3.2. Synthesis and Spectral Data
3.2.1. Synthesis of 1-Phenylphosphol-2-ene 1-oxide (1)
To a 300 mL (4.02 mol) of butadiene condensed in a 2 L round-bottom flask cooled to −78 °C was added 2 g of 2,6-di-tert-butyl-2-methylphenol (polymerization inhibitor) followed by a solution of 136 mL (179 g, 1 mol) of P,P-dichlorophenylphosphine dissolved in 650 mL of dry petroleum ether. The flask was tightly closed and left at rt for 3 months. After this time, 300 mL of H2O was slowly added to the stirred reaction mixture. Then, 70 mL of 5% aqueous solution of NaHCO3 and ca. 160 mL of 30% of aqueous solution of NaOH were gradually added with care to reach the pH range of 6.5–7.0. Then, the resulting layers were separated and the water layer was washed four times with CHCl3 (4 × 100 mL). The combined organic layers were dried over MgSO4, concentrated under reduced pressure and distilled at 167–192 °C/2 mmHg to afford 110 g (61.8%) of 1-phenylphosphol-2-ene 1-oxide (1) which solidified after cooling into a white solid: mp = 79 °C. 1H NMR (300 MHz) δ 2.05–2.25 (m, 2H), 2.65–2.8 (m, 1H), 2.9–3.05 (m, 1H), 6.2–6.4 (dm, 1H), 7.0–7.25 (dm, 1H), 7.45–7.55 (m, 3H), 7.6–7.75 (m, 1H), 13C NMR (75 MHz) δ 26.1 (d, J = 71.77), 30.45 (d, J = 10.57), 126.0 (s), 127.3 (s), 129 (d, J = 12.09), 130.85 (d, J = 10.43), 132.15 (d, J = 2.94), 133.3 (s), 134.6 (s), 153.1(d, J = 24.38), 31P NMR (121 MHz) δ 61.2. Elemental Anal. Calcd. for C10H11OP: C 67.41, H 6.22, found C 67.3, H 6.31. This product typically contains a small amount of isomeric 1-phenylphosphol-3-lene 1-oxide (typically less then 3–5% as revealed by its 1H and 31P NMR spectra by the presence of pertinent signals: 1H NMR (300 MHz) δ 6.0 (d, 3JP-H = 40 H); 31P NMR (121 MHz) δ 56.3). It can be recrystallized from toluene when needed, but, for the purpose of resolution, it has been used as such.
3.2.2. Resolution of 1-Phenylphosphol-2-ene 1-oxide (1)
Step 1. Reduction of rac-1.
In a 250 mL three-neck round-bottom flask equipped with a reflux condenser and an addition funnel was placed 32.5 g (0.183 mol) of racemic 1-phenylphosphol-2-ene 1-oxide (1) dissolved in 20 mL of dry benzene. The solution was degassed, purged with argon, and heated to 60 °C. Then, 25.15 mL (0.2 mol) of PhSiH3 dissolved in 20 mL of benzene was added dropwise over a period of 2 h. After the addition of PhSiH3, the reaction mixture was heated at 60 °C for additional 40 h. After this time, the reaction mixture was allowed to cool to room temperature, the volatiles were removed under reduced pressure and the residue was distilled under reduced pressure at 80–100 °C/0.8 mmHg to yield 26 g (88%) of 1-phenylphosphol-2-ene (2), as a colorless oil which was directly used for preparation of salts in step 2.
Step 2. Quaternization of 1-Phenylphosphol-2-ene (2) by L-menthyl bromoacetate.
In a 2 L round-bottom flask was dissolved 44.5 g (0.16 mol) of L-menthyl bromoacetate in 25 mL of dry ethanol and 780 mL of dry ethyl acetate. The solution was degassed and placed under argon atmosphere. Then, 26 g (0.16 mol) of rac-2 dissolved in 40 mL of ethyl acetate was added dropwise over a period of 8 h with magnetic stirring. During the addition, after ca. 2 h, a white precipitate started to accumulate slowly. After the addition was completed, the reaction mixture was stirred at room temperature overnight. The formed crystalline precipitate was filtered off and was found to contain the (SP) epimer of 3 in great predominance (>90%). Crystallization of the precipitate from AcOEt-EtOH (10:1) was repeated (1–4 times) until 1H NMR monitoring showed that the resulting crystals contained only a single, diastereomerically pure, salt (SP)-3. The second P-epimer, (RP)-3, was obtained by repeated recrystallizations of the residue from dry benzene-hexane 10:1 (or from toluene), until 1H NMR monitoring showed that the resulting crystals contained only the single, diastereomerically pure, salt (RP)-3:
(SP)-3: 25.9 g (37%), white crystals: mp = 197–199 °C, [α]D = −13.65 (c 2.15, CHCl3). 1H NMR (300 MHz) δ 0.59 (d, J = 6.9, 3H), 0.77 (d, J = 6.9, 3H), 0.83 (d, J = 6.5, 3H), 0.8–1.0 (m, 2H), 1.2–1.3 (m, 1H), 1.3–1.4 (m, 1H), 1.45–1.55 (m, 1H), 1.6–1.7 (m, 2H), 1.7–1.75 (m, 1H), 2.7–2.85 (m, 1H), 3.1–3.2 (m, 1H), 3.45–3.6 (m,1H), 3.75–3.9 (m, 1H), 4.64 (dt, J = 4.4, J = 10.9, 1H), 4.67 (dd, J = 14.0, J = 17.3, 1H), 4.9 (dd, J = 13.5, J = 17.3, 1H), 6.75 (dddd, J = 2.2, J = 2.3, J = 8, J = 20.3, 1H), 7.53 (dddd, J = 2.6, J = 2.7, J = 8.0, J = 36.4, 1H), 7.55–7.65 (m, 2H), 7.7–7.75 (m, 1H), 7.9–8.1 (m, 2H); 13C NMR (75 MHz) δ 15.8 (s), 20.8 (s), 21.3 (s), 21.8 (s), 22.9 (s), 25.8 (s), 31.4 (s), 33.8 (d, J = 34), 33.83 (s), 34.1 (d, J = 12), 40.5 (s), 113.35 (d, J = 81), 120.3 (d, J = 88), 129.95 (d, J = 14), 132.18 (d, J = 11), 134.5 (s), 162.9 (d, J = 23), 164.93 (d, J = 4); 31P NMR (121 MHz) δ 52.91, Elemental Anal. for C22H32BrO2P: calcd. C 60.14, H 7.34, found C 59.95, H 7.4.
(RP)-3: 23.1 g (33%), white tiny needles: mp = 76–77 °C, [α]D = −55.55 (c 2.14, CHCl3). 1H NMR (300 MHz) δ 0.52 (d, J = 6.9, 3H), 0.78 (d, J = 6.9, 3H), 0.86 (d, J = 6.3, 3H), 0.8–1.0 (m, 2H), 1.2–1.3 (m, 1H), 1.3–1.45 (m, 1H), 1.55–1.55 (m, 1H), 1.6–1.7 (m, 2H), 1.75–1.8 (m, 1H), 2.7–2.85 (m, 1H), 3.1–3.2 (m, 1H), 3.45–3.55 (m,1H), 3.8–3.9 (m, 1H), 4.55 (dt, J = 4.5, J = 11.2, 1H), 4.8 (d, J = 13.5, 2H), 6.73 (dddd, J = 1.8, J = 2.1, J = 8, J = 28, 1H), 7.58 (dddd, J = 2.1, J = 2.5, J = 8.4, J = 37.3, 1H), 7.6–7.7 (m, 2H), 7.7–7.75 (m, 1H), 7.95–8.1 (m, 2H). 13C NMR (75 MHz) δ 15.8 (s), 20.7 (s), 20.9 (d, J = 57), 21.85 (s), 22.9 (s), 25.9 (s), 31.4 (s), 33.75 (d, J = 34), 33.83 (s), 34.09 (d, J = 13), 40.4 (s), 46.6 (s), 113.0 (d, J = 80), 120.3 (d, J = 84), 129.95 (d, J = 12.5), 132.1 (d, J = 11), 134.6 (s), 163.1 (d, J = 22), 165.02 (d, J = 4). 31P NMR (121 MHz) δ 53.03. Elemental Anal. For C22H32BrO2P: calcd. C 60.14, H 7.34, found C 59.86, H 7.41.
Step 3. Hydrolysis of the resolved phosphonium salts (SP)-3 and (RP)-3.
In a 500 mL round-bottom flask was dissolved 25 g (56.9 mmol) of (SP)-3 in 45 mL of CH2Cl2. To the solution was added 130 mL of H2O and 120 mL of 1M NaOH, and the reaction mixture was stirred at rt for 12 h. The two phases were separated and the water phase was washed twice with CH2Cl2 (2 × 30 mL). The combined dichloromethane layers were dried over MgSO4 and concentrated. The residue was purified by crystallization from toluene which afforded 9.1 g (90%) of (SP)-1-phenylphosphol-2-ene-1-oxide (SP-1) as white crystals, mp = 83–84 °C, [α]D = +306.6 (c 1.3, CHCl3).
The (RP)-1-phenylphosphol-2-ene-1-oxide (RP-1) was prepared from (RP)-3 analogously as described for the SP isomer. White crystals: mp = 84–85 °C, [α]D = −302.4 (c 1.42, CHCl3).
The enantiomeric purity of the synthesized enantiomers of phospholene oxide
1 was determined by running their
1H and
31P NMR spectra in the presence of equimolar amounts of (
S)-3,5-dinitro-
N-α-phenylethyl-benzamide as a chiral solvating agent (Kagan’s reagent) according to the described procedure [
56]. As no presence of signals of the opposite enantiomer could be detected in either spectrum, the enantiomeric purity of both (
SP)-
1 and (
RP)-
1 was assigned to be at least 98% ee.
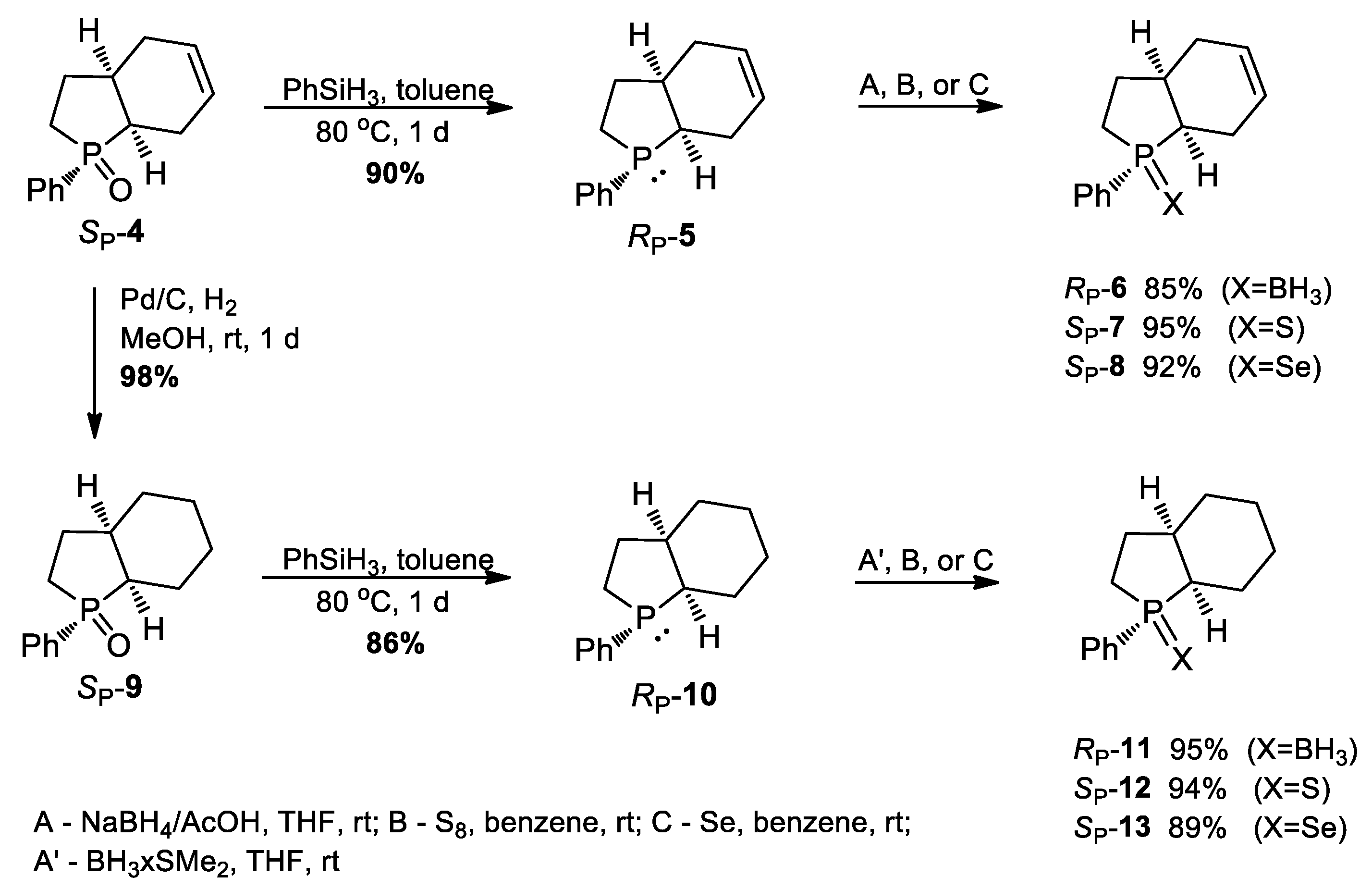
3.2.3. Synthesis of (SP)-1-Phenyl-2,3,3a,4,7,7a-heksahydrophosphindole 1-oxide (SP-4): Typical Procedure
Two grams (0.011 mol) of (SP)-1, 3 g (0.056 mol) of 1.3-butadiene, and 0.019 g (0.089 mmol) of 2,6-di-tert-butyl-4-methylphenol (polymerization inhibitor) dissolved in 7 mL of toluene were placed in a tightly closed glass ampoule and heated at 200 °C for 48 h. During that time, two additional portions of the diene were added after controlling the progress of cycloaddition by TLC. After completion of heating, the reaction mixture was dissolved in 75 mL of methanol and filtered to remove the polymeric side-products formed. Then, 150 mL of 15% hydrochloric acid was added and the resulting mixture was washed with benzene (2 × 75 mL). The organic layers were combined, dried over anhydrous MgSO4, filtered, and evaporated under reduced pressure. The residue was purified on silica gel column using ethyl acetate/methanol (25:1) as eluent to give a white solid which was recrystallized from toluene/hexane mixture to yield 1.3 g (51%) of cycloadduct (SP)-4 as white crystals. Mp = 95–96 °C, [α]D = −42.30 (c 1.13, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 7.80–7.73 (m, 2H); 7.52–7.28 (m, 3H); 5.81–5.70 (m, 2H); 2.52–2.03 (m, 10H). 13C NMR (CDCl3, 75.5 MHz): δ 134.3 (d, J = 88.1 Hz); 131.5 (d, J = 2.8 Hz); 130.0 (d, J = 9.2 Hz); 128.5 (d, J = 11.2 Hz); 124.7 (d, J = 0.9 Hz); 124.5 (d, J = 6.7 Hz); 35.7 (d, J = 69 Hz); 34.0 (d, J = 12.2 Hz); 27.9 (d, J = 5.6 Hz); 26.7 (d, J = 8.1 Hz); 25.8 (d, J = 65 Hz); 19.2 (d, J = 3.2 Hz). 31P NMR (CDCl3, 121.5 MHz): δ 63.29 (s). Elemental Anal. for C14H17OP: calcd. C, 72.39; H, 7.37; found C, 72.47; H, 7.34.
