
Interactions between Artificial Channel Protein, Water Molecules, and Ions Based on Theoretical Approaches


Abstract
:1. Introduction
2. Results and Discussion
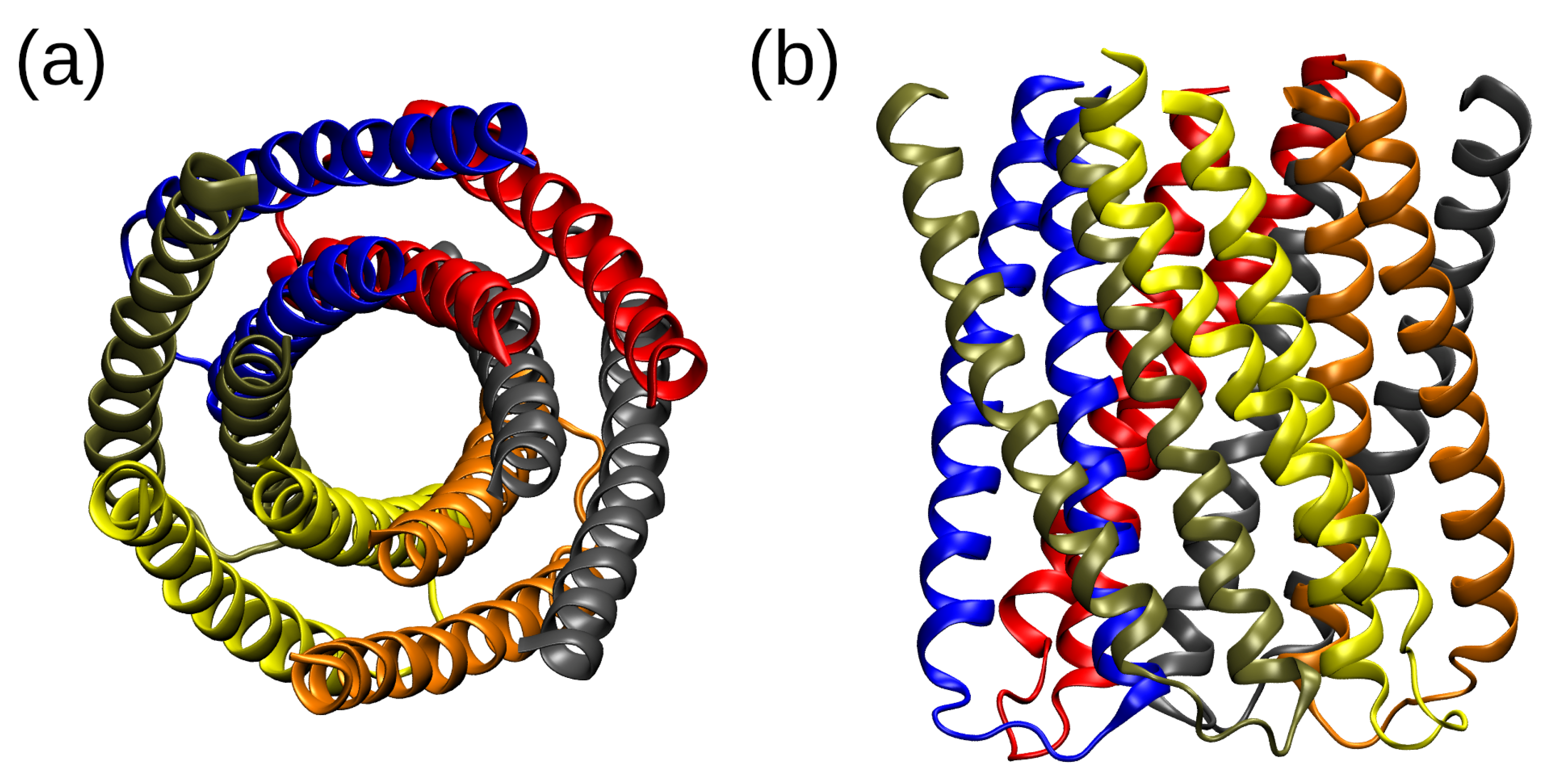
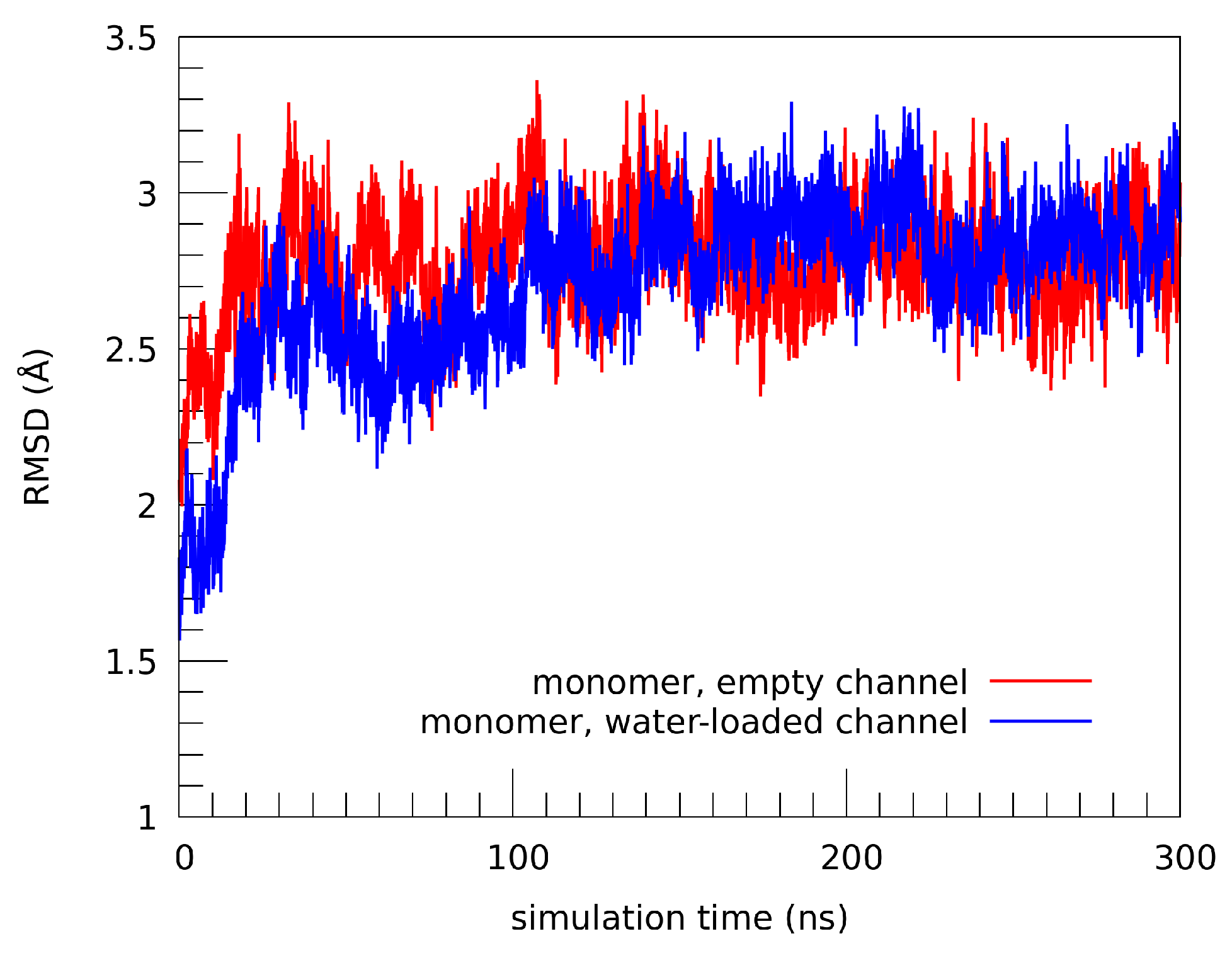
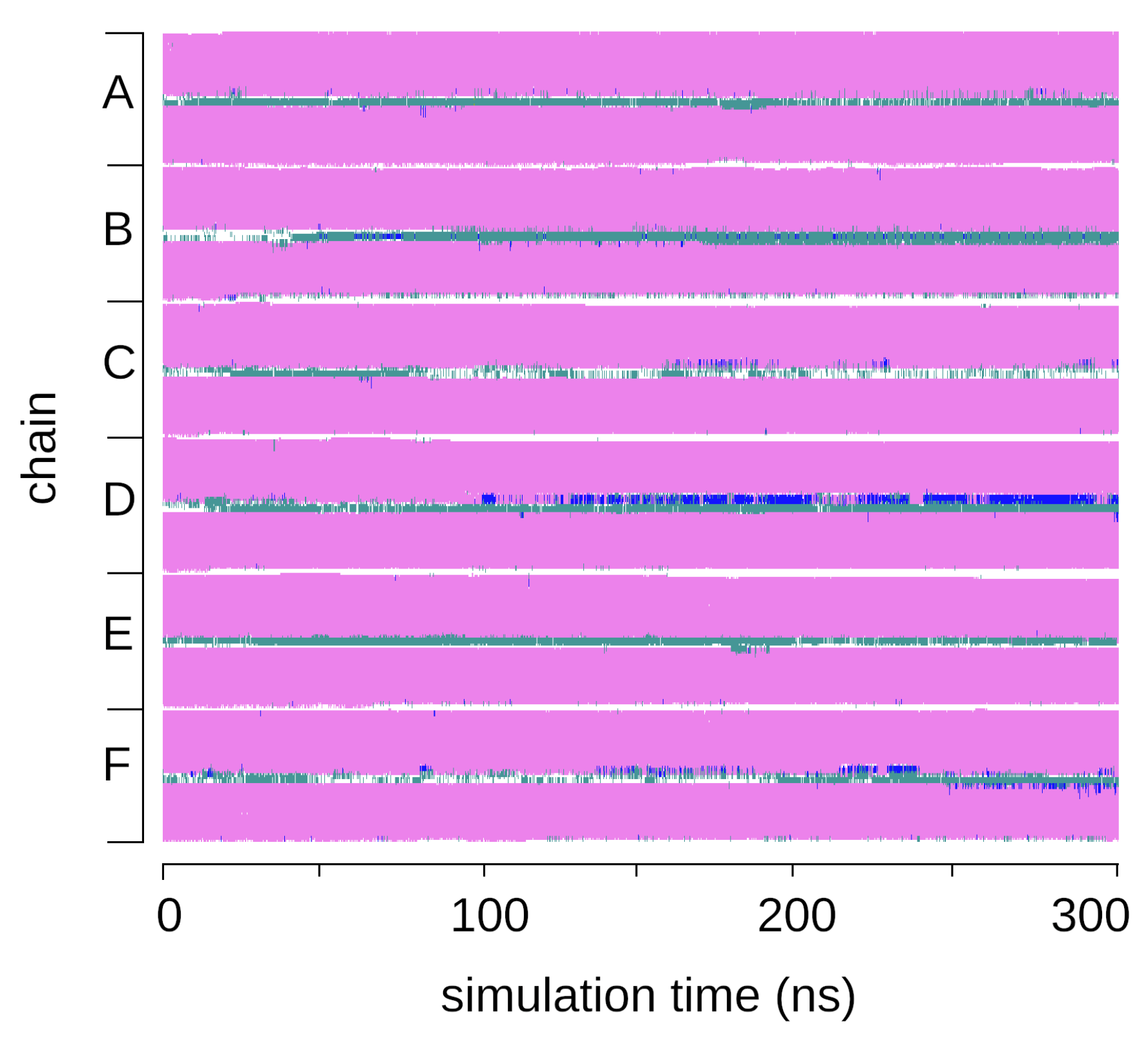
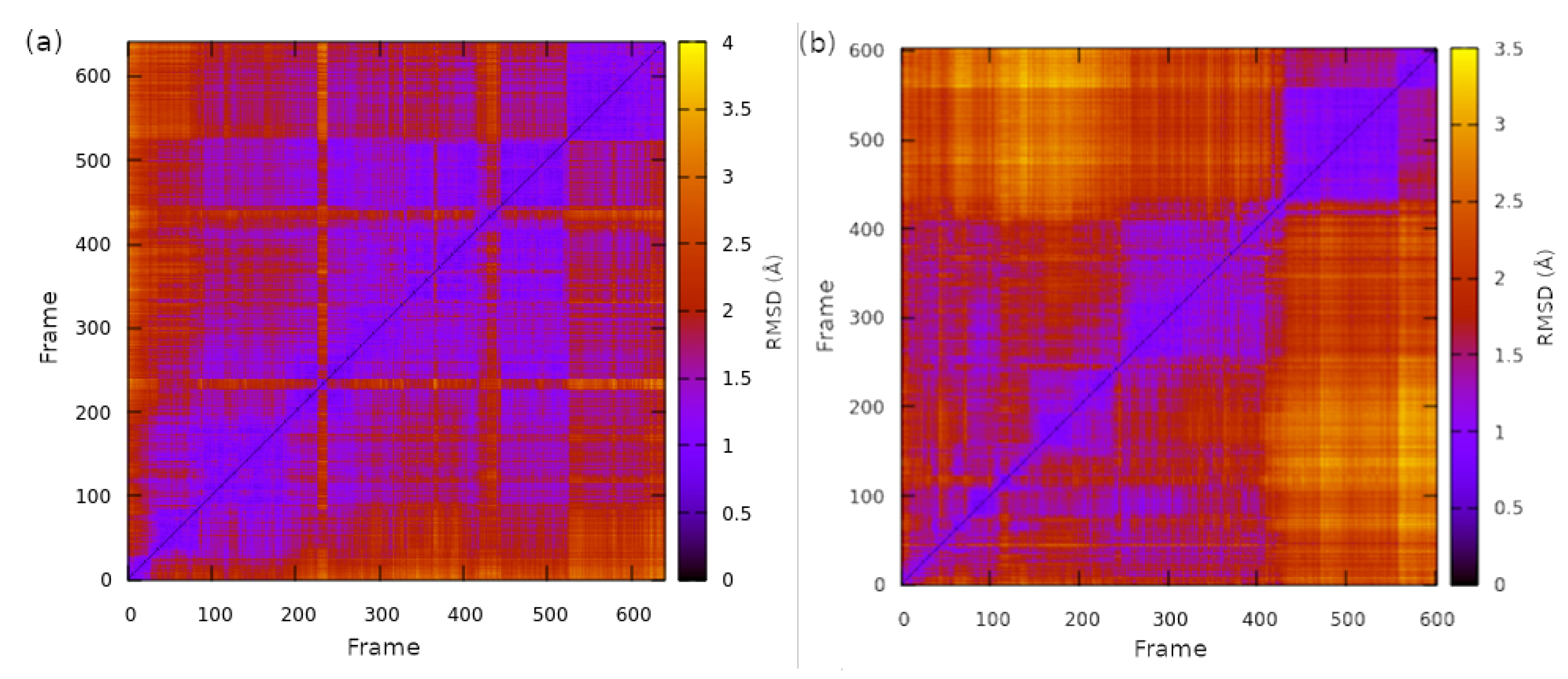
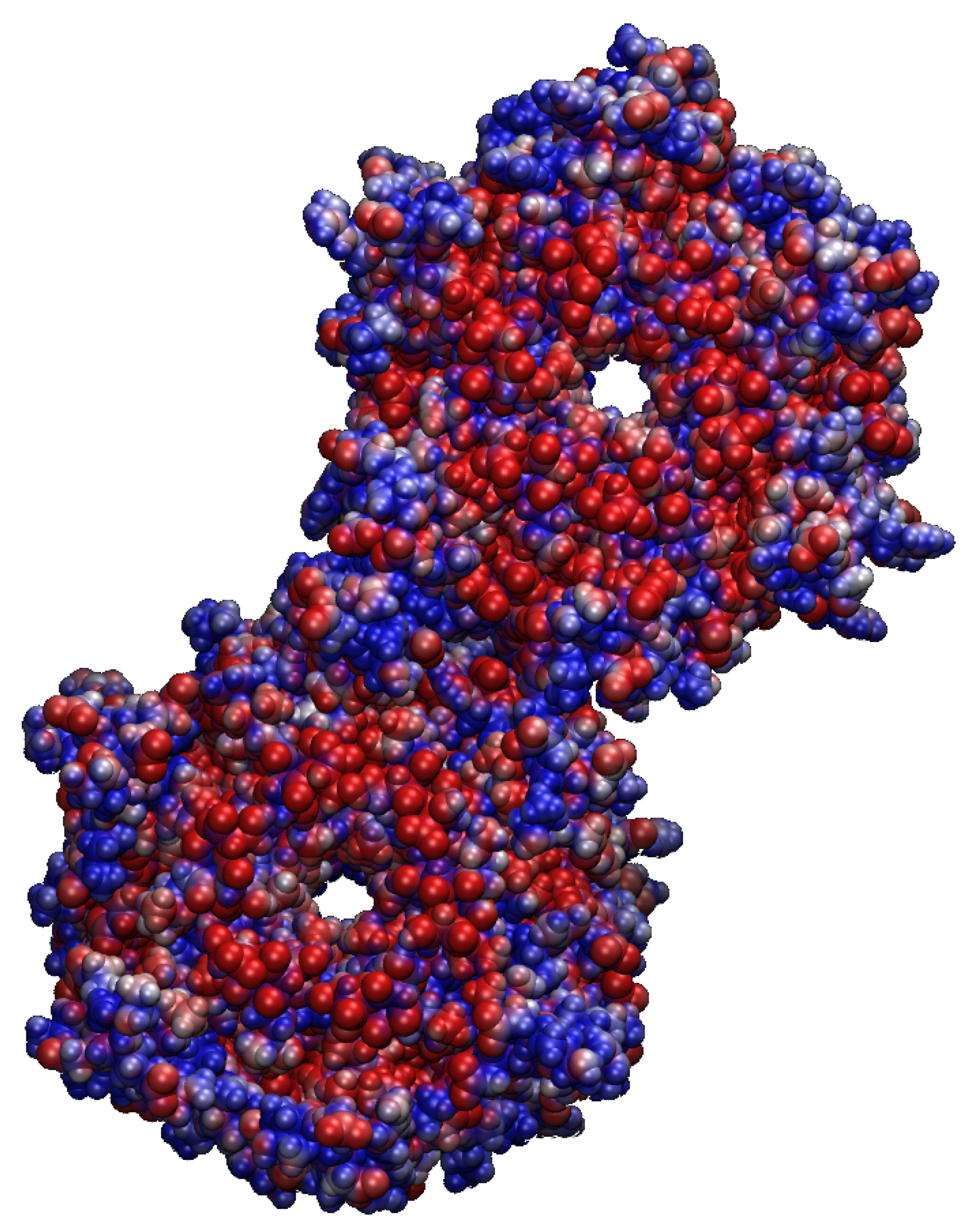
2.1. Structural Properties of the TMHC6 Protein Channel in the Light of the Classical Molecular Dynamics (MD) Simulations
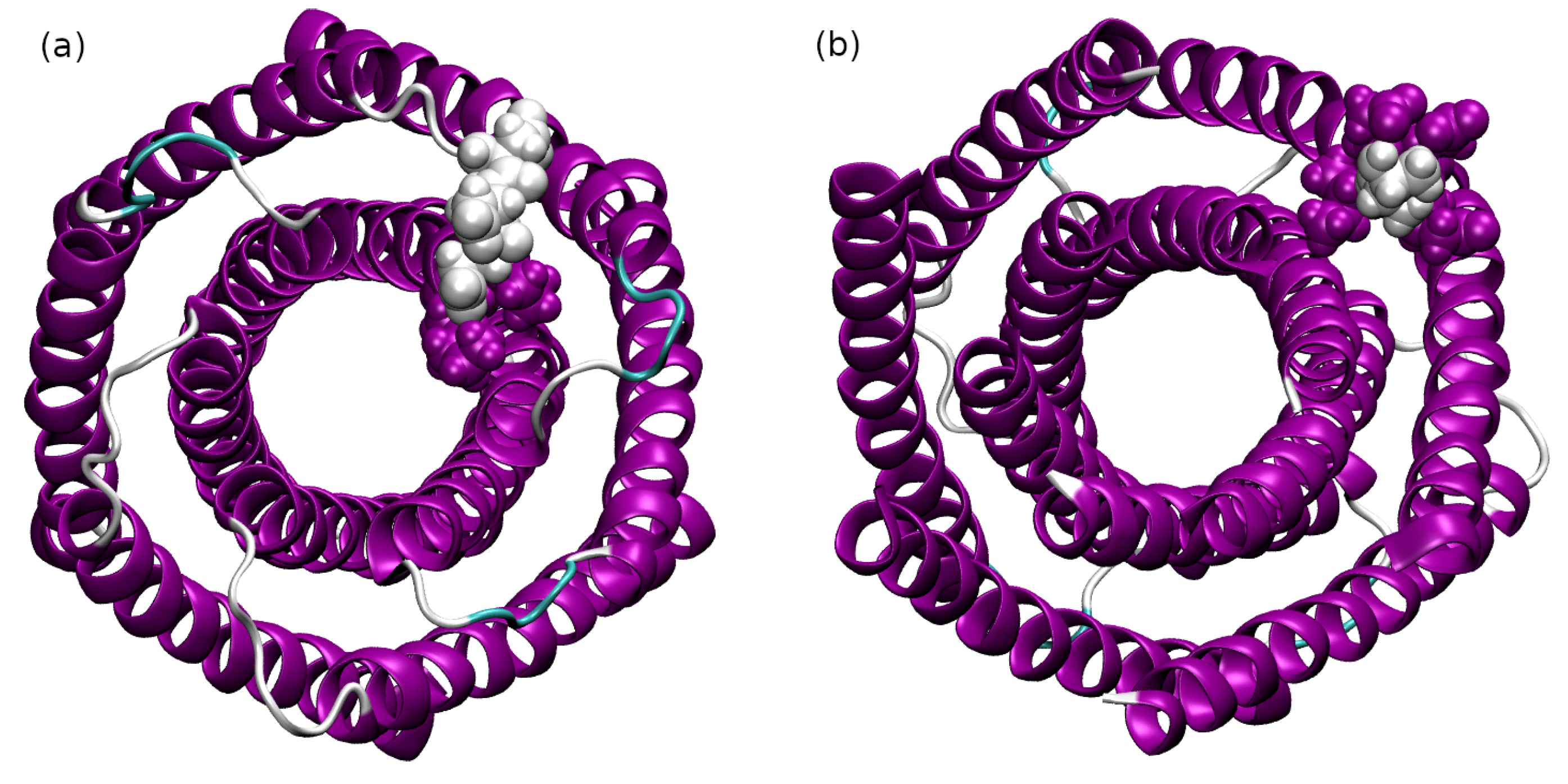
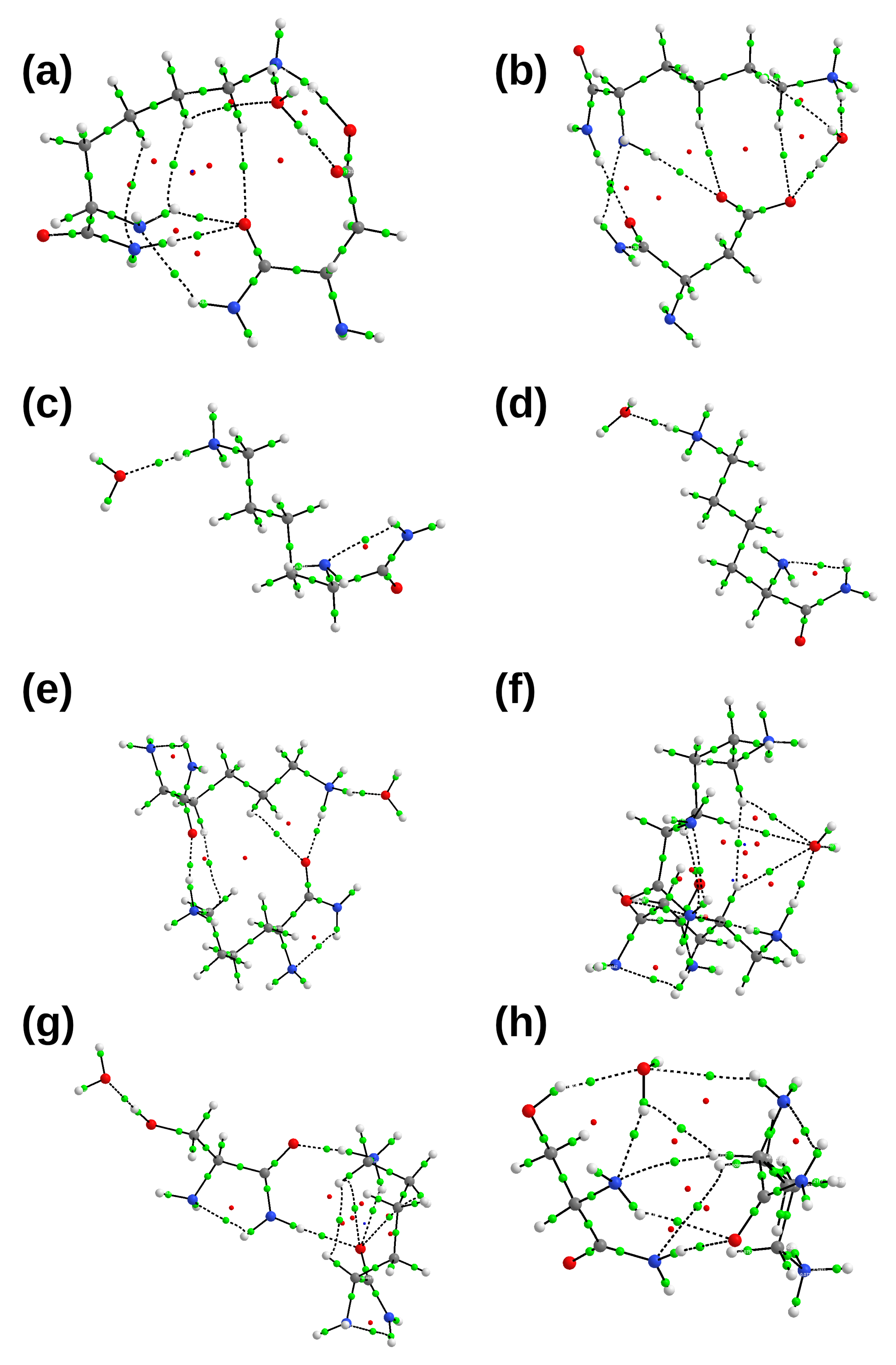
2.2. Interactions between the TMHC6 Protein Channel and Water Molecules or Ions
3. Computational Methodology
3.1. Classical Molecular Dynamics (MD) Simulations Protocol for the TMHC6 Protein
3.2. The Computational Protocol for Interaction Energy Estimation between the Protein, Water Molecules, and Ions
3.2.1. Symmetry-Adapted Perturbation Theory (SAPT)
3.2.2. Atoms in Molecules (AIM)
4. Conclusions
- (i)
- The single channel assemblies are stable and the presence or absence of water molecules in the channel does not affect its stability;
- (ii)
- The SAPT method showed that electrostatic interactions play a dominant role in the intermolecular interactions of the channel and ions, providing means to direct the ions into the channel entry;
- (iii)
- The AIM analysis revealed the intermolecular hydrogen bond presence in the studied complexes as well as other interactions, which cannot be classified as hydrogen bonds. However, their presence stabilizes the structure of the investigated residues;
- (iv)
