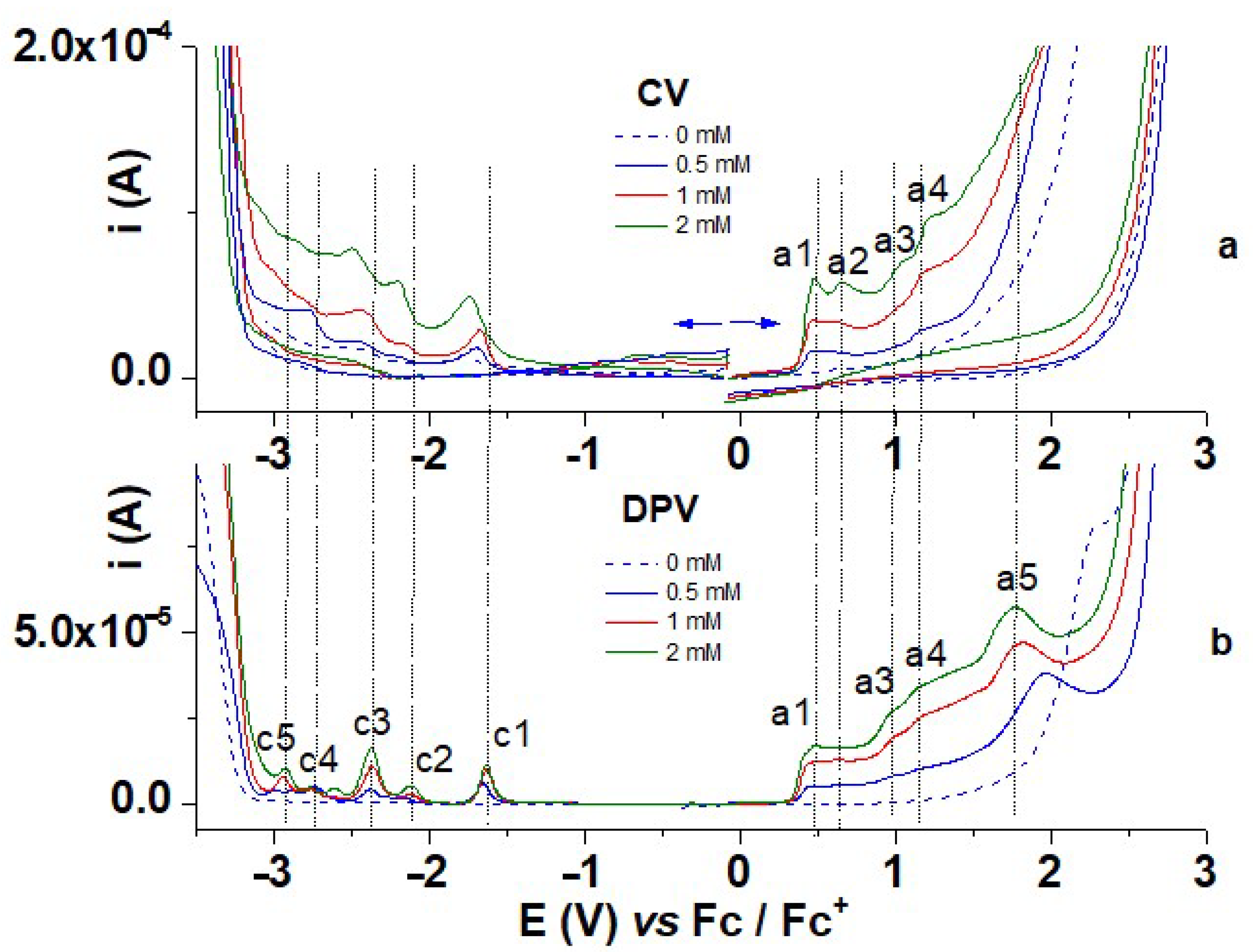
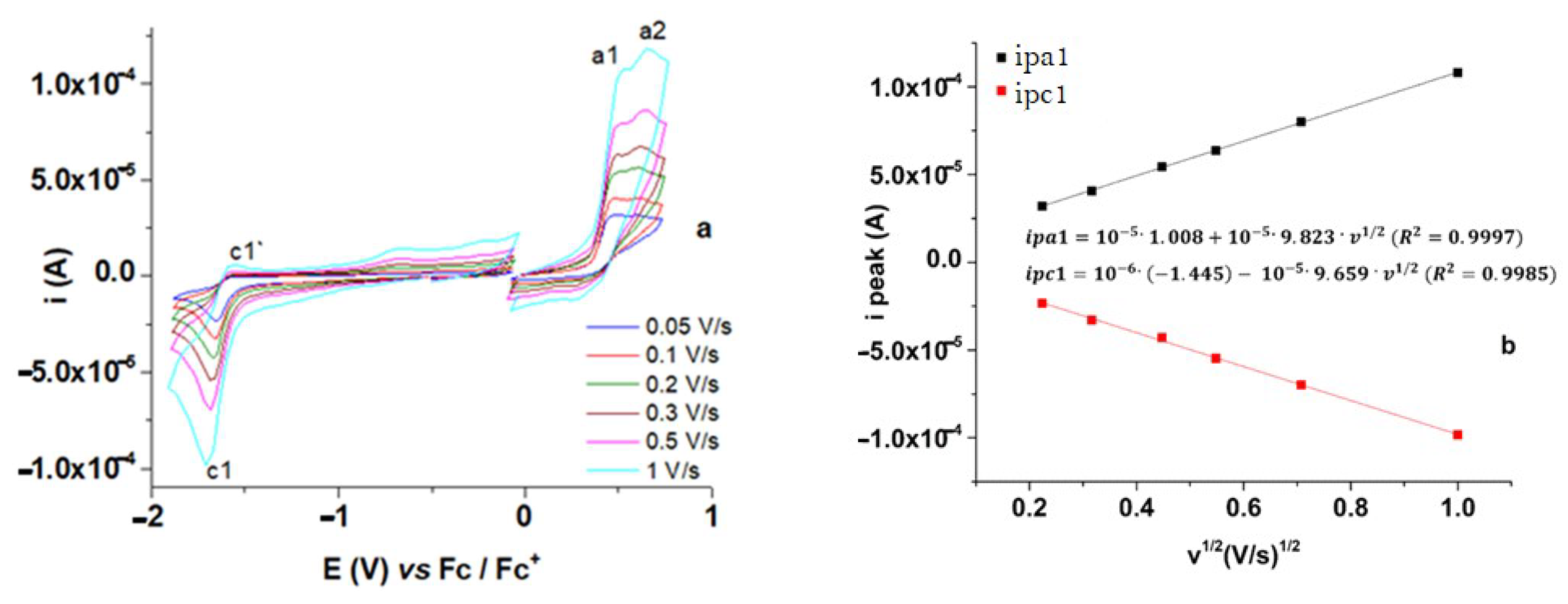
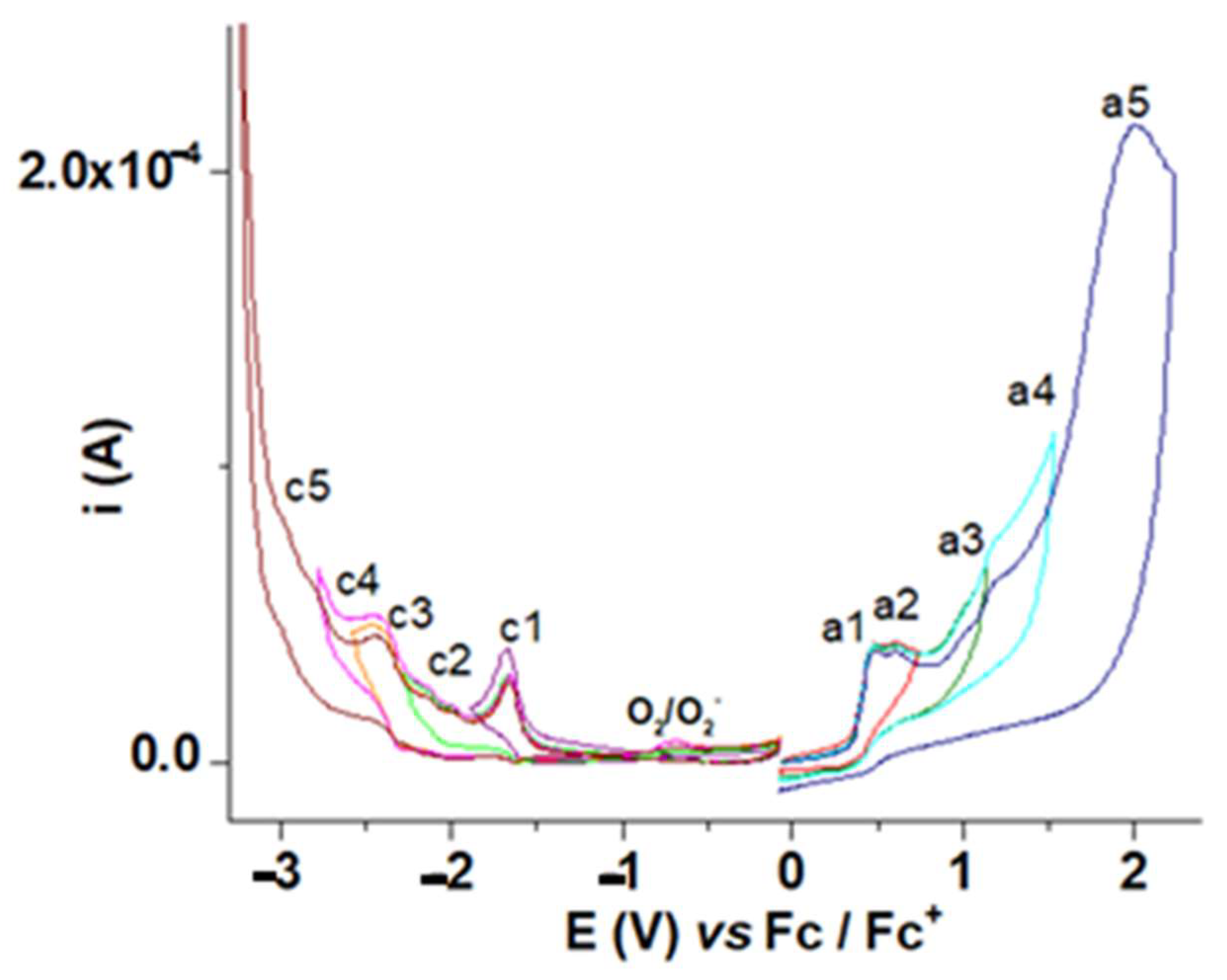
3.1. Electrochemical Characterization of M
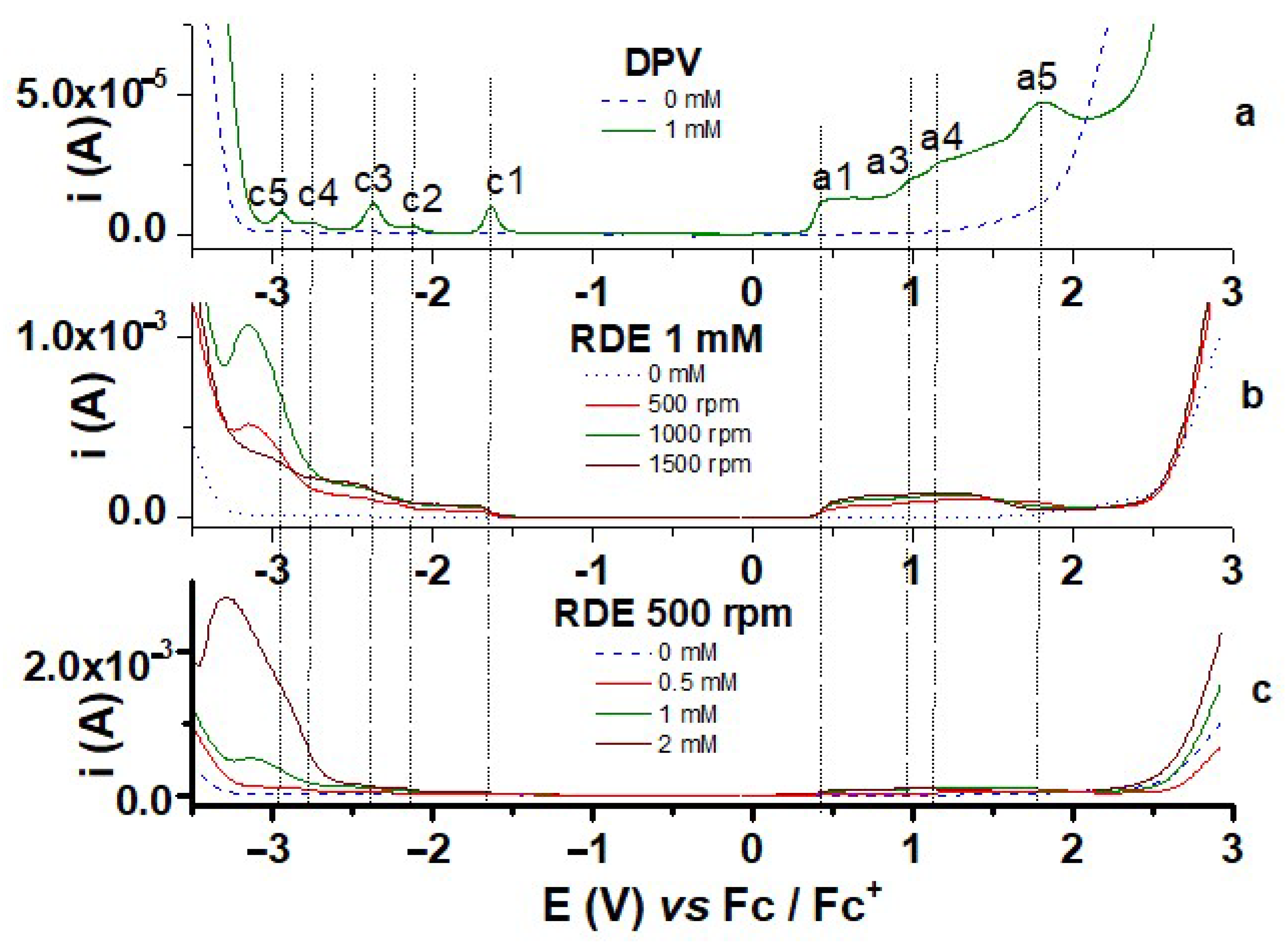
3.1.1. Characterization by CV and DPV
3.1.2. Characterization by RDE
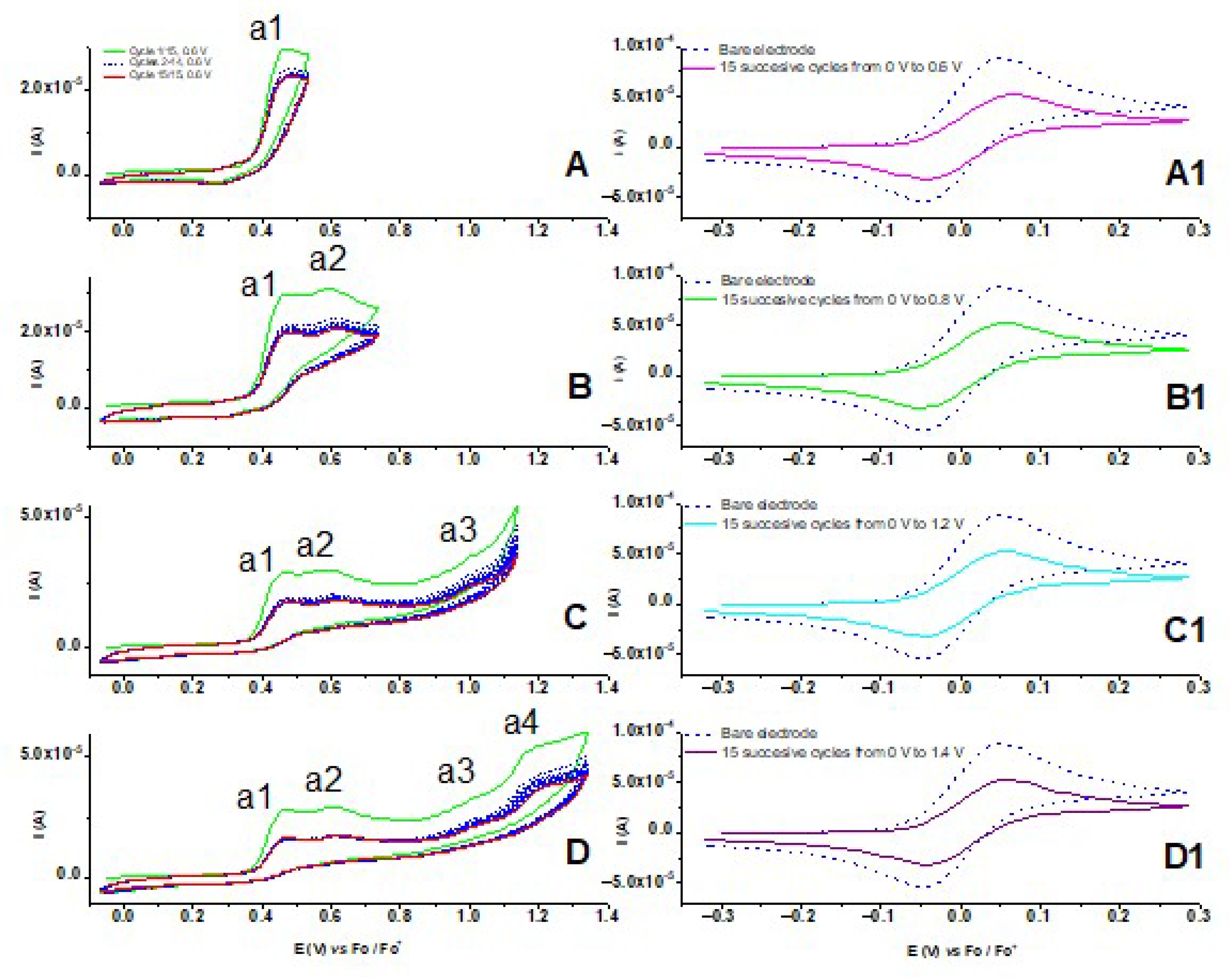
3.2. Preparation of the Modified Electrodes
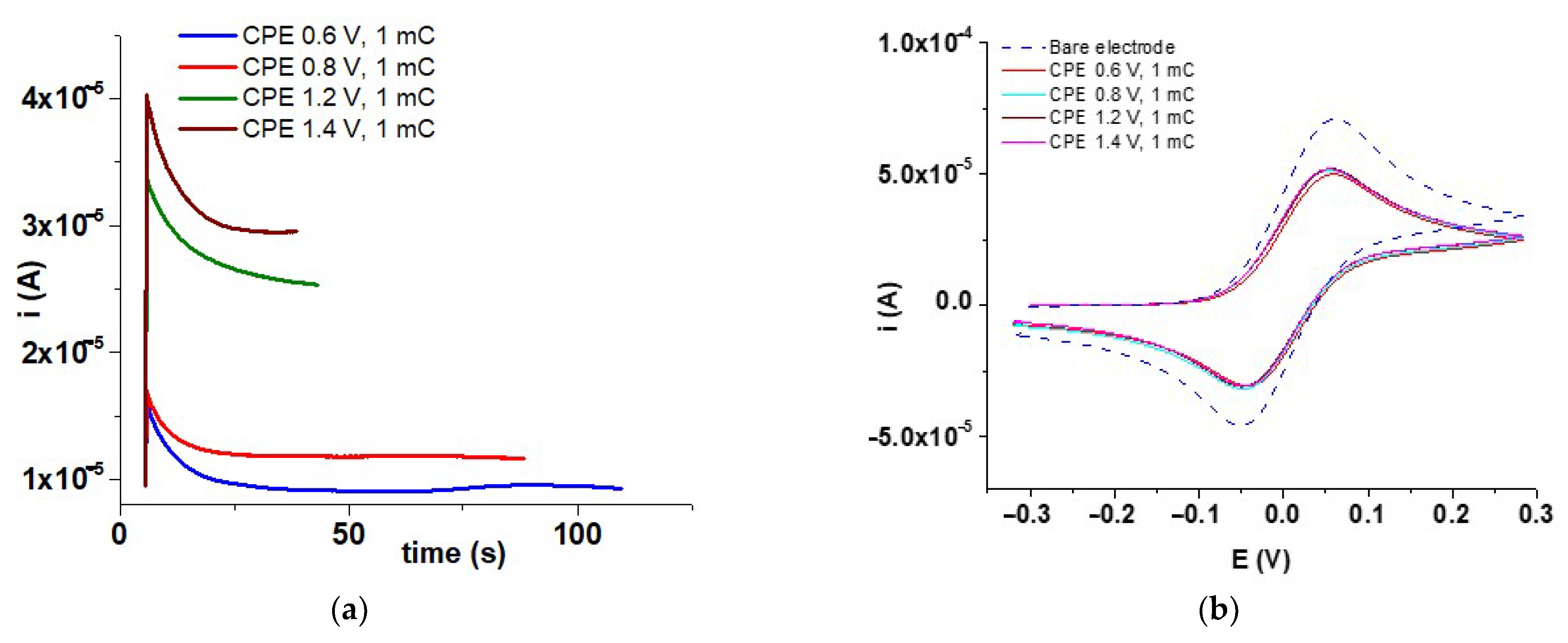
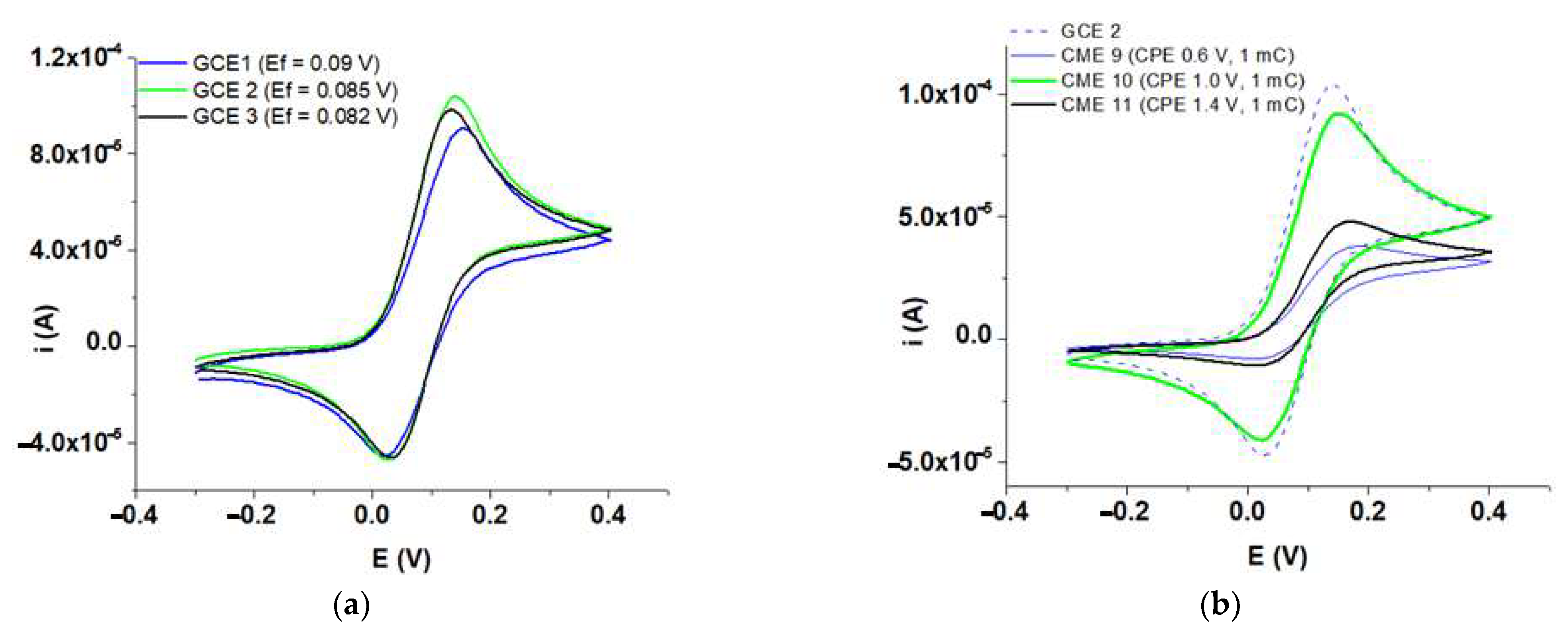
3.3. Evidence for M Films Formation by Fc Redox Probe
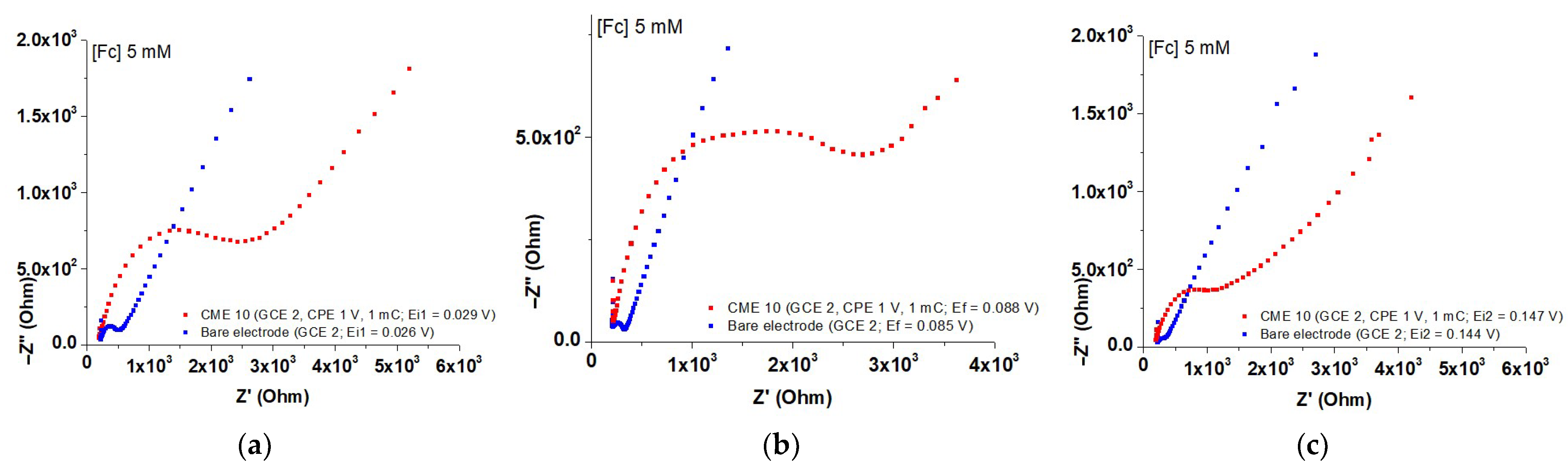
3.4. Characterization of M-CMEs by EIS
3.5. Characterization of M-CMEs by SEM
3.6. Characterization of M-CMEs by XPS
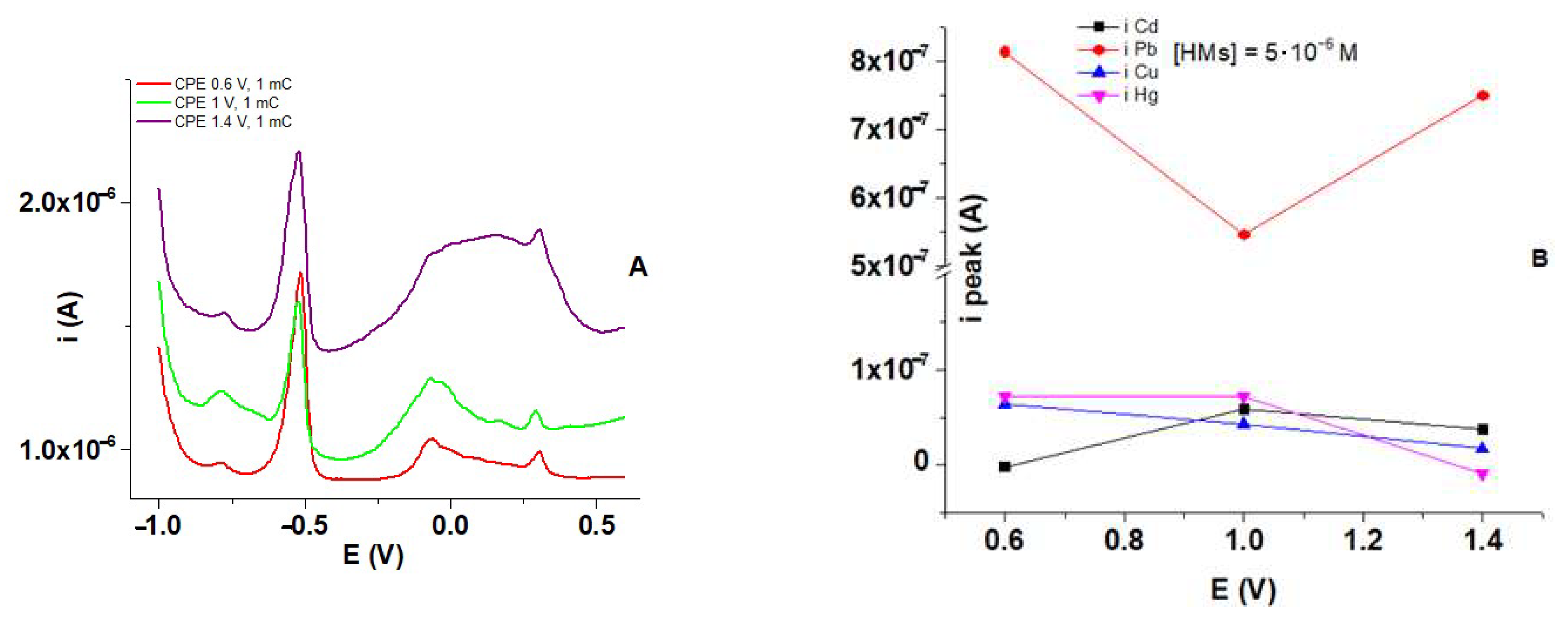
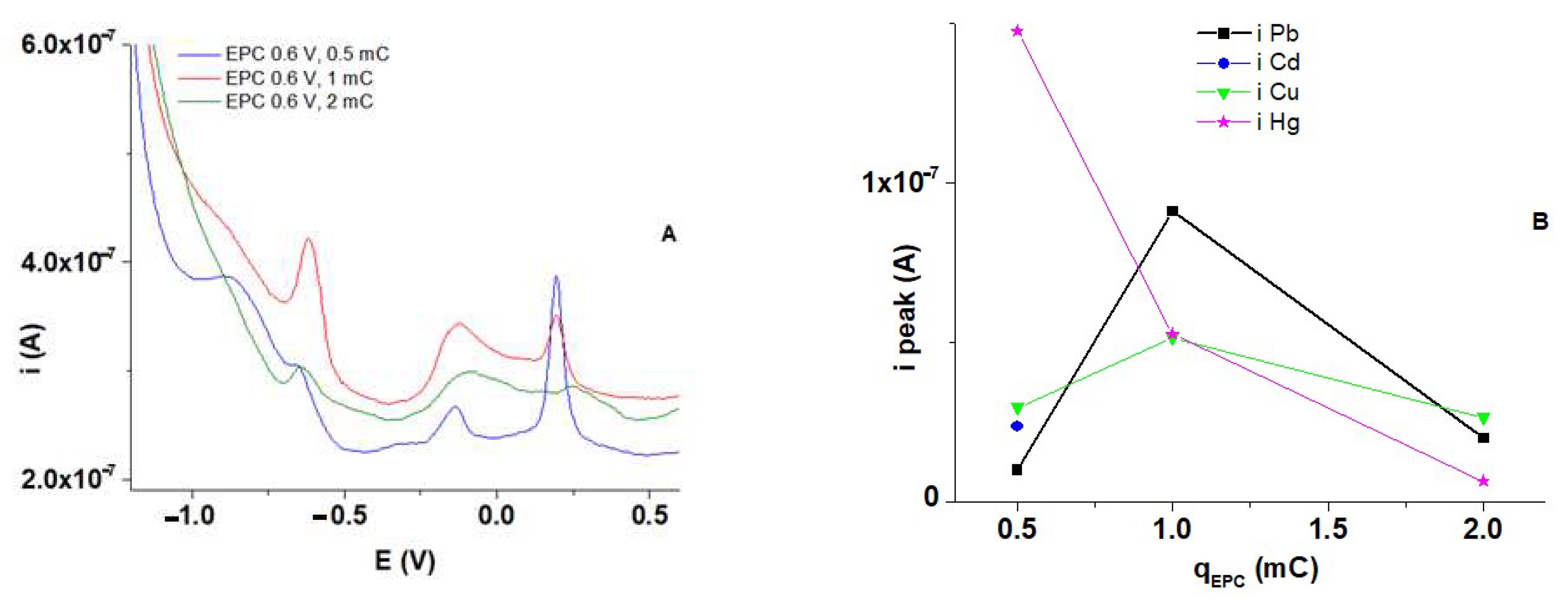
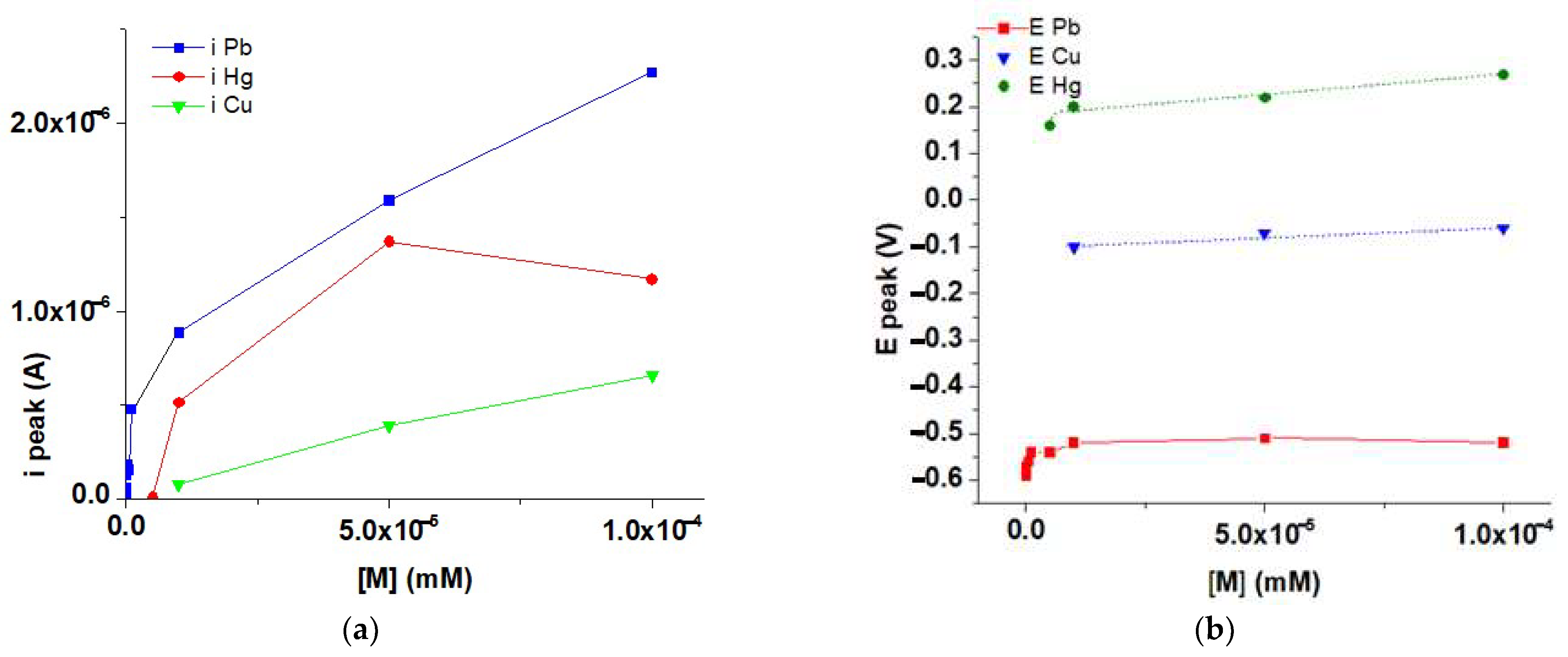
3.7. HMs Recognition Experiments Using polyM Films
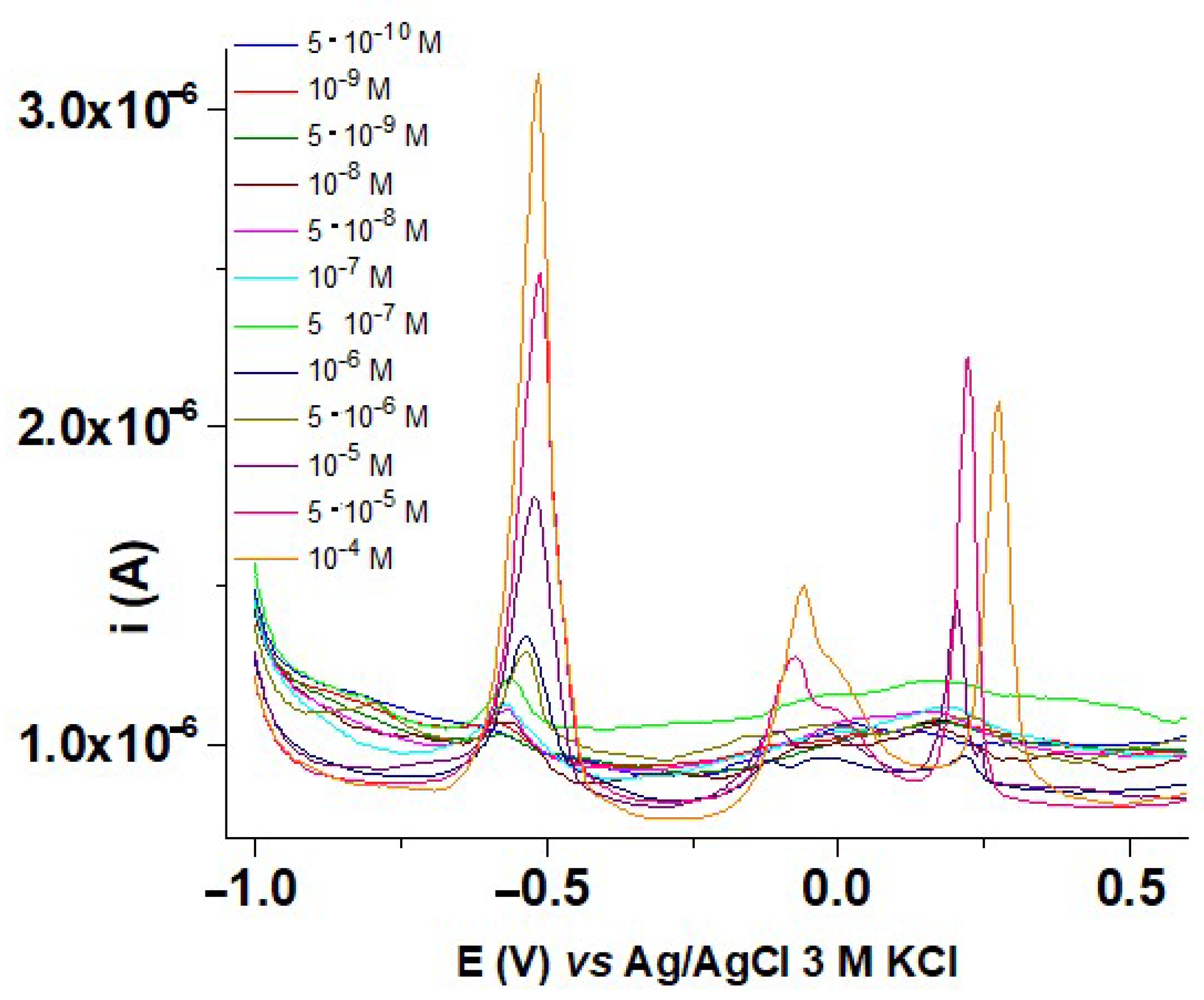
3.8. Influence of HMs Ions Concentrations on DPV Response
4. Discussion
Evidence for M Films Formation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Castro, P.P.; Carpanez, A.G.; Amarante, G.W. Azlactone Reaction Developments. Chem. Eur. J. 2016, 22, 10294–10318. [Google Scholar] [CrossRef] [PubMed]
- Zayas-Gonzalez, Y.M.; Ortiz, B.J.; Lynn, D.M. Layer-by-Layer Assembly of Amine-Reactive Multilayers Using an Azlactone-Functionalized Polymer and Small-Molecule Diamine Linkers. Biomacromolecules 2017, 18, 