3.2.4. Synthesis of (RP)-1-Phenyl-2,3,3a,4,7,7a-heksahydrophosphindole (5): Typical Procedure
In a Schlenk flask protected from air was placed 1 g (0.0043 mol) of (SP)-4 and 5 mL of toluene and then 0.7 g (0.0064 mol) of PhSiH3 was added. The mixture was heated under argon atmosphere at 75–80 °C for 24 h. Then, the reaction mixture was concentrated and distilled under reduced pressure to give 0.83 g (90%) of phosphine (RP)-5 as a colorless oil. Bp = 160 °C/0.2 mmHg, [α]D = +75.22 (c 0.9, CHCl3). 31P NMR (CDCl3, 121.5 MHz): δ 3.84 (s). 1H NMR (CDCl3, 300 MHz): δ 7.50–7.38 (m, 2H); 7.34–7.21 (m, 3H); 5.77–5.53 (m, 2H); 2.40–1.97 (m, 9H); 1.74–1.39 (m, 1H). 13C NMR (CDCl3, 75.5 MHz): δ 141.8 (d, J = 23 Hz); 130.7 (d, J = 15.4 Hz); 128.3 (d, J = 5.1 Hz); 127.5 (s); 125.4 (d, J = 13.8 Hz); 125.4 (d, J = 0.9 Hz); 38.3 (d, J = 8 Hz); 37.6 (d, J = 2.8 Hz); 31.4 (d, J = 3.4 Hz); 26.5 (d, J = 2.4 Hz); 25.7 (d, J = 28.6 Hz); 22.7 (d, J = 14 Hz). The configuration and high enantiomeric purity of (RP)-5 was confirmed by its oxidation by H2O2 which afforded back (SP)-4 of [α]D = −42.26 (c 0.97, CHCl3).
3.2.5. Synthesis of (RP)-1-Phenyl-2,3,3a,4,7,7a-heksahydrophosphindole-borane (6): Typical Procedure
To a solution of 1 g (0.0046 mol) of (RP)-5 in dry THF (4 mL) kept under argon at 0 °C was added 0.26 g (0.0069 mol) of solid NaBH4 in one portion. Then, 0.46 g (0.0077 mol) of acetic acid dissolved in THF (1.9 mL) was added dropwise within 30 min. The resulting mixture was left at room temperature for 1 h, and the conversion was checked by TLC. Then, water (4.5 mL) was added slowly, followed by 0.44 g (0.42 mL) of acetic acid dissolved in 5.6 mL of water. The reaction mixture was evaporated and the residue was passed through a silica gel column using CH2Cl2/hexane (2:3) to give 0.9 g (85%) of borane (RP)-6 as white crystals. Mp = 62–63 °C, [α]D = −10.46 (c 1.04, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 7.75–7.68 (m, 2H); 7.49–7.40 (m, 3H); 5.75–5.60 (m, 2H); 2.50–1.85 (m, 10H); 1.10–0.20 (m, 3H, BH3). 13C NMR (CDCl3, 75.5 MHz): δ 132 (d, J = 10.1 Hz); 131.6 (d, J = 8.4 Hz); 131.2 (d, J = 2.4 Hz); 129.2 (d, J = 9.4 Hz); 125 (d, J = 8.7 Hz); 37.4 (d, J = 3.1 Hz); 35.4 (d, J = 35 Hz); 30.6; 26.7 (d, J = 5.1 Hz); 23.0 (d, J = 35.2 Hz); 22.1 (d, J = 4.9 Hz). 31P NMR (CDCl3, 121.5 MHz): δ 40.11. Elemental Anal. For C14H20BP: calcd. C, 73.08; H, 8.76; found C, 72.99; H, 8.36.
3.2.6. Synthesis of (SP)-1-Phenyl-2,3,3a,4,7,7a-heksahydrophosphindole 1-sulfide (7): Typical Procedure
To a solution of 1 g (0.0046 mole) of phosphine (RP)-5 in 6 mL of benzene under argon was added 0.14 g (0.0046 mol) of sublimed sulfur. The reaction mixture was stirred at room temperature for 24 h. After this time, the reaction mixture was concentrated and the crude product was purified by column chromatography using CH2Cl2/hexane (2:3) followed by crystallization from methanol to yield 1.10 g (96%) of sulfide 7 as white crystals. Mp = 90–91 °C, [α]D = −19.08 (c 1.13, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 7.93–7.85 (m, 2H); 7.55–7.45 (m, 3H); 5.83–5.70 (m, 2H); 2.71–2.62 (m, 1H); 2.50–1.95 (m, 9H). 13C NMR (CDCl3, 75.5 MHz): δ 135 (d, J = 70.1 Hz); 131.7 (d, J = 3 Hz); 130.8 (d, J = 9.8 Hz); 129 (d, J = 11.5 Hz); 125.2 (d, J = 7.6 Hz); 125.1 (d, J = 1.3 Hz); 39.0 (d, J = 53 Hz); 36.3 (d, J = 10.3 Hz); 32.5 (d, J = 52.7 Hz); 29.9 (d, J = 3.7 Hz); 27.3 (d, J = 7.2 Hz). 31P NMR (CDCl3, 121.5 MHz): δ 65.56 (s). Elemental Anal. For C14H17SP: calcd. 67.71; H, 6.89; found C, 67.57; H, 6.85.
3.2.7. Synthesis of (SP)-1-Phenyl-2,3,3a,4,7,7a-heksahydrophosphindole 1-Selenide (8): Typical Procedure
To a solution of 1 g (0.0046 mole) of phosphine (RP)-5 in 6 mL of benzene under argon was added 0.36 g (0.0046 mol) of selenium. The reaction mixture was magnetically stirred at room temperature for 24 h. After this time, the reaction mixture was concentrated and the crude product was purified by column chromatography using CH2Cl2/hexane (2:3) followed by crystallization from methanol to yield 1.25 g (92%) of selenide 8 as white crystals. Mp = 63 °C, [α]D = −15.05 (c 1.65, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 7.92–7.84 (m, 2H); 7.50–7.45 (m, 3H); 5.82–5.69 (m, 2H); 2.89–2.80 (m, 1H); 2.52–2.04 (m, 9H). 13C NMR (CDCl3, 75.5 MHz): δ 132 (d, J = 70.1 Hz); 131.7 (d, J = 3 Hz); 131.1 (d, J = 9.8 Hz); 129 (d, J = 11.5 Hz); 125.1 (d, J = 8.5 Hz); 125.0 (d, J = 1.5 Hz); 38.5 (d, J = 45.8 Hz); 36.6 (d, J = 9.5 Hz); 32.9 (d, J = 46.3 Hz); 30.4 (d, J = 2.7 Hz); 27.2 (d, J = 7.2 Hz); 23.3. 31P NMR (CDCl3, 121.5 MHz): δ 53.81 (s). Elemental Anal. For C14H17SeP: calcd. C, 56.95; H, 5.80; found C, 56.79; H, 5.70.
3.2.8. Synthesis of (SP)-1-Phenyl-octahydrophosphindole 1-oxide (9): Typical Procedure
To a flask containing 1 g (0.0043 mol) of (SP)-5 dissolved in 15 mL of methanol was added 0.045 g (0.00043 mol) of Pd/C. Argon was passed through the flask for 10 min., and the flask was capped with a balloon filled with hydrogen. Then, the reaction mixture was magnetically stirred at room temperature for 24 h, filtered through Celite and the filtrate was evaporated. The resulting solid residue was recrystallized from toluene/hexane to give 0.99 g (98%) of saturated oxide (SP)-9 as white crystals. Mp = 96–98 °C, [α]D = −27.65(c 1.34, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 7.79–7.71 (m, 2H); 7.54–7.43 (m, 3H); 2.29–2.03 (m, 5H); 1.93–1.59 (m, 7H); 1.44–1.37 (m, 2H). 13C NMR (CDCl3, 75.5 MHz): δ 135.1 (d, J = 87 Hz); 131.4 (d, J = 2.8 Hz); 129.9 (d, J = 9.2 Hz); 128.5 (d, J = 11.1 Hz); 39.7 (d, J = 68.2 Hz); 38.2 (d, J = 11.4 Hz); 28.10 (d, J = 6.8 Hz); 27.5 (d, J = 64.7 Hz); 27.2 (d, J = 5.8 Hz); 24.3 (d, J = 6.9 Hz); 22.6; 21.50 (d, J = 3.3 Hz). 31P NMR (CDCl3, 121.5 MHz): δ 60.01 (s). Elemental Anal. For C14H19OP: calcd. C, 71.77; H, 8.17; found C, 71.62; H, 8.10.
3.2.9. Synthesis of (RP)-1-Phenyl-octahydrophosphindole (10)
(
RP)-
10 was obtained in 86% yield according to typical procedure described in
Section 3.2.4. (
RP)-
10: a colorless oil, bp = 136 °C/0.2 mmHg, [α]
D = +26.61 (
c 1.1, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.50–7.43 (m, 2H); 7.36–7.26 (m, 3H); 2.32–2.17 (m, 3H); 2.05–1.78 (m, 5H); 1.60–1.41 (m, 6H).
13C NMR (CDCl
3, 75.5 MHz):
δ 135(d,
J = 11.6 Hz); 131.0 (d,
J = 16.5 Hz); 128.6 (d,
J = 5.4 Hz); 127.8 (s); 44.5 (d,
J = 8.9 Hz); 41.4 (d,
J = 1.4 Hz); 31.6 (d,
J = 3.1 Hz); 28.1 (d,
J = 2.2 Hz); 27.8 (d,
J = 26.1 Hz); 25.5 (d,
J = 13.1 Hz); 23.8 (d,
J = 12.1 Hz); 23.1.
31P NMR (CDCl
3, 121.5 MHz):
δ -1.38 (s). The configuration and high enantiomeric purity of (
RP)-
10 was confirmed by its oxidation by H
2O
2 which afforded back (
SP)-
9 of [α]
D = −27.71 (
c 1.1, CHCl
3).
3.2.10. Synthesis of (RP)-1-Phenyl-octahydrophosphindole-borane (11): Typical Procedure
To a solution of 1 g (0.0046 mol) of phosphine (RP)-10 in 6 mL of benzene under argon atmosphere was added 0.50 g (0.0066 mol) of BH3-SMe2 complex and the reaction mixture was stirred at room temperature for 24 h. At the end of the reaction, the mixture was concentrated and the crude product was passed through a silica gel column using CH2Cl2/hexane (2:3) as eluent to give 0.96 g (95%) of phosphine-borane (RP)-11 white solid. Mp = 68–69 °C, [α]D = −5.04 (c 1.15, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 7.74–7.86 (m, 2H); 7.47–7.45 (m, 3H); 2.29–1.32 (m, 14H); 1.25–0.40 (m, 3H, BH3). 13C NMR (CDCl3, 75.5 MHz): δ 132.7 (d, J = 46.5 Hz); 131.6 (d, J = 8.4 Hz); 131.2 (d, J = 2.5 Hz); 129.1 (d, J = 9.4 Hz); 40.9 (d, J = 3.0 Hz); 39.7 (d, J = 33.8 Hz); 29.5; 27.8 (d, J = 5 Hz); 25.2 (d, J = 10.5 Hz); 24.3 (d, J = 35.7 Hz); 24.0 (d, J = 3.9 Hz); 22.3. 31P NMR (CDCl3, 121.5 MHz): δ 37.3. Elemental Anal. For C14H22BP: calcd. C, 72.44; H, 9.55; found C, 72.23; H, 9.55.