- The presence of the polar environment affected the conformations of the studied complexes and the formation of the intra- and intermolecular interactions. As a further noticeable consequence, the qualitative changes in the electron density distribution were observed.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MD | molecular dynamics |
| DFT | density functional theory |
| PES | potential energy surface |
| PCM | polarizable continuum model |
| SAPT | symmetry-adapted perturbation theory |
| AIM | atoms in molecules |
| RMSD | root mean square deviation |
| RMSF | root mean square fluctuation |
| BCP | bond critical point |
| RCP | ring critical point |
| HB | hydrogen bond |
| BSSE | basis set superposition error |
References
- Whitford, D. Proteins: Structure and Function; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2005. [Google Scholar]
- McLachlan, A.D. Protein Structure and Function. Annu. Rev. Phys. Chem. 1972, 23, 165–192. [Google Scholar] [CrossRef]
- Huang, P.S.; Boyken, S.E.; Baker, D. The coming of age of de novo protein design. Nature 2016, 537, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.J.; Lim, H.Y.; Huang, J.; Kazlauskas, R.J. Comparison of Five Protein Engineering Strategies for Stabilizing an α/β-Hydrolase. Biochemistry 2017, 56, 6521–6532. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, R.; Wang, J.; Wilson, L.M.; Yan, Y. Protein Engineering for Improving and Diversifying Natural Product Biosynthesis. Trends Biotechnol. 2020, 38, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Applying and improving AlphaFold at CASP14. Proteins 2021, 89, 1711–1721. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Consortium. Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019, 47, D520–D528. [Google Scholar] [CrossRef] [Green Version]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Carpenter, E.P.; Beis, K.; Cameron, A.D.; Iwata, S. Overcoming the challenges of membrane protein crystallography. Curr. Opin. Struct. Biol. 2008, 18, 581–586. [Google Scholar] [CrossRef] [Green Version]