1499–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Davis, J.L.; Aden, B.M.; Lokitz, B.S.; Kilbey, S.M. Versatile Synthesis of Amine-Reactive Microgels by Self-Assembly of Azlactone-Containing Block Copolymers. Macromolecules 2018, 51, 3691–3701. [Google Scholar] [CrossRef]
- Chauveau, C.; Vanbiervliet, E.; Fouquay, S.; Michaud, G.; Simon, F.; Carpentier, J.-F.; Guillaume, S.M. Azlactone Telechelic Polyolefins as Precursors to Polyamides: A Combination of Metathesis Polymerization and Polyaddition Reactions. Macromolecules 2018, 51, 8084–8099. [Google Scholar] [CrossRef]
- Marra, I.F.S.; de Castro, P.P.; Amarante, G.W. Recent Advances in Azlactone Transformations. Eur. J. Org. Chem. 2019, 2019, 5830–5855. [Google Scholar] [CrossRef]
- Almalki, A.J.; Ibrahim, T.S.; Taher, E.S.; Mohamed, M.F.A.; Youns, M.; Hegazy, W.A.H.; Al-Mahmoudy, A.M.M. Synthesis, Antimicrobial, Anti-Virulence and Anticancer Evaluation of New 5(4H)-Oxazolone-Based Sulfonamides. Molecules 2022, 27, 671. [Google Scholar] [CrossRef]
- Mariappan, G.; Saha, B.P.; Datta, S.; Kumar, D.; Haldar, P.K. Design, synthesis and antidiabetic evaluation of oxazolone derivatives. J. Chem. Sci. 2011, 123, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.A.B.; Martinho, J.M.G.; Afonso, C.A.M. Synthesis of a Biologically Active Oxazol-5-(4H)-one via an Erlenmeyer–Plöchl Reaction. J. Chem. Educ. 2015, 92, 1543–1546. [Google Scholar] [CrossRef]