3.2.11. Synthesis of (SP)-1-Phenyl-octahydrophosphindole 1-sulfide (12)
(
SP)-
12 was obtained in 94% yield according to typical procedure described in
Section 3.2.6. (
SP)-
12: white crystals, mp = 82–84 °C, [α]
D = − 4.03 (
c 1.15, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.94–7.86 (m, 2H); 7.50–7.47 (m, 3H); 2.68–2.59 (m, 1H); 2.42–2.19 (m, 4H); 2.00–1.90 (m, 3H); 1.73–1.65 (m, 3H); 1.51–1.48 (m, 2H); 1.35–1.29 (m, 1H).
13C NMR (CDCl
3, 75.5 MHz):
δ 135.5 (d,
J = 69.5 Hz); 131.6 (d,
J = 3 Hz); 130.6 (d,
J = 10 Hz); 129 (d,
J = 11.4 Hz); 41.8 (d,
J = 52 Hz); 39.5 (d,
J = 9.9 Hz); 34.7 (d,
J = 52.2 Hz); 28.3 (d,
J = 8.2 Hz); 28.0 (d,
J = 3.7 Hz); 25.1 (d,
J = 10.4 Hz); 23.5 (d,
J = 1.6 Hz); 22.0.
31P NMR (CDCl
3, 121.5 MHz):
δ 64,87 (s). Elemental Anal. For C
14H
19SP: calcd. C, 67.17; H, 7.65; found C, 67.09; H, 7.55.
3.2.12. Synthesis of (SP)-1-Phenyl-octahydrophosphindole 1-selenide (13)
(
SP)-
13 was obtained in 89% yield according to typical procedure described in
Section 3.2.7. (
SP)-
13: white crystals, mp = 67–68 °C, [α]
D = −0.95 (
c 0.97, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.93–7.83 (m, 2H); 7.49–7.47 (m, 3H); 2.84–2.74 (m, 1H); 2.56–1.35 (m, 13H).
13C NMR (CDCl
3, 75,5 MHz):
δ 134.5 (d,
J = 61.4 Hz); 131.6 (d,
J = 3 Hz); 131.0 (d,
J = 9.8 Hz); 129.0 (d,
J = 11.3 Hz); 41.3 (d,
J = 44.8 Hz); 39.5 (d,
J = 8.8 Hz); 33.9 (d,
J = 45.9 Hz); 28.3 (d,
J = 2.8 Hz); 28.0 (d,
J = 8.3 Hz); 25.3 (d,
J = 11.7 Hz); 25.0; 21.7.
31P NMR (CDCl
3, 121.5 MHz):
δ 52.96 (s). Elemental Anal. For C
14H
19SeP: calcd C, 56.57; H, 6.44; found C, 56.40; H, 6.40.
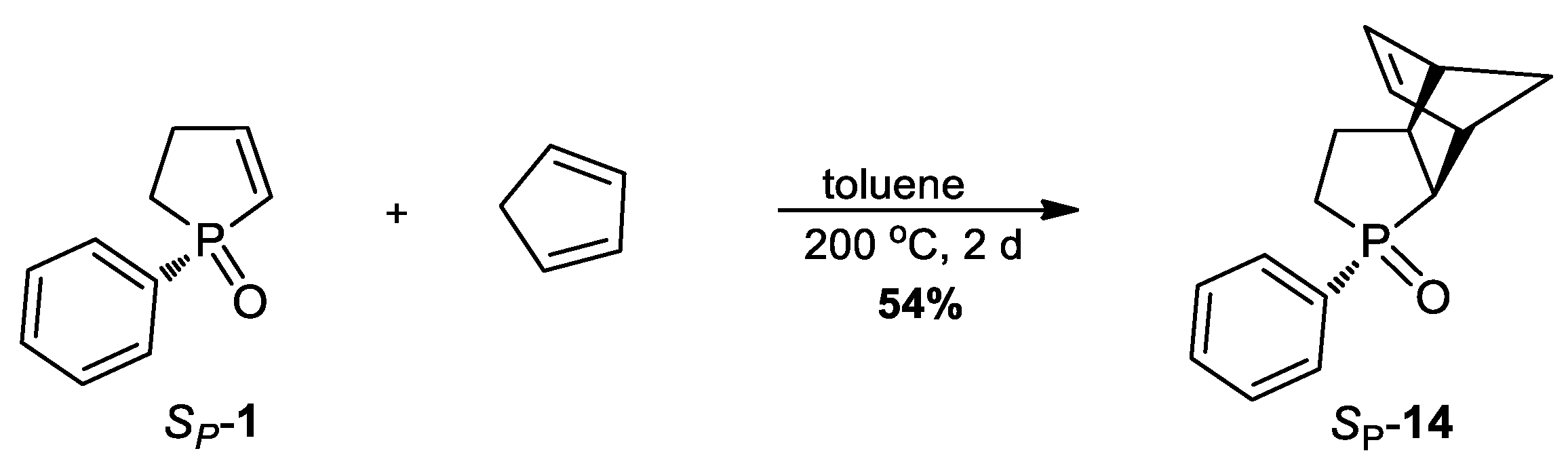
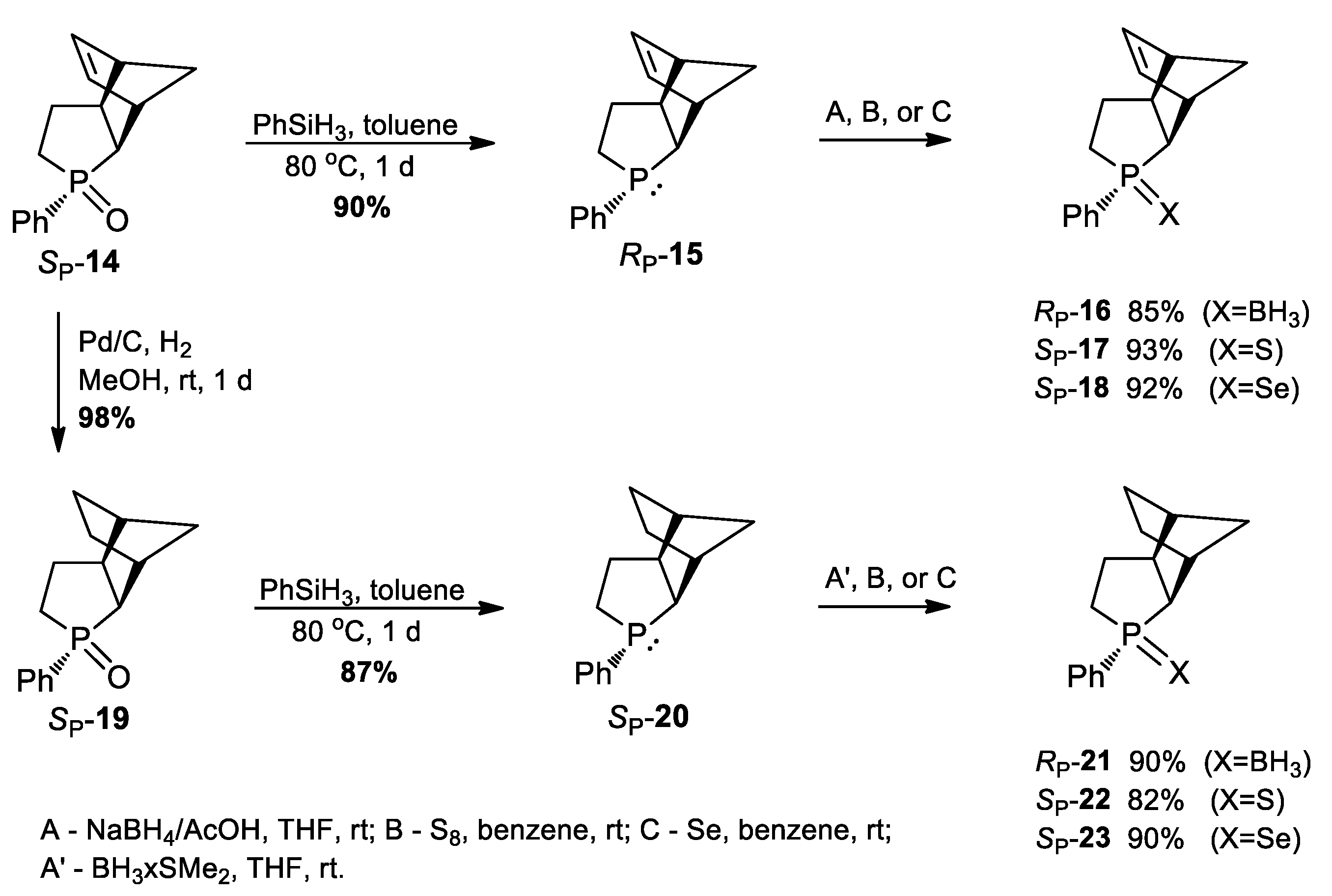
3.2.13. Synthesis of (SP)-3-Phenyl-3-phosphatricyclo[5.2.1.02,6]dec-8-ene 3-oxide (14)
(
SP)-
14 was obtained in 54% yield according to typical procedure described in
Section 3.2.3. (
SP)-1
4: white crystals, mp = 100–101 °C, [α]
D = −21.23 (
c 1.02, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.72–7.66 (m, 2H); 7.49–7.46 (m, 3H); 6.64–6.61 (m, 1H); 6.25–6.22 (m, 1H); 3.32 (s, 1H); 3.04 (m, 2H); 2.73–2.70 (m, 1H); 1.96–1.85 (m, 5H); 1.58–1.55 (m, 1H).
13C NMR (CDCl
3, 75.5 MHz):
δ 138.4 (d,
J = 4.97 Hz); 135.0 (d,
J = 89.1 Hz); 133.7(s); 131.5 (d,
J = 2.7 Hz); 129.8 (d,
J = 9.1 Hz); 128.7 (d,
J = 11.0 Hz); 53.0 (d,
J = 9.6 Hz); 48.2 (d,
J = 2.1 Hz); 46.4 (d,
J = 2.1 Hz); 45.1 (d,
J = 30.8 Hz); 44.5 (d,
J = 32.6 Hz); 29.4 (d,
J = 67.3 Hz); 23.5 (d,
J = 9.8 Hz).
31P NMR (CDCl
3, 121.5 MHz):
δ 61.43 (s). Elemental Anal. for C
15H
17OP: calcd. C, 73.75; H, 7.01; found C, 73.60; H, 6.98.
3.2.14. Synthesis of (RP)-3-Phenyl-3-phosphatricyclo[5.2.1.02,6]dec-8-ene (15)
(
RP)-
15 was obtained in 89% yield according to typical procedure described in
Section 3.2.4. (
RP)-
15: a colorless oil, bp = 150 °C/0.2 mmHg, [α]
D = −16.8 (
c 0.9, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.42–7.37 (m, 2H); 7.34–7.24 (m, 3H); 6.38–6.35 (m, 1H); 6.16–6.13 (m, 1H); 3.18–3.17 (m, 2H); 3.04–3.0 (m, 1H); 2.90 (s, 1H); 1.85–1.78 (m, 4H); 1.47–1.65 (m, 2H).
13C NMR (CDCl
3, 75.5 MHz):
δ 141.6 (d,
J = 19.4 Hz); 136.7 (d,
J = 9.2 Hz); 135.5(s); 131.1 (d,
J = 15.4 Hz); 128.3 (d,
J = 5.3 Hz); 127.3 (s); 52.9 (d,
J = 6.2 Hz); 51.2 (d,
J = 9.1 Hz); 50.4 (d,
J = 3.4 Hz); 48.5 (s); 47.3 (d,
J = 21.3 Hz); 30.1 (d,
J = 4.1 Hz); 28.8 (d,
J = 12.3 Hz).
31P NMR (CDCl
3, 121.5 MHz):
δ -6.84 (s). The configuration and high enantiomeric purity of (
RP)-
15 was confirmed by its oxidation by H
2O
2 which afforded back (
SP)-
14 of [α]
D = −21.2 (
c 1.21, CHCl
3).
3.2.15. Synthesis of (RP)-3-Phenyl-3-phosphatricyclo[5.2.1.02,6]dec-8-ene-borane (16)
(RP)-16: a colorless oil, [α]D = −32.1 (c 1.04, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 7.70–7.62 (m, 2H); 7.46–7.43 (m, 3H); 6.58–6.55 (m, 1H); 6.19–6.16 (m, 1H); 3.32–3.21 (m, 2H); 3.01–2.95 (m, 2H); 1.92–1.91 (m, 4H); 1.6–1.58 (m, 1H); 1.56–1.48 (m, 1H); 1.01–0.66 (m, 3H, BH3). 13C NMR (CDCl3, 75.5 MHz): δ 137.8 (d, J = 3.78 Hz); 134.4 (s); 131.3 (d, J = 7.8 Hz); 130.6 (d, J = 2.4 Hz); 128.8 (d, J = 9.1 Hz); 53.3 (d, J = 9.2 Hz); 48.4 (d, J = 1.9 Hz); 48.0 (d, J = 1.5 Hz);46.6 (d, J = 4.5 Hz); 46.0 (s); 28.1 (d, J = 33.6 Hz); 27.7 (d, J = 2.2 Hz). 31P NMR (CDCl3, 121.5 MHz): δ 35.89. Elemental Anal. For C15H20BP: calcd. C, 74.41; H, 8.32; found C, 74.31; H, 8.29.