- Mravic, M.; Thomaston, J.L.; Tucker, M.; Solomon, P.E.; Liu, L.; DeGrado, W.F. Packing of apolar side chains enables accurate design of highly stable membrane proteins. Science 2019, 363, 1418–1423. [Google Scholar] [CrossRef]
- Joh, N.H.; Wang, T.; Bhate, M.P.; Acharya, R.; Wu, Y.; Grabe, M.; Hong, M.; Grigoryan, G.; DeGrado, W.F. De novo design of a transmembrane Zn2+-transporting four-helix bundle. Science 2014, 346, 1520–1524. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Kortemme, T. Recent advances in de novo protein design: Principles, methods, and applications. J. Biol. Chem. 2021, 296, 100558. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Sesterhenn, F.; Bonet, J.; van Aalen, E.A.; Scheller, L.; Abriata, L.A.; Cramer, J.T.; Wen, X.; Rosset, S.; Georgeon, S.; et al. Bottom-up de novo design of functional proteins with complex structural features. Nat. Chem. Biol. 2021, 17, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Kuhlman, B.; Dantas, G.; Ireton, G.C.; Varani, G.; Stoddard, B.L.; Baker, D. Design of a Novel Globular Protein Fold with Atomic-Level Accuracy. Science 2003, 302, 1364–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, J.B.; Zanghellini, A.; Lovick, H.M.; Kiss, G.; Lambert, A.R.; St.Clair, J.L.; Gallaher, J.L.; Hilvert, D.; Gelb, M.H.; Stoddard, B.L.; et al. Computational Design of an Enzyme Catalyst for a Stereoselective Bimolecular Diels-Alder Reaction. Science 2010, 329, 309–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Lu, P.; El-Din, T.M.G.; Pei, X.Y.; Johnson, M.C.; Uyeda, A.; Bick, M.J.; Xu, Q.; Jiang, D.; Bai, H.; et al. Computational design of transmembrane pores. Nature 2020, 585, 129–134. [Google Scholar] [CrossRef]