- Mesaik, M.A.; Rahat, S.; Khan, K.M.; Zia, U.; Choudhary, M.I.; Murad, S.; Ismail, Z.; Attaur, R.; Ahmad, A. Synthesis and immunomodulatory properties of selected oxazolone derivatives. Bioorg. Med. Chem. 2004, 12, 2049–2057. [Google Scholar] [CrossRef]
- Tandel, R.; Mammen, D. Synthesis and Study of Some Compounds Containing Oxazolone Ring, Showing Biological Activity. Indian J. Chem. 2008, 47, 932–937. [Google Scholar] [CrossRef]
- Hassanein, H.H.; Khalifa, M.M.; El-Samaloty, O.N.; El-Rahim, M.A.; Taha, R.A.; Magda; Ismail, M.F. Synthesis and biological evaluation of novel imidazolone derivatives as potential COX-2 inhibitors. Arch. Pharmacal. Res. 2008, 31, 562. [Google Scholar] [CrossRef] [PubMed]
- Razus, A.C. Azulene Moiety as Electron Reservoir in Positively Charged Systems; A Short Survey. Symmetry 2021, 13, 526. [Google Scholar] [CrossRef]
- Ghazvini Zadeh, E.H.; Tang, S.; Woodward, A.W.; Liu, T.; Bondar, M.V.; Belfield, K.D. Chromophoric Materials Derived from a Natural Azulene: Syntheses, Halochromism and One-Photon and Two-Photon Microlithography. J. Mater. Chem. C 2015, 3, 8495. [Google Scholar] [CrossRef]
- Koch, M.; Blacque, O.; Venkatesan, K.; Mater, J. Impact of 2,6-connectivity in azulene: Optical properties and stimuli responsive behavior. J. Mater. Chem. C 2013, 1, 7400. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Morita, N. Creation of Stabilized Electrochromic Materials by Taking Advantage of Azulene Skeletons. Eur. J. Org. Chem. 2009, 27, 4567–4579. [Google Scholar] [CrossRef]