3.2.16. Synthesis of (SP)-3-Phenyl-3-phosphatricyclo[5.2.1.02,6]dec-8-ene 3-sulfide (17)
(
SP)-
17 was obtained in 93% yield according to typical procedure described in
Section 3.2.6. (
SP)-
17: white crystals, mp = 103–105 °C, [α]
D = −33.01 (
c 1.03, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.87–7.79 (m, 2H); 7.48–7.47 (m, 3H); 6.81–6.78 (m, 1H); 6.23–6.21 (m, 1H); 3.36 (s, 1H); 3.20–3.14 (m, 1H); 3.07–3.02 (m, 2H); 2.10–1.91 (m, 4H); 1.64–1.49 (m, 2H).
13C NMR (CDCl
3, 75.5 MHz):
δ 138.1 (d,
J = 5.1 Hz); 135.1 (d,
J = 71.1 Hz); 133.75 (s); 131.2 (d,
J = 2.85 Hz); 130.4 (d,
J = 9.54 Hz); 128.7 (d,
J = 11.2 Hz); 52.6 (d,
J = 10.3 Hz); 49.5 (d,
J = 60.89 Hz); 47.6 (d,
J = 10.8 Hz); 47.2 (s); 35.7 (d,
J = 52.8 Hz); 25.9 (d,
J = 6.0 Hz).
31P NMR (CDCl
3, 121.5 MHz):
δ 62.19 (s). Elemental Anal. For C
15H
17SP: calcd. C, 69.20; H, 6.58; found C, 69.10; H, 6.55.
3.2.17. Synthesis of (SP)-3-Phenyl-3-phosphatricyclo[5.2.1.02,6]dec-8-ene 3-selenide (18)
(
SP)-
17 was obtained in 92% yield according to typical procedure described in
Section 3.2.7. (1.2:1) as eluent followed by crystallization from methanol to yield 1.24 g (92%) of selenide (
SP)-
18: white crystals, mp = 93–94 °C, [α]
D = −39.98 (
c 1.09, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.87–7.80 (m, 2H); 7.46–7.45 (m, 3H); 6.90–6.88 (m, 1H); 6.23–6.20 (m, 1H); 3.39 (s, 1H); 3.25–3.23 (m, 1H); 3.22–3.10 (m, 2H); 2.21–2.14 (m, 2H); 1.95–1.90 (m, 2H); 1.58–1.52 (m, 2H).
13C NMR (CDCl
3, 75.5 MHz):
δ 137.7 (d,
J = 5.3 Hz); 133.9 (d,
J = 63 Hz); 133.8 (s); 131.3 (d,
J = 2.9 Hz); 130.9 (d,
J = 9.5 Hz); 128.7 (d,
J = 11.1 Hz); 52.6 (d,
J = 10.5 Hz); 49.8 (d,
J = 53.6 Hz); 49.4 (d,
J = 1.9 Hz); 48.7 (d,
J = 9.2 Hz); 47.7 (s); 36.1 (d,
J = 45.9 Hz); 27.1 (d,
J = 4.2 Hz).
31P NMR (CDCl
3, 121.5 MHz):
δ 47.22 (s). Elemental Anal. For C
15H
17SeP: calcd. C, 58.63; H, 5.57; found C, 58.45; H, 5.53.
3.2.18. Synthesis of (SP)-3-Phenyl-3-phosphatricyclo[5.2.1.02,6]decane 3-oxide (19)
(
SP)-
19 was obtained in 98% yield according to typical procedure described in
Section 3.2.8. The resulting solid residue was recrystallized from toluene/hexane to give 0.98 g (98%) of saturated oxide (
SP)-
19: white crystals, mp = 105–106 °C, [α]
D = −20.57 (
c 1.08, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.73–7.65 (m, 2H); 7.54–7.43 (m, 3H); 2.79–2.67 (m, 2H); 2.40–2.30 (m, 3H); 2.12–2.00 (m, 3H); 1.85–1.75 (m, 2H); 1.69–1.52 (m, 5H).
13C NMR (CDCl
3, 75.5 MHz):
δ 135.7 (d,
J = 88.9 Hz); 131.7 (d,
J = 2.6 Hz); 129.9 (d,
J = 8.9 Hz); 129.0 (d,
J = 10.8 Hz); 44.2 (d,
J = 15.7 Hz); 43.8 (d,
J = 12.0 Hz); 43.4 (d,
J = 2.3 Hz); 42.6 (d,
J = 73.6 Hz); 40.7 (d,
J = 2.4 Hz); 29.4 (d,
J = 65.3 Hz); 25.4 (d,
J = 6.2 Hz); 23.5; 21.45 (d,
J = 10.4 Hz).
31P NMR (CDCl
3, 121.5 MHz):
δ 60.03 (s). Elemental Anal. For C
15H
19OP: calcd. C, 73.15; H, 7.77; found C, 73.15; H, 7.74.
3.2.19. Synthesis of (RP)-3-Phenyl-3-phosphatricyclo[5.2.1.02,6]decane (20)
(
RP)-
20 was obtained in 87% yield according to typical procedure described in
Section 3.2.4. (
RP)-
20: a colorless oil, bp = 210 °C/0.3 mmHg, [α]
D = −11.62 (
c 0.9, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.62–7.12 (m, 5H); 2.78–2.67 (m, 2H); 2.44 (s, 1H); 2.16 (s, 1H); 1.99–1.82 (m, 2H); 1.77–1.67 (m, 2H); 1.52–1.44 (m, 2H); 1.40–1.18 (m, 4H).
13C NMR (CDCl
3, 75.5 MHz):
δ 142.0 (d,
J = 16.2 Hz); 131.4 (d,
J = 15.46 Hz); 128.6 (d,
J = 5.4 Hz); 127.69 (s); 50.8 (d,
J = 10.6 Hz); 48.9 (d,
J = 4.1 Hz); 43.6 (d,
J = 6.5 Hz); 43.4 (s); 42.1 (d,
J = 18.1 Hz); 29.2 (d,
J = 12.5 Hz); 28.7 (d,
J = 4.9 Hz); 25.7 (d,
J = 17.7 Hz); 23.9 (s).
31P NMR (CDCl
3, 121.5 MHz):
δ -9.90 (s). The configuration and high enantiomeric purity of (
RP)-
20 was confirmed by its oxidation by H
2O
2 which afforded back (
SP)-
19 of [α]
D = −20.52 (
c 1.2, CHCl
3).
3.2.20. Synthesis of (RP)-3-Phenyl-3-phosphatricyclo[5.2.1.02,6]decane-borane (21)
To a solution of 1 g (0.004 mol) of phosphine (RP)-20 in 6 mL of benzene under argon atmosphere was added 0.49 g (0.0065 mol) of the BH3-SMe2 complex, and the reaction mixture was stirred at room temperature for 24 h. At the end of the reaction, the mixture was concentrated and the crude product was passed through a silica gel column using CH2Cl2/hexane (1:2) as eluent to yield 0.95 g (90%) of phosphine-borane (RP)-21 as an oil. [α]D = −42.24 (c 0.98, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 7.65–7.55 (m, 2H); 7.47–7.37 (m, 3H); 2.92–2.84 (m, 1H); 2.72–2.66 (m, 2H); 2.36 (s, 1H); 2.15–1.95 (m, 5H); 1.58–1.36 (m, 5H); 1.25–0.30 (m, 3H, BH3). 13C NMR (CDCl3, 75.5 MHz): δ 132 (d, J = 49.1 Hz); 131.3 (d, J = 7.8 Hz); 130.9 (d, J = 2.3 Hz); 129.1 (d, J = 9.2 Hz); 47.3 (d, J = 2.6 Hz); 45.1 (d, J = 35.7 Hz); 43.8 (d, J = 10.8 Hz); 43.1 (d, J = 1.4 Hz); 40.8 (d, J = 3.7 Hz); 28.2 (d, J = 32.3 Hz); 25.9 (d, J = 2.0 Hz); 25.3 (d, J = 6.0 Hz); 23.3 (s). 31P NMR (CDCl3, 121.5 MHz): δ 33.16. Elemental Anal. For C15H22BP: calcd. C, 73.80; H, 9.08; found C, 73.70; H, 9.01.
3.2.21. Synthesis of (SP)-3-Phenyl-3-phosphatricyclo[5.2.1.02,6]decane 3-sulfide (22)
(
SP)-
22 was obtained in 82% yield according to typical procedure described in
Section 3.2.6. (
SP)-2
2: white crystals, mp = 95–96 °C, [α]
D = −22.61 (
c 1.72, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.82–7.75 (m, 2H); 7.50–7.42 (m, 3H); 2.85–2.74 (m, 2H); 2.70–2.65 (m, 1H); 2.56–2.53 (m, 1H); 2.44 (s, 1H); 2.31–2.22 (m, 1H); 2.19–2.07 (m, 2H); 1.95–1.79 (m, 1H); 1.66–1.46 (m, 5H).
13C NMR (CDCl
3, 75.5 MHz):
δ 135.2 (d,
J = 72.2 Hz); 131.5 (d,
J = 2.9 Hz); 130.3 (d,
J = 9.5 Hz); 129.0 (d,
J = 11.2 Hz); 46.3 (d,
J = 48.6 Hz); 45.9 (d,
J = 3.8 Hz); 44.1 (d,
J = 2.1 Hz); 43.4 (d,
J = 12.9 Hz); 41.3 (s); 35.6 (d,
J = 51.0 Hz); 24.9 (d,
J = 6.7 Hz); 23.9 (d,
J = 6.6 Hz); 23.4 (s).
31P NMR (CDCl
3, 121.5 MHz):
δ 59.32 (s). Elemental Anal. For C
15H
19SP: calcd. C, 68.67; H, 7.30; found C, 68.57; H, 7.27.
3.2.22. Synthesis of (SP)-3-Phenyl-3-phosphatricyclo[5.2.1.02,6]decane 3-selenide (23)
(
SP)-
23 was obtained in 90% yield according to typical procedure described in
Section 3.2.7. (
SP)-
23: white crystals, mp = 118–119 °C, [α]
D = −45.87 (
c 1.09, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.82–7.75 (m, 2H); 7.45–7.42 (m, 3H); 2.78–2.76 (m, 3H); 2.61–2.56 (m, 2H); 2.51–2.38 (m, 1H); 2.26–1.92 (m, 2H); 1.82–1.75 (m, 1H); 1.61–1.53 (m, 5H).
13C NMR (CDCl
3, 75.5 MHz):
δ 133.9 (d,
J = 64.4 Hz); 131.6 (d,
J = 2.9 Hz); 130.7 (d,
J = 9.5 Hz); 129.0 (d,
J = 11.1 Hz); 46.5 (d,
J = 12.7 Hz); 46.1 (d,
J = 26.1 Hz); 44.3 (d,
J = 2.0 Hz); 43.4 (d,
J = 13.0 Hz); 41.7 (s); 36.1 (d,
J = 44.4 Hz); 25.1 (d,
J = 4.7 Hz); 24.6 (d,
J = 7.0 Hz); 23.4 (s).
31P NMR (CDCl
3, 121.5 MHz):
δ 42.60 (s). Elemental Anal. For C
15H
19SeP: calcd. C, 58.25; H, 6.19; found C, 58.11; H, 6.15.