- Carrillo-Tripp, M.; Saint-Martin, H.; Ortega-Blake, I. A comparative study of the hydration of Na+ and K+ with refined polarizable model potentials. J. Chem. Phys. 2003, 118, 7062–7073. [Google Scholar] [CrossRef]
- Dudev, T.; Lim, C. Ion Selectivity Strategies of Sodium Channel Selectivity Filters. Acc. Chem. Res. 2014, 47, 3580–3587. [Google Scholar] [CrossRef]
- Dudev, T.; Mazmanian, K.; Lim, C. Competition between Li+ and Na+ in sodium transporters and receptors: Which Na+-Binding sites are “therapeutic” Li+ targets? Chem. Sci. 2018, 9, 4093–4103. [Google Scholar] [CrossRef] [Green Version]
- Jing, Z.; Rackers, J.A.; Pratt, L.R.; Liu, C.; Rempe, S.B.; Ren, P. Thermodynamics of ion binding and occupancy in potassium channels. Chem. Sci. 2021, 12, 8920–8930. [Google Scholar] [CrossRef]
- Chaudhari, M.I.; Vanegas, J.M.; Pratt, L.; Muralidharan, A.; Rempe, S.B. Hydration Mimicry by Membrane Ion Channels. Annu. Rev. Phys. Chem. 2020, 71, 461–484. [Google Scholar] [CrossRef] [Green Version]
- Ngo, V.; Li, H.; MacKerell, A.D.; Allen, T.W.; Roux, B.; Noskov, S. Polarization Effects in Water-Mediated Selective Cation Transport across a Narrow Transmembrane Channel. J. Chem. Theory Comput. 2021, 17, 1726–1741. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Nguyen-Dang, T.T.; Tal, Y. Quantum topology of molecular charge distributions. II. Molecular structure and its change. J. Chem. Phys. 1979, 70, 4316–4329. [Google Scholar] [CrossRef]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins Struct. Funct. Bioinf. 1995, 23, 566–579. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Sherrill, C.D. Density fitting of intramonomer correlation effects in symmetry-adapted perturbation theory. J. Chem. Phys. 2010, 133, 014101. [Google Scholar] [CrossRef] [Green Version]