- Tsurui, K.; Murai, M.; Ku, S.-Y.; Hawker, C.J.; Robb, M.J. Modulating the Properties of Azulene-Containing Polymers through Controlled Incorporation of Regioisomers. Adv. Funct. Mater. 2014, 24, 7338–7347. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Kok-Peng Ng, J.; Jia, P.; Lin, T.; Mui Cho, C.; Xu, J.; Lu, X.; He, C. Synthesis, Electronic, and Emission Spectroscopy, and Electrochromic Characterization of Azulene-Fluorene Conjugated Oligomers and Polymers. Macromolecules 2009, 42, 5534–5544. [Google Scholar] [CrossRef]
- Amir, E.; Amir, R.J.; Campos, L.M.; Hawker, C.J. Stimuli-Responsive Azulene-Based Conjugated Oligomers with Polyaniline-like Properties. J. Am. Chem. Soc. 2011, 133, 10046–10049. [Google Scholar] [CrossRef]
- Bakun, P.; Czarczynska-Goslinska, B.; Goslinski, T.; Lijewski, S. In vitro and in vivo biological activities of azulene derivatives with potential applications in medicine. Med. Chem. Res. 2021, 30, 834–846. [Google Scholar] [CrossRef]
- Vasile (Corbei), A.-A.; Stefaniu, A.; Pintilie, L.; Stanciu, G.; Ungureanu, E.-M. In Silico characterization and preliminary anticancer assessment of some 1,3,4-Thiadiazoles. UPB Sci. Bull. B 2021, 83, 3–12. [Google Scholar]