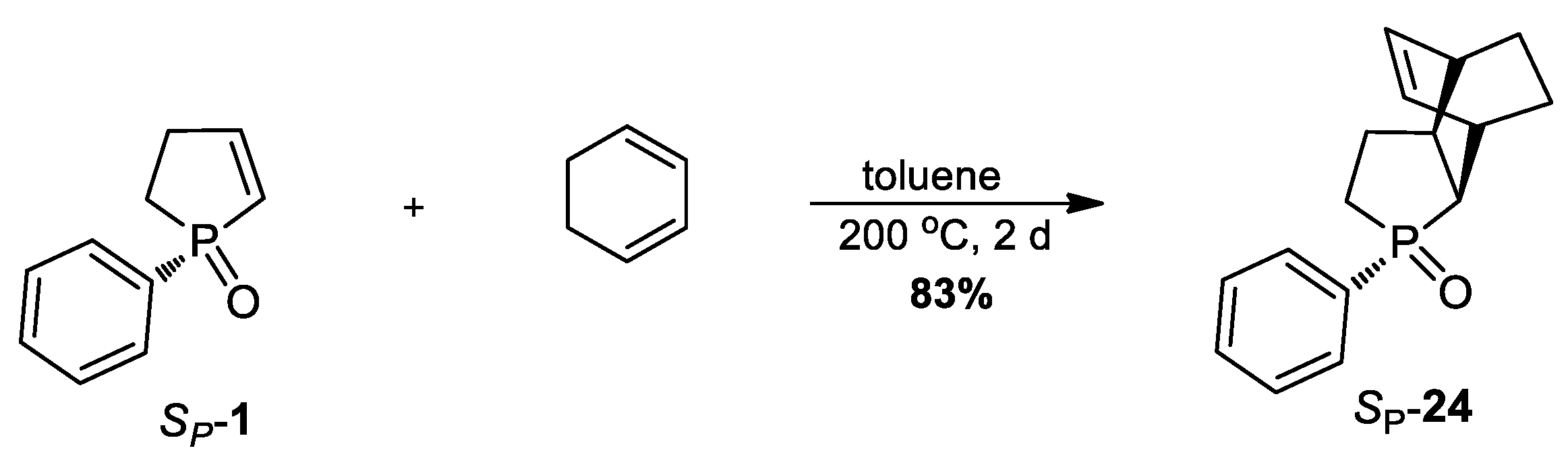
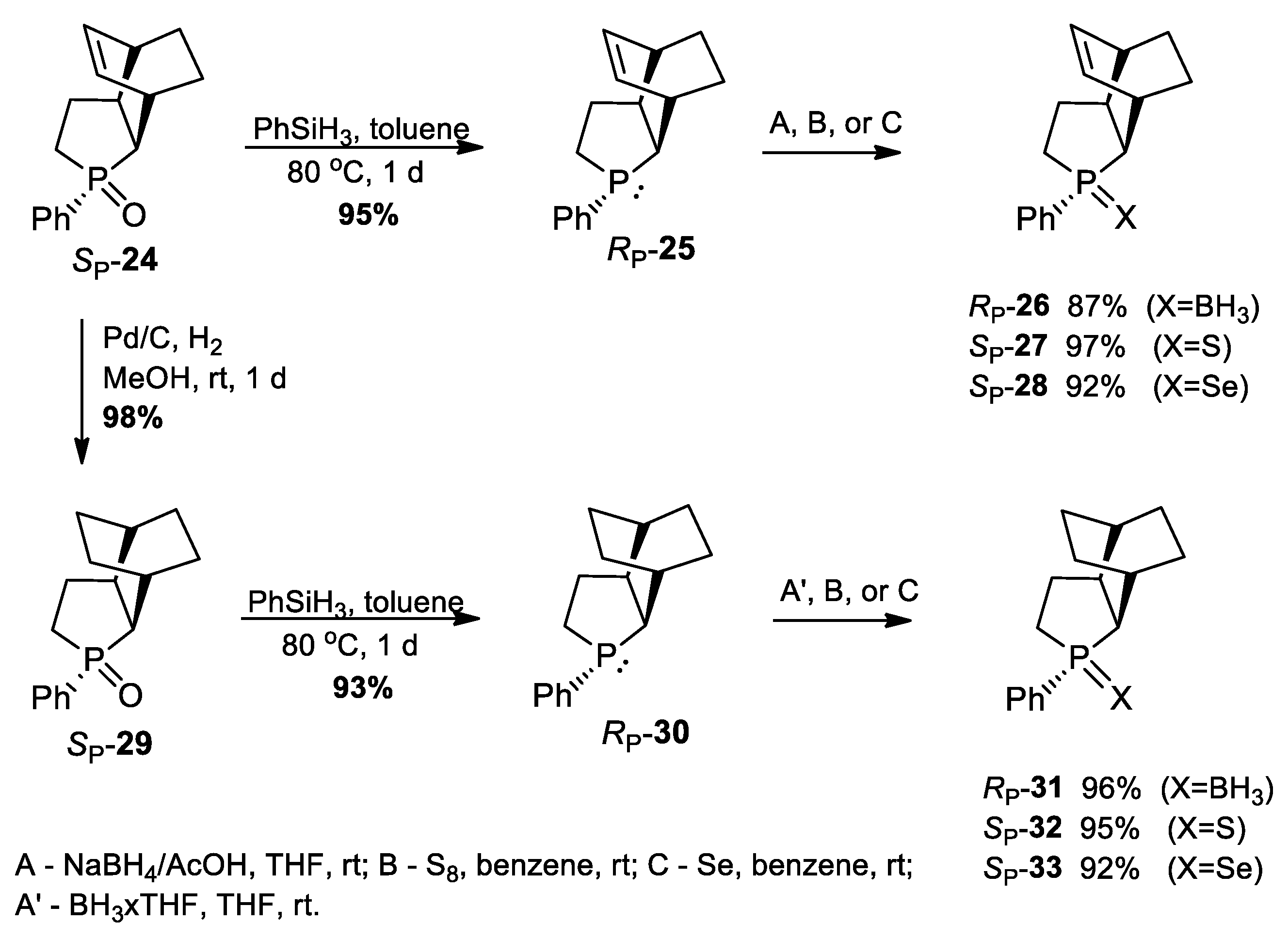
3.2.23. Synthesis of (SP)-3-Phenyl-3-phosphatricyclo[5.2.2.02,6]undec-8-ene 3-oxide (24)
(
SP)-
24 was obtained in 83% yield according to typical procedure described for (
SP)-
4 in
Section 3.2.3. This time, however, addition of cyclohexadiene was repeated three times (4+2+1 equiv.) and the reaction mixture was heated for 72 h. (
SP)-
24: white crystals, mp = 88–89 °C, [α]
D = −18.3 (
c 1.12, CHCl
3).
31P NMR (CDCl
3, 121.5 MHz):
δ 61.45 (s).
1H NMR (CDCl
3, 300 MHz):
δ 7.75–7.68 (m, 2H); 7.51–7.43 (m, 3H); 6.56 (ddd, 1H,
J = 1.1 Hz; 6.6 Hz; 18 Hz); 6.28 (t, 1H,
J = 7.4 Hz); 3.02 (s, 1H); 2.71–2.69 (m, 1H); 2.58–2.49 (m, 1H); 2.29 (dt, 1H,
J = 2.5 Hz; 10.2 Hz); 2.08–1.88 (m, 4H); 1.54–1.49 (m, 2H); 1.36–1.31 (m, 2H).
13C NMR (CDCl
3, 75.5 MHz):
δ 135.6 (d,
J = 2.3 Hz); 134.8 (d,
J = 87.2 Hz); 132.4; 131.9 (d,
J = 2.8 Hz); 130.5 (d,
J = 9 Hz); 129.0 (d,
J = 10.9 Hz); 44.7 (d,
J = 25.6 Hz); 44.2 (d,
J = 36.5 Hz); 35.9 (d,
J = 2.1 Hz); 30.4 (d,
J = 2.8 Hz); 28.7 (d,
J = 48.3 Hz); 28.2 (d,
J = 5.4 Hz); 26.3 (d,
J = 12.3 Hz); 25.1. Elemental Anal. For C
16H
19OP: calcd. C, 74.40; H, 7.41; found C, 74.25; H, 7.38.
3.2.24. Synthesis of (RP)-3-Phenyl-3-phosphatricyclo[5.2.2.02,6]undec-8-ene (25)
(
RP)-
25 was obtained in 89% yield according to typical procedure described in
Section 3.2.4. (
RP)-
25: a colorless oil, bp = 150 °C/0.2 mmHg, [α]
D = −3.6 (
c 1.67, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.39–7.34 (m, 2H); 7.31–7.18 (m, 3H); 6.38–6.26 (m, 2H); 2.97–2.94 (m, 1H); 2.61–2.48 (m, 3H); 2.05–1.96 (m, 1H); 1.85–1.69 (m, 3H); 1.58–1.51 (m, 2H); 1.31–1.19 (m, 2H).
13C NMR (CDCl
3, 75.5 MHz):
δ 142.1 (d,
J = 21.2 Hz); 135.0; 134.2 (d,
J = 4.4 Hz); 130.8 (d,
J = 15.1 Hz); 128.2 (d,
J = 5.1 Hz); 127.4; 50.0 (d,
J = 8.3 Hz); 47.3 (d,
J = 2.9 Hz); 36.8 (s); 35.3 (d,
J = 26.4 Hz); 35.0 (s); 26.6 (d,
J = 10.1 Hz); 26.0 (d,
J = 12.9 Hz).
31P NMR (CDCl
3, 121.5 MHz):
δ 7.88. The configuration and high enantiomeric purity of (
RP)-
25 was confirmed by its oxidation by H
2O
2 which afforded back (
SP)-
24 of [α]
D = −18.35 (
c 0.97, CHCl
3).
3.2.25. Synthesis of (RP)-3-Phenyl-3-phosphatricyclo[5.2.2.02,6]undec-8-ene-borane (26)
(
RP)-
26 was obtained in 87% yield according to typical procedure described in
Section 3.2.5. (
RP)-
26: white crystals, mp = 90–91 °C, [α]
D = −31.98 (
c 1, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.68–7.63 (m, 2H); 7.46–7.44 (m, 3H); 6.48–6.44 (m, 1H); 6.26–6.21 (m, 1H); 3.05–3.03 (m, 1H); 2.68–2.51 (m, 3H); 2.13–2.06 (m, 1H); 1.94–1.88 (m, 3H); 1.57–1.54 (m, 2H); 1.33–1.29 (m, 2H); 1.10–0.25 (m, 3H, BH
3).
13C NMR (CDCl
3, 75.5 MHz):
δ 135.4 (d,
J = 1.49 Hz); 132.8 (s); 132.3 (d,
J = 46.5 Hz); 131.7 (d,
J = 8.0 Hz); 131.1 (d,
J = 2.4 Hz); 129.1 (d,
J = 9.1 Hz); 47.0 (s); 44.5 (d,
J = 35.9 Hz); 36.3 (s); 32.2 (d,
J = 3.0 Hz); 32.0 (d,
J = 4.7 Hz); 26.8 (d,
J = 12.4 Hz); 26.4 (d,
J = 35.1 Hz); 25.1 (s).
31P NMR (CDCl
3, 121.5 MHz):
δ 44.11. Elemental Anal. for C
16H
22BP: calcd. C, 75.03; H, 8.65; found C, 74.92; H, 8.61.
3.2.26. Synthesis of (SP)-3-Phenyl-3-phosphatricyclo[5.2.2.02,6]undec-8-ene 3-sulfide (27)
(
SP)-
27 was obtained in 96% yield according to typical procedure described in
Section 3.2.6. (
SP)-
27: white crystals, mp = 13–131 °C, [α]
D = +6.36 (
c 1.03, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.88–7.79 (m, 2H); 7.51–7.42 (m, 3H); 6.58 (t, 1H,
J = 7.2 Hz); 6.25 (t, 1H,
J = 7.2 Hz); 3.13 (m, 1H); 2.71–2.65 (m, 2H); 2.59–2.54 (m, 1H); 2.17–1.87 (m, 4H); 1.57–1.47 (m, 2H); 1.36–1.30 (m, 2H).
13C NMR (CDCl
3, 75.5 MHz):
δ 135.0 (d,
J = 2.4 Hz); 134.5 (d,
J = 78.1 Hz); 131.8 (s); 131.2 (d,
J = 2.9 Hz); 130.4 (d,
J = 9.6 Hz); 128.6 (d,
J = 11.2 Hz); 47.19 (d,
J = 54.8 Hz); 46.2 (d,
J = 6.4 Hz); 36.4 (d,
J = 1.9 Hz); 34.0 (d,
J = 52.7 Hz); 31.6 (s); 29.6 (d,
J = 10.6 Hz); 25.8 (d,
J = 13.5 Hz); 24.6 (d,
J = 1.1 Hz).
31P NMR (CDCl
3, 121.5 MHz):
δ 67.98 (s). Elemental Anal. For C
16H
19SP: calcd. C, 70.04; H, 6.97; found C, 69.95; H, 6.95.
3.2.27. Synthesis of (SP)-3-Phenyl-3-phosphatricyclo[5.2.2.02,6]undec-8-ene 3-selenide (28)
(
SP)-
28 was obtained in 92% yield according to typical procedure described in
Section 3.2.7. (
SP)-
28: white crystals, mp = 130–131 °C, [α]
D = +8.2 (
c 1.09, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.88–7.79 (m, 2H); 7.51–7.42 (m, 3H); 6.58 (t, 1H,
J = 7.2 Hz); 6.25 (t, 1H,
J = 7.2 Hz); 3.13 (m, 1H); 2.71–2.65 (m, 2H); 2.59–2.54 (m, 1H); 2.17–1.87 (m, 4H); 1.57–1.47 (m, 2H); 1.36–1.30 (m, 2H).
13C NMR (CDCl
3, 75.5 MHz):
δ 135.0 (d,
J = 2.4 Hz); 134.5 (d,
J = 78.1 Hz); 131.8 (s); 131.2 (d,
J = 2.9 Hz); 130.4 (d,
J = 9.6 Hz); 128.6 (d,
J = 11.2 Hz); 47.19 (d,
J = 54.8 Hz); 46.2 (d,
J = 6.4 Hz); 36.4 (d,
J = 1.9 Hz); 34.0 (d,
J = 52.7 Hz); 31.6 (s); 29.6 (d,
J = 10.6 Hz); 25.8 (d,
J = 13.5 Hz); 24.6 (d,
J = 1.1 Hz).
31P NMR (CDCl
3, 121.5 MHz):
δ 59.8 (s). Elemental Anal. For C
16H
19SeP: calcd. C, 59.82; H, 5.96; found C, 59.75; H, 5.94.
3.2.28. Synthesis of (SP)-3-Phenyl-3-phosphatricyclo[5.2.2.02,6]undecane 3-oxide (29)
(
SP)-
29 was obtained in 98% yield according to typical procedure described in
Section 3.2.8. (
SP)-
29: white crystals, mp = 119–120 °C, [α]
D = −5.6 (
c 1.08, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.77–7.70 (m, 2H); 7.54–7.45 (m, 3H); 2.33–2.00 (m, 7H); 1.95–1.82 (m, 2H); 1.79–1.63 (m, 3H); 1.56–1.46 (m, 4H).