- Bader, R. Atoms in Molecules: A Quantum Theory; International Series of Monographs on Chemistry; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Spicher, S.; Grimme, S. Robust Atomistic Modeling of Materials, Organometallic, and Biochemical Systems. Angew. Chem. Int. Ed. 2020, 132, 15795–15803. [Google Scholar] [CrossRef]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef]
- Sobczyk, L.; Grabowski, S.J.; Krygowski, T.M. Interrelation between H-Bond and Pi-Electron Delocalization. Chem. Rev. 2005, 105, 3513–3560. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Vener, M.V.; Egorova, A.N.; Churakov, A.V.; Tsirelson, V.G. Intermolecular hydrogen bond energies in crystals evaluated using electron density properties: DFT computations with periodic boundary conditions. J. Comput. Chem. 2012, 33, 2303–2309. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Rozas, I.; Alkorta, I.; Elguero, J. Behavior of Ylides Containing N, O, and C Atoms as Hydrogen Bond Acceptors. J. Am. Chem. Soc. 2000, 122, 11154–11161. [Google Scholar] [CrossRef]
- Case, D.; Aktulga, H.; Belfon, K.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; III, T.C.; Cisneros, G.; Cruzeiro, V.; Darden, T.; et al. Amber 2021; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD–Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2017, 27, 112–128. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.; Kelley, C.; Lang, R.; Kotz, D.; Campbell, J.; Elber, G.; Woo, A. Gnuplot 5.8.2: An Interactive Plotting Program. 2019. Available online: http://www.gnuplot.info (accessed on 10 December 2021).