- Zeller, K.P. Houben Weyl, Methoden der Organischen Chemie. Thieme Stuttgart 1985, 5/2c, 127. [Google Scholar]
- McDonald, R.N.; Stewart, W.S. Nonbenzenoid Aromatic Systems. I. Synthesis of 1-Vinylazulene and Certain Substituted 1-Vinylazulenes. J. Org. Chem. 1965, 30, 270. [Google Scholar] [CrossRef]
- Hafner, K. Neuere Ergebnisse der Azulen-Chemie. Angew. Chem. 1958, 70, 419. [Google Scholar] [CrossRef]
- Hunig, S.; Ort, B. Mehrstufige reversible Redoxsysteme, XXXVII. Biazulenyle und ω, ω′-Biazulenylpolyene: Synthesen und spektroskopische Eigenschaften. Liebigs Ann. Chem. 1984, 12, 1905–1935. [Google Scholar] [CrossRef]
- Briquet, A.A.S.; Hansen, H.J. New Results in the Synthesis of Styrylazulene Derivatives: Application of the ‘Anil synthesis’ to the preparation of azulenes substituted with styryl groups at the seven-membered ring. Helv. Chim. Acta 1994, 77, 1921–1939. [Google Scholar] [CrossRef]
- Razus, A.C.; Birzan, L. Synthesis of azulenic compounds with a homo-or hetero-atomic double bond at position 1. Arkivoc 2018, part VI, 1–56. [Google Scholar] [CrossRef]
- Wang, F.; Lai, Y.-H.; Han, M.-Y. Stimuli-Responsive Conjugated Copolymers Having Electro-Active Azulene and Bithiophene Units in the Polymer Skeleton: Effect of Protonation and p-Doping on Conducting Properties. Macromolecules 2004, 37, 3222–3230. [Google Scholar] [CrossRef]