13C NMR (CDCl
3, 75,5 MHz):
δ 134.9 (d,
J = 87.4 Hz); 131.8 (d,
J = 2.6 Hz); 130.4 (d,
J = 9.1 Hz); 128.9 (d,
J = 11.0 Hz); 41.7 (d,
J = 8.1 Hz); 41.1 (d,
J = 68.9 Hz); 29.9 (d,
J = 65.4 Hz); 28.8 (d,
J = 3.0 Hz); 27.6 (d,
J = 13.6 Hz); 27.3 (s); 26.5 (d,
J = 11.5 Hz); 25.1 (d,
J = 3.3 Hz); 22.7 (d,
J = 1.7 Hz); 20.9.
31P NMR (CDCl
3, 121.5 MHz):
δ 61.86 (s). Elemental Anal. For C
16H
21OP: calcd. C, 73.82; H, 8.13; found C, 73.72; H, 8.10.
3.2.29. Synthesis of (RP)-3-Phenyl-3-phosphatricyclo[5.2.2.02,6]undecane (30)
(
RP)-
30 was obtained in 93% yield according to typical procedure described in
Section 3.2.4. (
RP)-
30: a colorless oil, bp = 180 °C/0.1 mmHg, [α]
D = −8.79 (
c 1.8, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.66–7.30 (m, 2H); 7.26–7.13 (m, 3H); 2.42–2.31 (m, 2H); 2.04–1.81 (m, 6H); 1.61–1.28 (m, 8H).
13C NMR (CDCl
3, 75.5 MHz):
δ 142.6 (d,
J = 20.7 Hz); 131.2 (d,
J = 15.4 Hz); 128.6 (d,
J = 5.4 Hz); 127.8; 46.8 (d,
J = 9.9 Hz); 43.8 (d,
J = 2 Hz); 33.0 (d,
J = 2.3 Hz); 30.12; 29.5 (d,
J = 23.2 Hz); 28.0 (d,
J = 10.8 Hz); 27.7; 27.6 (d,
J = 9 Hz); 22.8 (d,
J = 10.8 Hz); 21.3.
31P NMR (CDCl
3, 121.5 MHz):
δ 2.85. The configuration and high enantiomeric purity of (
RP)-
30 was confirmed by its oxidation by H
2O
2 which afforded back (
SP)-
29 of [α]
D = −5.38 (
c 1.24, CHCl
3).
3.2.30. Synthesis of (RP)-3-Phenyl-3-phosphatricyclo[5.2.2.02,6]undecane-borane (31)
(
RP)-
31 was obtained in 96% yield according to typical procedure described in
Section 3.2.9. with using 1M BH
3-THF instead of BH
3-SMe
2.
(RP)-31: a white solid, mp = 60–62 °C, [α]
D = −9.3 (
c 1.2, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.73–7.63 (m, 2H); 7.48–7.39 (m, 3H); 2.73–2.37 (m, 2H); 2.20–2.01 (m, 6H); 1.76–1.68 (m, 4H); 1.64–1.51 (m, 3H); 1.47–1.29 (m, 1H); 1.29–0.30 (m, 3H, BH
3).
13C NMR (CDCl
3, 75.5 MHz):
δ 135.4 (d,
J = 1.49 Hz); 132.8 (s); 132.3 (d,
J = 46.5 Hz); 131.7 (d,
J = 8.0 Hz); 131.1 (d,
J = 2.4 Hz); 129.1 (d,
J = 9.1 Hz); 47.0 (s); 44.5 (d,
J = 35.9 Hz); 36.3 (s); 32.2 (d,
J = 3.0 Hz); 32.0 (d,
J = 4.7 Hz); 26.8 (d,
J = 12.4 Hz); 26.4 (d,
J = 35.1 Hz); 25.1 (s).
31P NMR (CDCl
3, 121.5 MHz):
δ 39.35. Elemental Anal. For C
16H
24BP: calcd. C, 74.44; H, 9.37; found C, 74.22; H, 9.34.
3.2.31. Synthesis of (SP)-3-Phenyl-3-phosphatricyclo[5.2.2.02,6]undecane 3-sulfide (32)
(
SP)-
32 was obtained in 95% yield according to typical procedure described in
Section 3.2.6. (
RP)-32: white crystals, mp = 124–125 °C, [α]
D = +2.8 (
c 1.03, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.90–7.82 (m, 2H); 7.50–7.47 (m, 3H); 2.52–2.49 (m, 1H); 2.41–2.06 (m,7H); 1.74–1.44 (m, 8H).
13C NMR (CDCl
3, 75.5 MHz):
δ 135.2 (d,
J = 70.0 Hz); 131.5 (d,
J = 2.9 Hz); 130.6 (d,
J = 9.7 Hz); 129.0 (d,
J = 11.3 Hz); 43.1 (d,
J = 24.7 Hz); 42.6 (d,
J = 20.2 Hz); 35.6 (d,
J = 52.0 Hz); 30.0 (d,
J = 2.73 Hz); 28.1 (d,
J = 10.6 Hz); 27.5 (d,
J = 14.6 Hz); 26.4 (d,
J = 70.8 Hz); 22.1 (d,
J = 2.1 Hz); 20.6.
31P NMR (CDCl
3, 121,5 MHz):
δ 65.93. Elemental Anal. For C
16H
21SP: calcd. C, 69.53; H, 7.65; found C, 69.43; H, 7.63.
3.2.32. Synthesis of (SP)-3-Phenyl-3-phosphatricyclo[5.2.2.02,6]undecane 3-selenide (33)
(
SP)-
33 was obtained in 95% yield according to typical procedure described in
Section 3.2.7. (
SP)-
33: white crystals, mp = 146–147 °C, [α]
D = −0.95 (
c 0.97, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.87–7.84 (m, 2H); 7.52–7.46 (m, 3H); 2.53–2.34 (m, 5H); 2.22–2.05 (m, 3H); 1.74–1.42 (m, 8H).
13C NMR (CDCl
3, 75.5 MHz):
δ 134.0 (d,
J = 62.2 Hz); 131.6 (d,
J = 2.9 Hz); 131.0 (d,
J = 9.7 Hz); 129.0 (d,
J = 11.2 Hz); 43.1 (d,
J = 6.0 Hz); 42.6 (d,
J = 44.6 Hz); 36.0 (d,
J = 45.7 Hz); 30.2 (d,
J = 2.5 Hz); 29.0 (d,
J = 9.3 Hz); 27.5 (d,
J = 14.9 Hz); 26.9; 26.8 (d,
J = 1.2 Hz); 21.8 (d,
J = 2.3 Hz); 20.5 (s).
31P NMR (CDCl
3, 121.5 MHz):
δ 52.94. Elemental Anal. For C
16H
21SeP: calcd. C, 59.44; H, 6.54; found C, 59.22; H, 6.50.
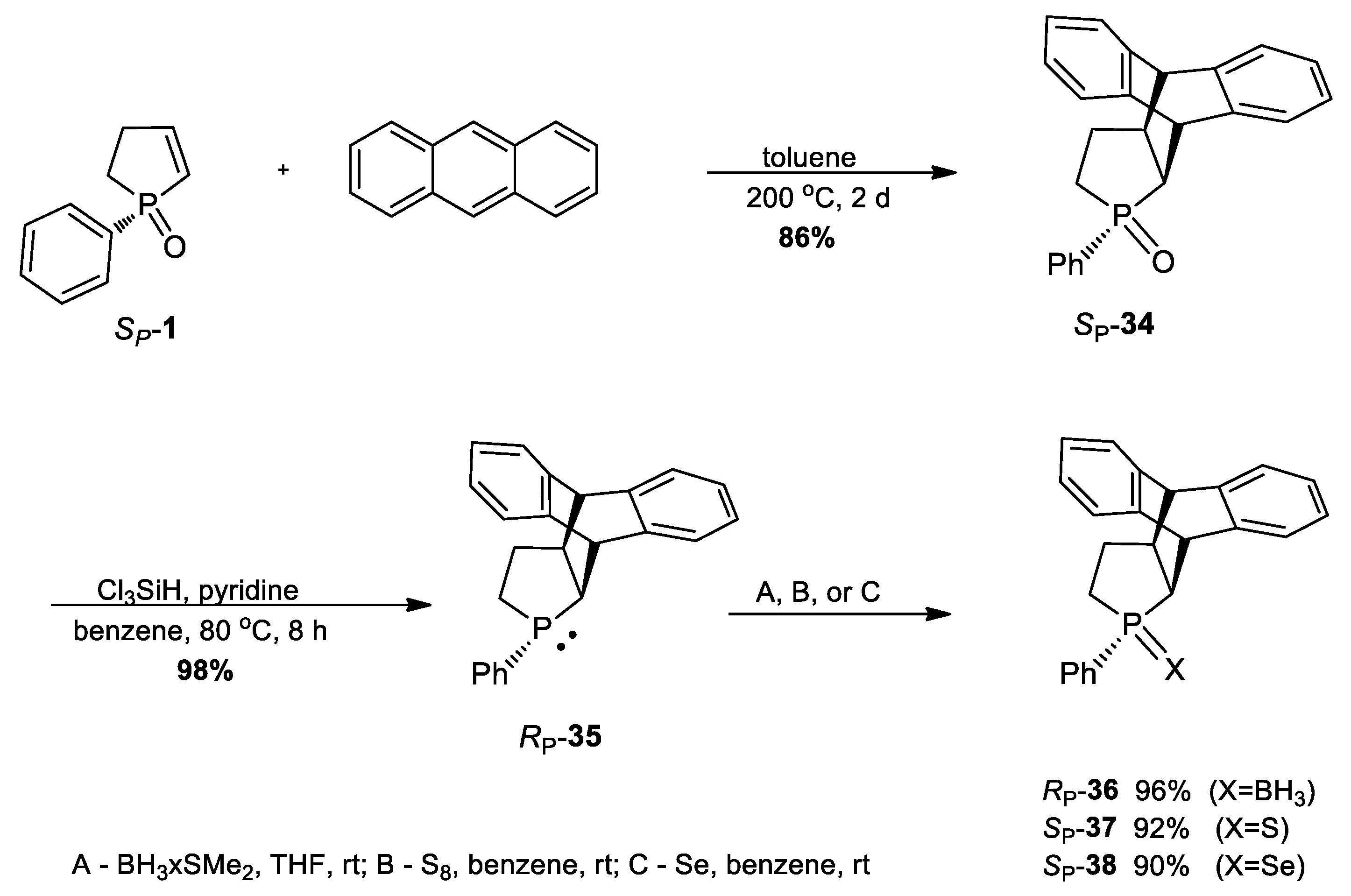
3.2.33. Synthesis of (SP)-Dibenzo[a,d]-3-Phenyl-3-phosphatricyclo[5.2.2.02.6]undeca-8,10-diene 3-oxide (34)
In a tightly closed glass ampoule was placed 2 g (0.011 mol) of (SP)-4, 3.9 g (0.022 mol) of anthracene, and 0.019g (0.089 mmol) of 2.6-di-tert-butyl-4-methylphenol in 7 mL of toluene. The reaction mixture was then heated at 200 °C for 48 h. After that time, the reaction mixture was evaporated under reduced pressure and the residue was passed through a silica gel column using AcOEt/CH3OH (30:1) as eluent to give a white solid which was recrystallized from toluene/hexane to yield 3.4 g (86%) of (SP)-34: white crystals, mp = 251–252 °C, [α]D = −12.3 (c 1.1, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 7.63–7.56 (m, 3H); 7.5–7.37 (m, 3H); 7.32–7.08 (m, 7H); 4.87 (t, 1H, J = 2.9 Hz); 4.29 (bs, 1H); 2.93–2.81 (m, 1H); 2.61–2.55 (m, 1H); 1.87–1.70 (m, 2H); 1.52–1.23 (m, 1H); 0.28–0.13 (m, 1H). 13C NMR (CDCl3, 75.5 MHz): δ 143.6 (d, J = 1.6 Hz); 143.4; 141.0; 140.6 (d, J = 2.6 Hz);134.3 (d, J = 88.3 H); 131.7 (d, J = 2.7 Hz); 129.8 (d, J = 9 Hz); 128.8 (d, J = 11 Hz); 126.9; 126.6; 126.4; 126.2; 126.0; 124.6; 123.8; 123.0; 51.1 (d, J = 1.5 Hz); 44.1 (d, J = 1.6 Hz); 42.4 (d, J = 13.8 Hz); 41.9 (d, J = 70 Hz); 26.4 (d, J = 66 Hz); 24.5 (d, J = 11.6 Hz). 31P NMR (CDCl3, 121.5 MHz): δ 58.65 (s). Elemental Anal. For C24H21OP: calcd. C, 80.88; H, 5.94; found C, 80.70; H, 5.90.