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. [Google Scholar] [CrossRef] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian˜16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Papajak, E.; Zheng, J.; Xu, X.; Leverentz, H.R.; Truhlar, D.G. Perspectives on Basis Sets Beautiful: Seasonal Plantings of Diffuse Basis Functions. J. Chem. Theory Comput. 2011, 7, 3027–3034. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, A.E.; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Remigio, R.D.; Richard, R.M.; et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Sokalski, W.A.; Dyguda, E.; Leszczyński, J. Quantitative Classification of Covalent and Noncovalent H-Bonds. J. Phys. Chem. B 2006, 110, 6444–6446. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll Version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Gas Phase | PCM (Water) | Gas Phase | PCM (Water) |
|---|---|---|---|---|
| Asp–Lys–Water | −16.75 | −30.80 | ||
| Lys–Water | −15.76 | −14.20 | ||
| Lys–Lys–Water | −14.18 | −12.32 | ||
| Ser–Lys–Water | −7.35 | −6.29 | ||
| Atom replacement | Optimization | |||
| Asp–Lys–Na+ | −4.96 | +4.97 | −58.15 | −33.29 |
| Lys–Na+ | +111.57 | +111.69 | divergent | +79.71 |
| Lys–Lys–Na+ | +135.78 | +167.92 | divergent | +126.35 |
| Ser–Lys–Na+ | +57.05 | +42.30 | +13.74 | +12.59 |
| Asp–Lys–Cl− | −5.32 | −21.34 | −45.67 | −27.14 |
| Lys–Cl− | −112.81 | −114.13 | −131.52 | −121.11 |
| Lys–Lys–Cl− | −140.98 | −182.10 | −187.31 | −197.01 |
| Ser–Lys–Cl− | −58.51 | −58.36 | −107.01 | −81.17 |
| System | Asp–Lys–Water | Lys–Lys–Water | ||
|---|---|---|---|---|
| Subsystem | Asp–Water | Lys–Water | Lys1–Water | Lys2–Water |
| Electrostatics | −27.30 | −20.77 | −19.41 | −2.63 |
| Exchange | 28.79 | 22.71 | 14.85 | 3.13 |