- Lete, C.; Esteban, B.M.; Kvarnström, C.; Razus, A.C.; Ivaska, A. Electrosynthesis and characterization of poly(2-[(E)-2-azulen-1-ylvinyl] thiophene) using polyazulene as model compound. Electrochim. Acta 2007, 52, 6476–6483. [Google Scholar] [CrossRef]
- Iftime, G.; Lacroix, P.G.; Nakatani, K.; Razus, A.C. Push-pull azulene-based chromophores with nonlinear optical properties. Tetrahedron Lett. 1998, 39, 6853. [Google Scholar] [CrossRef]
- Asato, A.E.; Liu, R.S.H.; Rao, V.P.; Cai, Y.M. Azulene-containing donor-acceptor compounds as second-order nonlinear chromophores. Tetrahedron Lett. 1996, 37, 419–422. [Google Scholar] [CrossRef]
- Birzan, L.; Cristea, M.; Draghici, C.C.; Tecuceanu, V.; Maganu, M.; Hanganu, A.; Arnold, G.-L.; Ungureanu, E.-M.; Razus, A.C. 1-vinylazulenes–potential host molecules in ligands for metal ion detectors. Tetrahedron 2016, 72, 2316. [Google Scholar] [CrossRef]
- Cristea, M.; Bîrzan, L.; Dumitrascu, F.; Enache, C.; Tecuceanu, V.; Hanganu, A.; Draghici, C.; Deleanu, C.; Nicolescu, A.; Maganu, M.; et al. 1-Vinylazulenes with Oxazolonic Ring-Potential Ligands for Metal Ion Detectors; Synthesis and Products Properties. Symmetry 2021, 13, 1209. [Google Scholar] [CrossRef]
- Sharma, N.; Banerjee, J.; Shrestha, N.; Chaudhury, D. A review on oxazolone, it’ s method of synthesis and biological activity. Eur. J. Biomed. Pharm. Sci. 2015, 2, 964–987. Available online: https://www.researchgate.net/publication/280882247 (accessed on 26 January 2023).