3.2.34. Synthesis of (RP)-Dibenzo[a,d]-3-Phenyl-3-phosphatricyclo[5.2.2.02.6]undeca-8,10-diene (35)
A three-neck flask equipped with a reflux condenser and a septum was charged with 2.7 g (0.034 mol) of pyridine in 68 mL of benzene. Then, 1.54 g (0.011 mole) of Cl3SiH was added followed by 0.81 g (0.0022 mol) of oxide (SP)-34 dissolved in benzene, and the reaction mixture was heated at 80 °C for 8 h. After the reaction mixture was cooled to room temperature, 20 mL of 30% NaOH was added slowly over a period of 1 h. The organic layer was separated, dried over anhydrous MgSO4, and concentrated under reduced pressure. The crude product was crystallized from methanol and yielded 0.76 g (98%) of phosphine (RP)-35: white crystals, mp = 189–190 °C, [α]D = −6.38 (c 1.5, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 7.42–6.96 (m, 13H); 4.51 (dd, 1H, J = 2.4 Hz, 4.3 Hz); 4.12 (d, 1H, J = 2.5 Hz); 2.88–2.76 (m, 2H); 1.9–1.7 (m, 2H); 1.31 (ddd, 1H, J = 2 Hz, 6.5 Hz, 14 Hz); 0.07–0.12 (m, 1H). 13C NMR (CDCl3, 75.5 MHz): δ 144.5 (d, J = 12.9 Hz); 144.0; 141.7; 141.4 (d, J = 3.1 Hz); 140.2 (d, J = 23 Hz); 129.0 (d, J = 15.4 Hz,); 128.3; 128.2; 126.4; 125.9 (d, J = 12.7 Hz); 125.8;125.3 (d, J = 2 Hz); 124.9; 123.5; 123.2; 51.1; 49.0 (d, J = 27 Hz); 48.5 (d, J = 14.3 Hz); 47.4 (d, J = 4.4 Hz); 32.9 (d, J = 4.4 Hz); 26.4 (d, J = 12.5 Hz). 31P NMR (CDCl3, 121.5 MHz): δ 4.12. The configuration and high enantiomeric purity of (RP)-30 was confirmed by its oxidation by H2O2 which afforded back (SP)-29 of [α]D = −11.95 (c 0.88, CHCl3).
3.2.35. Synthesis of (RP)-Dibenzo[a,d]-3-Phenyl-3-phosphatricyclo[5.2.2.02.6]undeca-8,10-diene -borane (36)
(
RP)-
36 was obtained in 96% yield according to typical procedure described in
Section 3.2.9.
(RP)-36: a crystalline solid, mp = 243–244 °C, [α]
D = −25.95 (
c 1.3, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.63 (m, 1H); 7.57–7.5 (m, 1H); 7.42–7.13 (m, H); 4.83 (dd, 1H,
J = 2.2 Hz, 3.9 Hz); 4.29 (d, 1H,
J = 3.3 Hz); 3.07–3.00 (m, 1H); 2.86–2.81 (m, 1H); 2.01–1.84 (m, 2H); 1.65 (m, 1H); 0.39–0.26 (m, 1H); 1.50–0.6 (m, 3H, BH
3).
13C NMR (CDCl
3, 75.5 MHz):
δ 144.7 (d,
J = 12.4 Hz);143.7; 141.6; 140.2 (d,
J = 1.5 Hz); 131.4 (d,
J = 44.6 Hz); 131.3 (d,
J = 8 Hz); 131.1 (d,
J = 2.4 Hz); 129.2 (d,
J = 9.2 Hz); 126.8; 126.6; 126.5; 125.1; 124.4; 123.4; 50.9; 45.8 (d,
J = 1.5 Hz); 45.3 (d,
J = 3.1 Hz); 44.1 (d,
J = 31.6 Hz); 30.0; 25.5 (d,
J = 32 Hz).
31P NMR (CDCl
3, 121.5 MHz):
δ 42.56.
3.2.36. Synthesis of (SP)-Dibenzo[a,d]-3-Phenyl-3-phosphatricyclo[5.2.2.02.6]undeca-8,10-diene 3-sulfide (37)
(
SP)-
37 was obtained in 92% yield according to typical procedure described in
Section 3.2.6. (
RP)-
37: white crystals, mp = 259–260 °C, [α]
D = +14.02 (
c 1.32, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.75–7.65 (m, 3H); 7.43–7.05 (m, 11H); 5.04 (dd, 1H,
J = 1.7 Hz, 4.5 Hz); 4.30 (s, 1H); 3.10–2.95 (m, 1H); 2.85–2.75 (m, 1H); 1.97–1.70 (m, 3H); 0.36–0.25 (m, 1H).
13C NMR (CDCl
3, 75.5 MHz):
δ 143.8 (d,
J = 13 Hz); 143.5; 141.3; 139.8 (d,
J = 2.3 Hz); 134.5 (d,
J = 71.8 Hz); 131.3 (d,
J = 2.8 Hz); 130.1 (d,
J = 9.6 Hz); 128.7 (d,
J = 11.3 Hz); 128.6; 126.4; 126.4; 126.1; 124.6; 123.9; 123.2; 51.2 (d,
J = 1.5 Hz); 45.0 (d,
J = 21.5 Hz); 44.7 (d,
J = 31.2 Hz); 44.3 (d,
J = 10.7 Hz); 32.2 (d,
J = 50.6 Hz); 27.2 (d,
J = 7.6 Hz).
31P NMR (CDCl
3, 121.5 MHz):
δ 63.30. Elemental Anal. for C
24H
21SP: calcd. C, 77.39; H, 5.68; found C, 77.29; H, 5.64.
3.2.37. Synthesis of (SP)-Dibenzo[a,d]-3-Phenyl-3-phosphatricyclo[5.2.2.02.6]undeca-8,10-diene 3-selenide (38)
(
SP)-
38 was obtained in 95% yield according to typical procedure described in
Section 3.2.7. To the solution of 2 g (0.0058 mol) of (
RP)-
35 in 10 mL of benzene was added 0.46 g (0.0058 mol) of selenium under argon and the reaction mixture was stirred at room temperature for 24 h. After this time, the reaction mixture was concentrated and the crude product was purified by column chromatography using CH
2Cl
2/hexane (1:1) as eluent followed by crystallization from methanol to yield 2.21 g (90%) of selenide (
SP)-
38: white crystals, mp = 248–249 °C, [α]
D = +15.07 (
c 1.04, CHCl
3)).
1H NMR (CDCl
3, 300 MHz):
δ 7.74–7.66 (m, 3H); 7.41–7.15 (m, 10H); 5.10 (dd, 1H,
J = 2.4 Hz, 5.4 Hz); 4.3 (dd, 1H,
J = 2.4 Hz, 2.7 Hz); 3.05–2.98 (m, 1H); 2.90–2.85 (m, 1H); 1.92–1.80 (m, 3H); 0.42–0.34 (m, 1H).
13C NMR (CDCl
3, 75.5 MHz):
δ 144.2 (d,
J = 13.4 Hz); 143.9; 141.7; 139.8 (d,
J = 2.2 Hz); 133.9 (d,
J = 63.7 Hz); 131.6 (d,
J = 3 Hz); 130.7 (d,
J = 9.5 Hz); 129.5; 129.1 (d,
J = 11.3 Hz); 128.8 (d,
J = 3 Hz); 126.8 (d,
J = 3 Hz); 126.8; 126.7; 126.4; 124.9; 124.3; 123.6; 51.4 (d,
J = 1.3 Hz); 46.0 (d,
J = 1.4 Hz); 45.3 (d,
J = 1.2 Hz); 44.8 (d,
J = 37 Hz); 33.0 (d,
J = 44 Hz); 28.8 (d,
J = 5.5 Hz
). 31P NMR (CDCl
3, 121.5 MHz):
δ 49.52 (s). Elemental Anal. For C
24H
21SeP: calcd. C, 77.39; H, 5.68; found C, 77.29; H, 5.64.
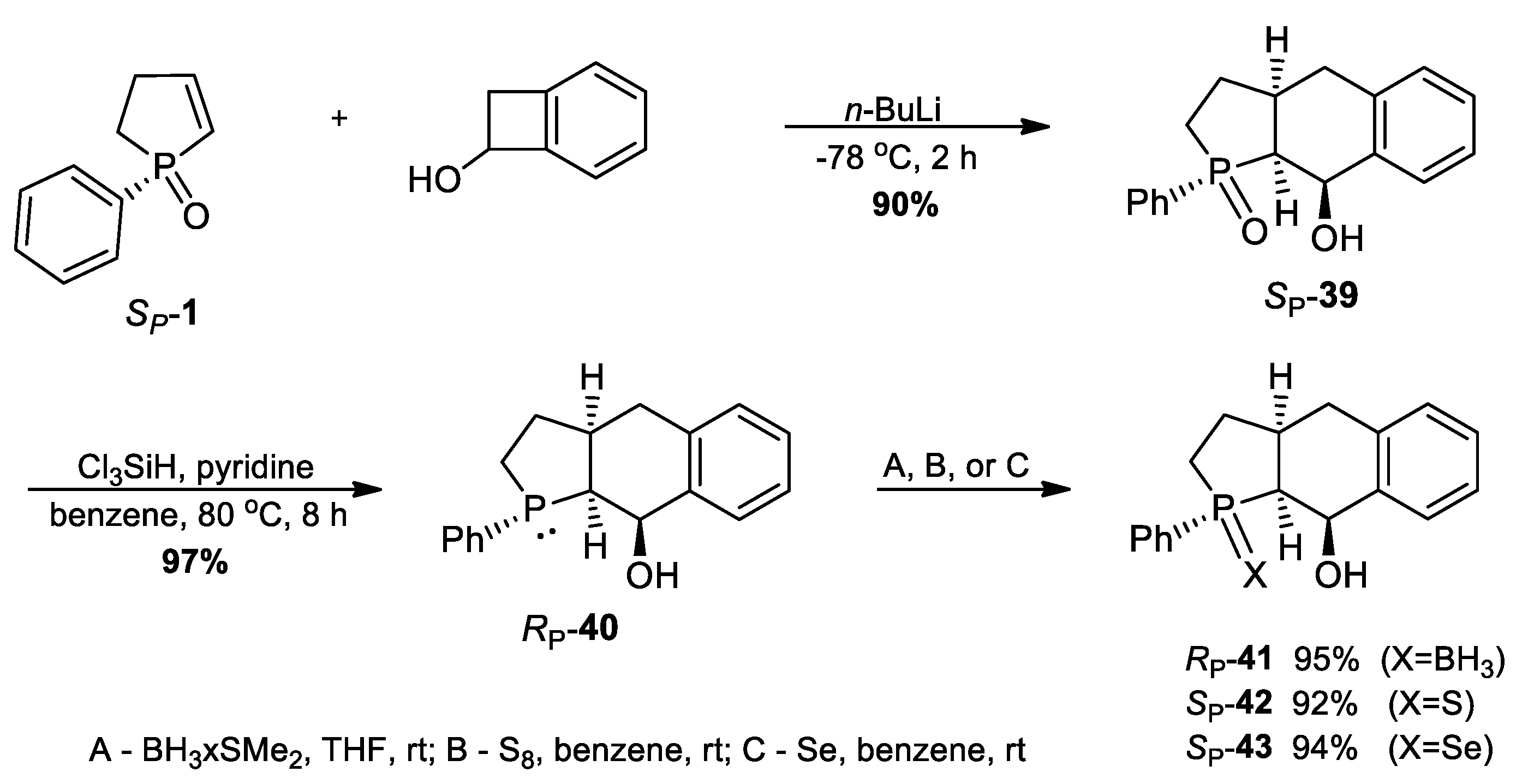
3.2.38. Synthesis of (SP)-1-Phenyl-2,3,3a,4,9,9a-hexahydrophosphacyclopenta[b]naphthalen-9-ol 1-oxide (39)
In a Schlenk flask under argon atmosphere was placed 0.8 g (0.0067 mol) of benzocyclobutenol dissolved in 120 mL of THF, and all of it was cooled down to −78 °C with stirring. After 1 h, 5 mL of 1.6 M n-BuLi (1.1 equiv.) was added dropwise and the resulting mixture was stirred at −78 °C for 0.5 h. Then, 1 g (0.0056 mol) of (SP)-4 in THF was added dropwise and the reaction mixture was stirred at −78 °C for 2 h, and then at 0 °C for 1 h. Next, the volatiles were removed from the reaction mixture and the residue was dissolved in CH2Cl2 (120 mL) and washed with saturated aq. NH4Cl (50 mL) and H2O (2 × 35 mL). The separated organic layer was dried over anhydrous MgSO4, filtered and evaporated under reduced pressure. The residue was purified by column chromatography using CHCl3/acetone (35:1) as eluent to yield 1.5 g (90%) of oxide (SP)-39: a white crystalline solid. mp = 145–147 °C, [α]D = −46.95 (c 1.22, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 7.8–7.72 (m, 2H); 7.55–7.51 (m, 3H); 7.31–7.2 (m, 4H); 5.22 (dd, 1H, J = 4 Hz; 8.8 Hz); 4.97 (d, 1H, J = 7.9 Hz); 3.14 (dd, 1H, J = 8 Hz; 14.5 Hz); 3.0(ddd, 1H, J = 1.3 Hz, 6.8 Hz, 15.7 Hz); 2.66–2.63 (m, 1H); 2.55–2.33 (m, 2H); 2.2–2.1 (m, 2H); 1.98–1.86 (m, 1H). 13C NMR (CDCl3, 75.5 MHz): δ 139.5 (d, J = 8.2 Hz); 137.7; 132.8 (d, J = 90 Hz); 132.6 (d, J = 2.8 Hz); 130.6 (d, J = 9.9 Hz); 129.4 (d, J = 11.4 Hz); 128.5; 128.4; 126.9; 126.8; 70.6 (d, J = 5.5 Hz); 43.7 (d, J = 67 Hz); 38.0 (d, J = 10.4 Hz); 34.3 (d, J = 4.4 Hz); 32.2 (d, J = 8.7 Hz); 30.0 (d, J = 65.3 Hz). 31P NMR (CDCl3, 121.5 MHz): δ 65.17 (s). Elemental Anal. For C18H19O2P: calcd. C, 72.47; H, 6.42; found C, 72.37; H, 6.40.