| Induction | −13.23 | −9.86 | −7.36 | −1.24 |
| Dispersion | −3.72 | −3.61 | −2.65 | −1.50 |
| Total SAPT2 | −15.47 | −11.52 | −14.57 | −2.23 |
| Ion/Molecule | Interaction Energy (kcal mol−1) | |||||
|---|---|---|---|---|---|---|
| X-ray | 30 ns | 60 ns | 90 ns | 120 ns | 150 ns | |
| K+ | −548.15 | −328.18 | −382.22 | −328.43 | −376.82 | −335.40 |
| Na+ | −543.07 | −314.68 | −317.13 | −315.82 | −318.58 | −356.17 |
| Cl− | 120.00 | 117.60 | 119.88 | 121.17 | 123.58 | 122.05 |
| Water | 0.12 | −3.51 | −2.61 | −0.35 | −3.05 | −3.88 |
| 180 ns | 210 ns | 240 ns | 270 ns | 300 ns | ||
| K+ | −326.43 | −331.65 | −326.32 | −340.27 | −332.66 | |
| Na+ | −313.50 | −318.70 | −317.40 | −317.46 | −321.73 | |
| Cl− | 117.86 | 123.49 | 119.55 | 121.58 | 125.33 | |
| Water | −2.17 | 0.65 | −3.28 | −6.33 | −0.72 | |
| System | BCP | VCP | GCP | E1HB | E2HB | ||
|---|---|---|---|---|---|---|---|
| Lys–Water (GAS) | Water–O...NH3+ | 0.0416 | 0.1357 | −0.03814 | 0.03603 | 11.967 | 9.700 |
| Lys–Water (PCM) | Water–O...NH3+ | 0.0408 | 0.1299 | −0.036450 | 0.03448 | 11.451 | 9.283 |
| Asp–Lys–Water (GAS) | Water–O...NH3+(Lys) | 0.0386 | 0.1276 | −0.03382 | 0.03286 | 10.612 | 8.846 |
| Water–H...COO−(Asp) | 0.0495 | 0.1530 | −0.04934 | 0.04380 | 15.480 | 11.791 | |
| Water–O...CH2(Lys) | 0.0055 | 0.0200 | −0.00337 | 0.00419 | 1.058 | 1.127 | |
| Asp–Lys–Water (PCM) | Water–O...NH3+(Lys) | 0.0479 | 0.1364 | −0.04457 | 0.03934 | 13.985 | 10.590 |
| Water–H...COO−(Asp) | 0.0561 | 0.1536 | −0.05701 | 0.04770 | 17.885 | 12.842 | |
| Water–O...CH2(Lys) | 0.0066 | 0.0209 | −0.00404 | 0.00464 | 1.268 | 1.248 | |
| Lys–Lys–Water (GAS) | Water–O...NH3+(Lys1) | 0.0360 | 0.1242 | −0.03118 | 0.03112 | 9.784 | 8.378 |