- Garcia-Sanz, C.; Andreu, A.; de las Rivas, B.; Jimenez, A.I.; Pop, A.; Silvestru, C.; Palomo, J.M. Pd-Oxazolone complexes conjugated to an engineered enzyme: Improving fluorescence and catalytic properties. Org. Biomol. Chem. 2021, 19, 2773–2783. [Google Scholar] [CrossRef] [PubMed]
- Anastasoaie, V.; Omocea, C.; Enache, L.-B.; Anicai, L.; Ungureanu, E.-M.; van Staden, J.F.; Enachescu, M. Surface Characterization of New Azulene-Based CMEs for Sensing. Symmetry 2021, 13, 2292. [Google Scholar] [CrossRef]
- Enache, L.-B.; Anastasoaie, V.; Lete, C.; Brotea, A.G.; Matica, O.-T.; Amarandei, C.-A.; Brandel, J.; Ungureanu, E.-M.; Enachescu, M. Polyazulene-Based Materials for Heavy Metal Ion Detection. 3. (E)-5-((6-t-Butyl-4,8-dimethylazulen-1-yl) diazenyl)-1H-tetrazole-Based Modified Electrodes. Symmetry 2021, 13, 1642. [Google Scholar] [CrossRef]
- Păun, A.-M.; Matica, O.-T.; Anăstăsoaie, V.; Enache, L.-B.; Diacu, E.; Ungureanu, E.-M. Recognition of Heavy Metal Ions by Using E-5-((5-Isopropyl-3,8-Dimethylazulen-1-yl) Dyazenyl)-1H-Tetrazole Modified Electrodes. Symmetry 2021, 13, 644. [Google Scholar] [CrossRef]
- Matica, O.-T.; Brotea, A.G.; Ungureanu, E.M.; Stefaniu, A. Electrochemical and Spectral Studies on Benzylidenerhodanine for Sensor Development for Heavy Metals in Waters. Appl. Sci. 2022, 12, 2681. [Google Scholar] [CrossRef]
- Matica, O.-T.; Brotea, A.G.; Ungureanu, E.M.; Mandoc, L.R.; Birzan, L. Electrochemical and spectral studies of rhodanine in view of heavy metals determination. Electrochem. Sci. Adv. 2022, 1–11. [Google Scholar] [CrossRef]
- Buica, G.-O.; Maior, I.; Ungureanu, E.M.; Vaireanu, D.I.; Diacu, E.; Bucher, C.; Saint-Aman, E. Electrochemical Impedance Characterization of Poly (Pyrrole-EDTA Like) Modified Electrodes. Rev. Chim. 2009, 60, 1205–1209. [Google Scholar]
- Wang, Y.; Rogers, E.I.; Compton, R.G. The measurement of the diffusion coefficients of ferrocene and ferrocenium and their temperature dependence in acetonitrile using double potential step microdisk electrode chronoamperometry. J. Electroanal. Chem. 2010, 648, 15–19. [Google Scholar] [CrossRef]
- Morikita, T.; Yamamoto, T. Electrochemical determination of diffusion coefficient of π-conjugated polymers containing ferrocene unit. J. Organomet. Chem. 2001, 637–639, 809–812. [Google Scholar] [CrossRef]
- Ikeuchi, H.; Kanakubo, M. Diffusion Coefficients of Ferrocene and Ferricinium Ion in Tetraethylammonium Perchlorate Acetonitrile Solutions, as Determined by Chronoamperometry. Electrochemistry 2001, 69, 34–36. [Google Scholar] [CrossRef] [Green Version]
- Krishna, D.N.G.; Philip, J. Review on surface-characterization applications of X-ray photoelectron spectroscopy (XPS): Recent developments and challenges. Appl. Surf. Sci. Adv. 2022, 12, 100332. [Google Scholar] [CrossRef]
- Chiticaru, E.A.; Pilan, L.; Damian, C.-M.; Vasile, E.; Burns, J.S.; Ionită, M. Influence of Graphene Oxide Concentration when Fabricating an Electrochemical Biosensor for DNA Detection. Biosensors 2019, 9, 113. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak | Method | Peak Characteristics | ||
|---|---|---|---|---|
| CV | DPV | RDE (E1/2) | ||
| a1 | 0.479 | 0.484 | 0.431 (500 rpm) 0.441 (1000 rpm) 0.440 (1500 rpm) | Quasireversible |
| a2 | 0.657 | - | 0.630 (500 rpm) | Irreversible |
| a3 | 1.120 | 0.981 | 1.070 (500 rpm) | Irreversible |
| a4 | 1.236 | 1.163 | - | Irreversible |
| a5 | 1.777 | - | Irreversible | |
| c1 | −1.743 | −1.629 | −1.651 (500 rpm) −1.651 (1000 rpm) −1.650 (1500 rpm) | Quasireversible |
| c2 | −2.201 | −2.119 | −2.247 (500 rpm) | Quasireversible |
| c3 | −2.496 | −2.371 | −2.412 (500 rpm) | Quasireversible |
| c4 | - | −2.763 | - | Irreversible |
| c5 | - | −2.926 | - | Irreversible |
| Sample | [M] (mM) | Preparation | Electrical Charge (mC) | Electrical Charge Density (mC/cm2) | Analysis |
|---|---|---|---|---|---|
| CME 1 | 2 | S * (0–0.6) | - | - | Fc redox probe *a |
| CME 2 | 2 | S * (0–0.8) | - | - | Fc redox probe *a |
| CME 3 | 2 | S * (0–1.2) | - | - | Fc redox probe *a |
| CME 4 | 2 | S * (0–1.4) | - | - | Fc redox probe *a |
| CME 5–5b | 2 | CPE at 0.6 V | 1 | 14 | Chrono *b, Fc redox probe *a |
| CME 6–6b | 2 | CPE at 0.8 V | 1 | 14 | Chrono *b, Fc redox probe *a |
| CME 7–7b | 2 | CPE at 1.2 V | 1 | 14 | Chrono *b, Fc redox probe *a |
| CME 8–8b | 2 | CPE at 1.4 V | 1 | 14 | Chrono *b, Fc redox probe *a |
| CME 9–9b | 2 | CPE at 0.6 V | 1 | 14 | Chrono *b, Fc redox probe *a, EIS |
| CME 10–10b | 2 | CPE at 1 V | 1 | 14 | Chrono *b, Fc redox probe *a, EIS |
| CME 11–11b | 2 | CPE at 1.4 V | 1 | 14 | Chrono *b, Fc redox probe *a, EIS |
| CME 12 | 1 | CPE at +1.4 V | 4 | 14 | Chrono *b, SEM, XPS |
| CME 13 | 1 | CPE at +1 V | 4 | 14 | Chrono *b, SEM, XPS |
| CME 14 | 1 | CPE at +0.6 V | 4 | 14 | Chrono *b, SEM, XPS |
| CME 15 | 1 | CPE at +0.6 V | 8 | 28 | Chrono *b, SEM, XPS |
| CME 16 | 1 | CPE at +0.6 V | 16 | 56 | Chrono *b, SEM, XPS |
| CME 17 | 1 | CPE at +0.6 V | 24 | 84 | Chrono *b, SEM, XPS |
| CME 18–18m | 2 | CPE at +0.6 V | 1 | 14 | Chrono *b, HMs *c, R *d |
| CME 19–19m | 2 | CPE at +1.0 V | 1 | 14 | Chrono *b, HMs *c, R *d |
| CME 20–20d | 2 | CPE at +1.4 V | 1 | 14 | Chrono *b, HMs *c, R *d |
| CME 21–21b | 2 | CPE at +0.6 V | 0.5 | 7 | Chrono *b, HMs *c, R *d |
| CME 22–22b | 2 | CPE at +0.6 V | 1 | 14 | Chrono *b, HMs *c, R *d |
| CME 23–23b | 2 | CPE at +0.6 V | 2 | 28 | Chrono *b, HMs *c, R *d |
| Crt. Nr. | ECPE (V) (M-CME) | Epa (V) | 105·ipa (A) | Epc (V) | 105·ipc (A) | ΔEp *1 (mV) | Ef *2 (V) |
|---|---|---|---|---|---|---|---|
| 1 | Bare electrode | 0.061 | 7.141 | −0.049 | −4.598 | 110 | 0.055 |
| 2 | 0.6 (CME 5) | 0.062 | 5.006 | −0.039 | −3.098 | 101 | 0.051 |
| 3 | 0.8 (CME 6) | 0.052 | 5.159 | −0.049 | −3.201 | 101 | 0.051 |
| 4 | 1.2 (CME 7) | 0.052 | 5.201 | −0.049 | −3.109 | 101 | 0.051 |
| 5 | 1.4 (CME 8) | 0.052 | 5.201 | −0.049 | −3.098 | 101 | 0.051 |
| Nr. Crt. | Modified Electrode | Characteristics of Film Synthesis | Eeq (V) | Ei1 (V) | Ei2 (V) |
|---|---|---|---|---|---|
| 1 | GCE 1 | - | 0.090 | 0.031 | 0.149 |
| 2 | GCE 2 | - | 0.085 | 0.026 | 0.144 |
| 3 | GCE 3 | - | 0.082 | 0.023 | 0.141 |
| 4 | CME 9 | GCE 1, 0.6 V, 1 mC | 0.096 | 0.037 | 0.155 |
| 5 | CME 10 | GCE 2, 1 V, 1 mC | 0.088 | 0.029 | 0.147 |
| 6 | CME 11 | GCE 3, 1.4 V, 1 mC | 0.090 | 0.031 | 0.149 |
| Nr. Crt. | Modified Electrode (Preparation Potential) | E (V) | 10−3·RGC (Ω) | 10−3·RMGC (Ω) | 10−3·(ΔR) (Ω) |
|---|---|---|---|---|---|
| 1 | CME 9 (0.6 V) | Ei1 = 0.037 | 0.654 | 6.823 | 6.169 |
| 2 | CME 9 (0.6 V) | Eeq = 0.096 | 1.452 | 2.740 | 1.288 |
| 3 | CME 9 (0.6 V) | Ei2 = 0.155 | 0.384 | 4.355 | 3.971 |
| 4 | CME 10 (1.0 V) | Ei1 = 0.029 | 0.307 | 2.421 | 2.114 |
| 5 | CME 10 (1.0 V) | Eeq = 0.088 | 0.121 | 2.502 | 2.381 |
| 6 | CME 10 (1.0 V) | Ei2 = 0.147 | 0.118 | 0.935 | 0.817 |
| 7 | CME 11 (1.4 V) | Ei1 = 0.031 | 0.495 | 5.530 | 5.035 |
| 8 | CME 11 (1.4 V) | Eeq = 0.090 | 1.462 | 3.366 | 1.904 |
| 9 | CME 11 (1.4 V) | Ei2 = 0.149 | 0.353 | 4.609 | 4.256 |
| Modified Electrode | Conditions for CPE | C1s XPS Core-Level Spectra |
|---|---|---|
| CME 12 | 1.4 V, 4 mC (14 mC/cm2) |  |
| CME 13 | 1 V, 4 mC (14 mC/cm2) |  |
| CME 14 | 0.6 V, 4 mC (14 mC/cm2) |  |
| CME 15 | 0.6 V, 8 mC (28 mC/cm2) |  |
| CME 16 | 0.6 V, 16 mC (56 mC/cm2) |  |
| CME 17 | 0.6 V, 24 mC (84 mC/cm2) |  |
| CME (Sample) | ECPE (V) | q (mC/cm2) | BE (eV) | Assignment | At% | Area (N) | C-C/ C-O | C-C/ O-C=O | C/O |
|---|---|---|---|---|---|---|---|---|---|
| CME 12 (P1) | +1.4 | 14 | 284.61 | C-C/C=C | 52.03 | 181.96 | 3.88 | ||
| 285.74 | C-O | 47.97 | 167.75 | ||||||
| CME 13 (P2) | +1 | 14 | 284.81 | C-C/C=C | 77.27 | 318.79 | 5.41 | ||
| 286.21 | C-O | 13.59 | 56.05 | 5.688 | |||||
| 288.79 | O-C=O | 9.14 | 37.72 | 8.451 | |||||
| CME 14 (P3) | +0.6 | 14 | 284.77 | C-C/C=C | 67.17 | 263.01 | 5.50 | ||
| 286.18 | C-O | 27.34 | 107.06 | 2.457 | |||||
| 288.92 | O-C=O | 5.49 | 21.50 | 12.233 | |||||
| CME 15 (P4) | +0.6 | 28 | 284.74 | C-C/C=C | 60.15 | 241.79 | 4.05 | ||
| 286.21 | C-O | 34.46 | 103.54 | 2.335 | |||||
| 288.68 | O-C=O | 5.39 | 20.12 | 12.017 | |||||
| CME 16 (P5) | +0.6 | 56 | 284.8 | C-C/C=C | 68.62 | 278.34 | 5.23 | ||
| 286.3 | C-O | 25.96 | 105.30 | 2.643 | |||||
| 288.89 | O-C=O | 5.42 | 22.00 | 12.652 | |||||
| CME 17 (P6) | +0.6 | 84 | 284.74 | C-C/C=C | 62.22 | 231.8 | 4.72 | ||
| 286.12 | C-O | 32.75 | 122.01 | 1.900 | |||||
| 288.67 | O-C=O | 5.03 | 187.5 | 1.236 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brotea, A.-G.; Matica, O.-T.; Musina, C.; Cristea, M.; Stefaniu, A.; Pandele, A.-M.; Ungureanu, E.-M. Advanced Materials Based on Azulenyl-Phenyloxazolone. Symmetry 2023, 15, 540. https://doi.org/10.3390/sym15020540
Brotea A-G, Matica O-T, Musina C, Cristea M, Stefaniu A, Pandele A-M, Ungureanu E-M. Advanced Materials Based on Azulenyl-Phenyloxazolone. Symmetry. 2023; 15(2):540. https://doi.org/10.3390/sym15020540
Chicago/Turabian StyleBrotea, Alina-Giorgiana, Ovidiu-Teodor Matica, Cornelia Musina (Borsaru), Mihaela Cristea, Amalia Stefaniu, Andreea-Madalina Pandele, and Eleonora-Mihaela Ungureanu. 2023. "Advanced Materials Based on Azulenyl-Phenyloxazolone" Symmetry 15, no. 2: 540. https://doi.org/10.3390/sym15020540