| (Lys1)NH3+...O(Lys2) | 0.0446 | 0.1490 | −0.04337 | 0.04031 | 13.607 | 10.851 | |
| (Lys2)O...CH2(Lys1) | 0.0078 | 0.0272 | −0.00493 | 0.00587 | 1.548 | 1.581 | |
| Lys–Lys–Water (PCM) | Water–O...NH3+(Lys1) | 0.0354 | 0.1170 | -0.02959 | 0.02942 | 9.282 | 7.920 |
| Water–O...CH2(Lys1) | 0.0063 | 0.0211 | −0.00395 | 0.00461 | 1.238 | 1.242 | |
| Water–O...CH2(Lys2) | 0.0065 | 0.0208 | −0.00395 | 0.00458 | 1.240 | 1.233 | |
| Water–O...CH2(Lys2) | 0.0069 | 0.0216 | −0.00410 | 0.00476 | 1.287 | 1.280 | |
| Ser–Lys–Water (GAS) | Water–O...HO(Ser) | 0.0309 | 0.1152 | −0.02605 | 0.02742 | 8.173 | 7.382 |
| (Ser)C=O...NH3+(Lys) | 0.0687 | 0.1544 | −0.07304 | 0.05582 | 22.917 | 15.026 | |
| Ser–Lys–Water (PCM) | Water–O...HO(Ser) | 0.0222 | 0.0755 | −0.01585 | 0.01737 | 4.972 | 4.675 |
| Water–O...NH(Lys-backbone) | 0.0073 | 0.0230 | −0.00448 | 0.00512 | 1.407 | 1.378 | |
| Water–H1...N(Ser) | 0.0365 | 0.0954 | −0.02865 | 0.02624 | 8.988 | 7.065 | |
| Water–H1...CH(Lys) | 0.0083 | 0.0280 | −0.00502 | 0.00602 | 1.576 | 1.619 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wojtkowiak, K.; Jezierska, A.; Panek, J.J. Interactions between Artificial Channel Protein, Water Molecules, and Ions Based on Theoretical Approaches. Symmetry 2022, 14, 691. https://doi.org/10.3390/sym14040691
Wojtkowiak K, Jezierska A, Panek JJ. Interactions between Artificial Channel Protein, Water Molecules, and Ions Based on Theoretical Approaches. Symmetry. 2022; 14(4):691. https://doi.org/10.3390/sym14040691
Chicago/Turabian StyleWojtkowiak, Kamil, Aneta Jezierska, and Jarosław J. Panek. 2022. "Interactions between Artificial Channel Protein, Water Molecules, and Ions Based on Theoretical Approaches" Symmetry 14, no. 4: 691. https://doi.org/10.3390/sym14040691