3.2.39. Synthesis of (RP)-1-Phenyl-2,3,3a,4,9,9a-hexahydrophosphacyclopenta[b]naphthalen-9-ol (40)
(
RP)-
40 was obtained in 97% yield according to procedure described in
Section 3.2.32. (
RP)-
40: a thick oil, [α]
D = −10.8 (
c 1.5, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.8–7.72 (m, 2H); 7.55–7.51 (m, 3H); 7.31–7.2 (m, 4H); 5.22 (dd, 1H,
J = 4 Hz; 8.8 Hz); 4.97 (d, 1H,
J = 7.9 Hz); 3.14 (dd, 1H,
J = 8 Hz; 14.5 Hz); 3.0(ddd, 1H,
J = 1.3 Hz, 6.8 Hz, 15.7 Hz); 2.66–2.63 (m, 1H); 2.55–2.33 (m, 2H); 2.2–2.1 (m, 2H); 1.98–1.86 (m, 1H).
13C NMR (CDCl
3, 75.5 MHz):
δ 139.5 (d,
J = 8.2 Hz); 137.7; 132.8 (d,
J = 90 Hz); 132.6 (d,
J = 2.8 Hz); 130.6 (d,
J = 9.9 Hz); 129.4 (d,
J = 11.4 Hz); 128.5; 128.4; 126.9; 126.8; 70.6 (d,
J = 5.5 Hz); 43.7 (d,
J = 67 Hz); 38.0 (d,
J = 10.4 Hz); 34.3 (d,
J = 4.4 Hz); 32.2 (d,
J = 8.7 Hz); 30.0 (d,
J = 65.3 Hz).
31P NMR (CDCl
3, 121.5 MHz):
δ -8.82. The configuration and high enantiomeric purity of (
RP)-
40 was confirmed by its oxidation by H
2O
2 which afforded back (
SP)-
39 of [α]
D = −46.6 (
c 1.05, CHCl
3).
It is important to note in this place that a primarily attempted reduction of (SP)-39 by PhSiH3 under our standard conditions (toluene, 80 °C, 24 h) led to the formation of two P-epimeric phosphine products as revealed by 31P NMR spectrum of the crude post-reduction mixture showing two signals at −8.35 and −3.67 ppm (7:3) indicating that a partial inversion at P in (RP)-40 had taken place already at 80 °C.
3.2.40. Synthesis of (RP)-1-Phenyl-2,3,3a,4,9,9a-hexahydrophosphacyclopenta[b]naphthalen-9-ol-borane (41)
(
RP)-
41 was obtained in 95% yield according to typical procedure described in
Section 3.2.9. with using 1M BH
3-THF instead of BH
3-SMe
2. (
RP)-
41: white crystals, mp = 104–105 °C, [α]
D = −34.86 (c 1.3, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.77–7.73 (m, 2H); 7.51–7.44 (m, 3H); 7.33–7.21 (m, 4H); 5.24–5.20 (m, 1H -OH); 3.1–2.92 (m, 2H); 2.85–2.72 (m, 3H); 2.50–2.0 (m, 4H); 1.29–0.30 (m, 3H, BH
3).
13C NMR (CDCl
3, 75.5 MHz):
δ 139.2 (d,
J = 8.2 Hz); 137.4; 132.0 (d,
J = 8.8 Hz); 131.8 (d,
J = 2.5 Hz); 131.0 (d,
J = 50.3 Hz); 129.4 (d,
J = 9.8 Hz); 128.6; 128.6; 127.0; 126.9; 70.8 (d,
J = 1.1 Hz, C-OH); 46.2 (d,
J = 34.5 Hz); 39.7 (d,
J = 5.4 Hz); 34.9 (d,
J = 4.5 Hz); 33.5 (d,
J = 3.4 Hz); 26.7 (d,
J = 37.3 Hz).
31P NMR (CDCl
3, 121.5 MHz):
δ 32.80. Elemental Anal. For C
18H
22BOP: calcd. C, 73.00; H, 7.48; found C, 72.91; H, 7.44.
3.2.41. Synthesis of (SP)-1-Phenyl-2,3,3a,4,9,9a-hexahydrophosphacyclopenta[b]naphthalen-9-ol 1-sulfide (42)
(
SP)-
42 was obtained in 92% yield according to typical procedure described in
Section 3.2.6. (
SP)-
42: white crystals, mp = 141–142 °C, [α]
D = +17.59 (
c 1.02, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.93–7.85 (m, 2H); 7.60–7.45 (m, 3H); 7.32–7.20 (m, 4H); 5.31 (dd, 1H,
J = 3.1 Hz, 8.7 Hz); 5.03 (bs, 1H-OH); 3.21 (dd, 1H,
J = 8 Hz, 14.6 Hz); 3.06 (ddd, 1H,
J = 2.4 Hz, 7.3 Hz, 14.7 Hz); 2.85–2.71 (m, 1H); 2.70–2.65 (m, 1H); 2.53–2.35 (m, 4H).
13C NMR (CDCl
3, 75.5 MHz):
δ 139 (d,
J = 10.9 Hz); 137,7; 133.2 (d,
J = 71.3 Hz); 132.2 (d,
J = 3 Hz); 130.9 (d,
J = 9.8 Hz); 129.2 (d,
J = 11.9 Hz); 128.7; 128.5; 127.2; 126.8; 70.8 (d,
J = 3.9 Hz); 47.2 (d,
J = 55 Hz); 39.6 (d,
J = 10.8 Hz); 36.3 (d,
J = 52.4 Hz); 34.3 (d,
J = 5.1 Hz); 33.9 (d,
J = 8.2 Hz).
31P NMR (CDCl
3, 121.5 MHz):
δ 61.08. Elemental Anal. For C
18H
19SOP: calcd. C, 68.76; H, 6.09; found C, 68.65; H, 6.05.
3.2.42. Synthesis of (SP)-1-Phenyl-2,3,3a,4,9,9a-hexahydrophosphacyclopenta[b]naphthalen-9-ol 1-selenide (43)
(
SP)-
42 was obtained in 92% yield according to typical procedure described in
Section 3.2.6. (
SP)-
43: white crystals, mp = 119–120 °C, [α]
D = +20.07 (
c 1.02, CHCl
3).
1H NMR (CDCl
3, 300 MHz):
δ 7.95–7.86 (m, 2H); 7.55–7.46 (m, 3H); 7.31–7.2 (m, 4H); 5.32 (dd, 1H,
J = 2.3 Hz, 8.1 Hz); 4.95 (bs, 1H-OH), 3.21 (dd, 1H,
J = 7.8 Hz, 15 Hz); 3.2–3.05 (m, 1H); 2.81–2.78 (m, 6H).
13C NMR (CDCl
3, 75.5 MHz):
δ 138.4 (d,
J = 10 Hz); 137.3 (d,
J = 0.7 Hz); 131.8 (d,
J = 62.9 Hz); 131.1 (d,
J = 10.5 Hz); 128.9 (d,
J = 11.8 Hz); 128.4; 128.1; 126.9; 126.4; 71.9 (d,
J = 3.0 Hz); 46.8 (d,
J = 46.3 Hz); 39.2 (d,
J = 10.3 Hz); 36.3 (d,
J = 46.5 Hz); 33.8 (d,
J = 1.2 Hz); 33.7 (d,
J = 3.9 Hz).
31P NMR (CDCl
3, 121.5 MHz):
δ 46.27. Elemental Anal. for C
18H
19SeOP: calcd. C, 59.84; H, 5.30; found C, 59.68; H, 5.28.
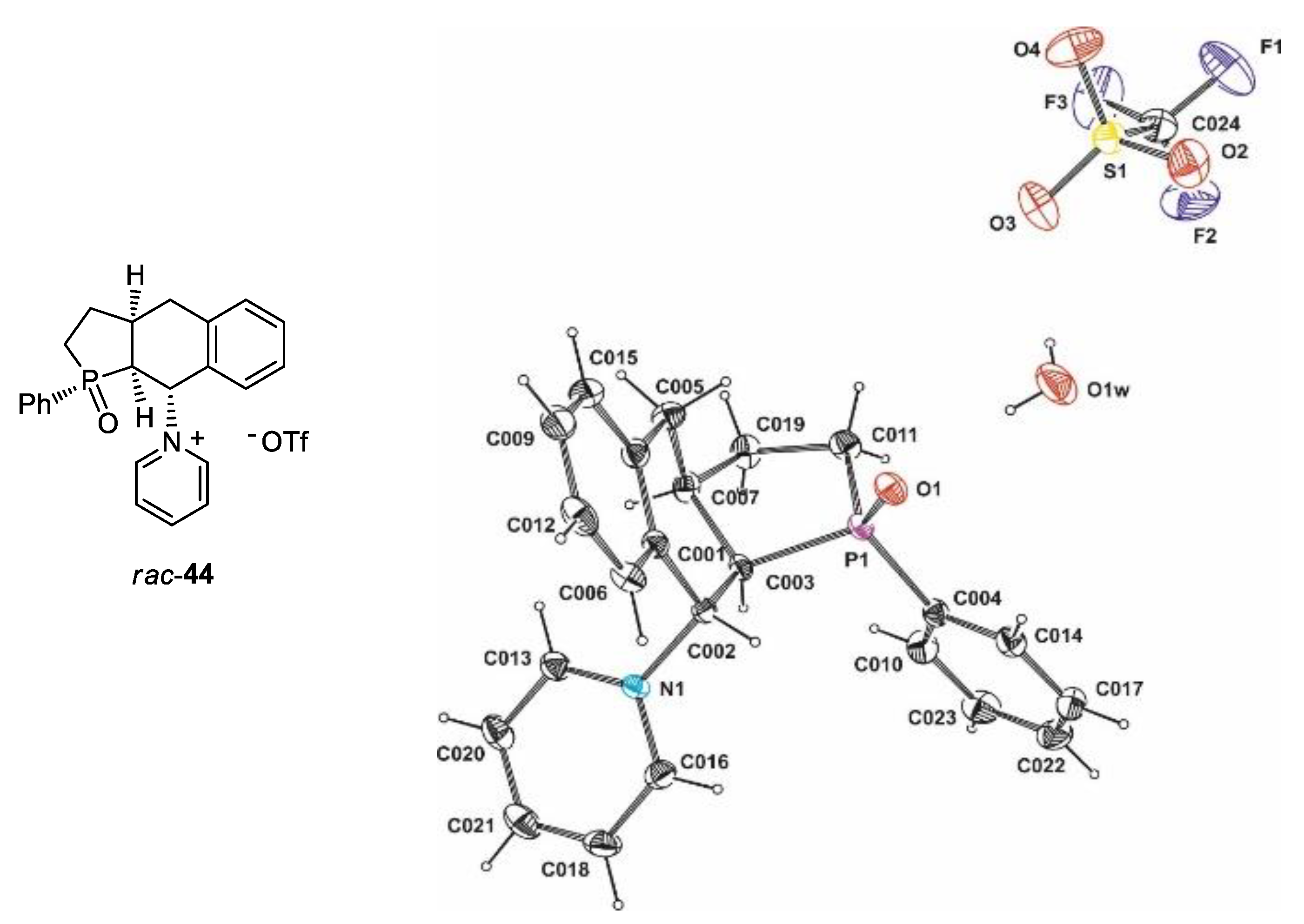
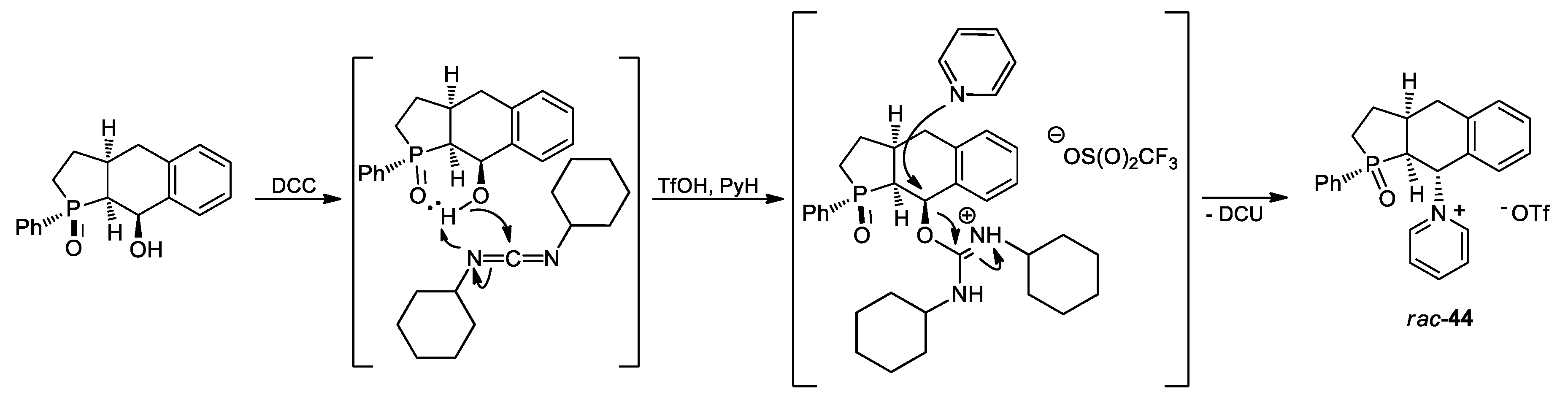
3.2.43. Synthesis of rac-1-[1-Oxido-1-phenyl-2,3,3a,4,9,9a-hexahydro-1H-benzo[f]phosphindol-9-yl]pyridinium Triflate (rac-44)
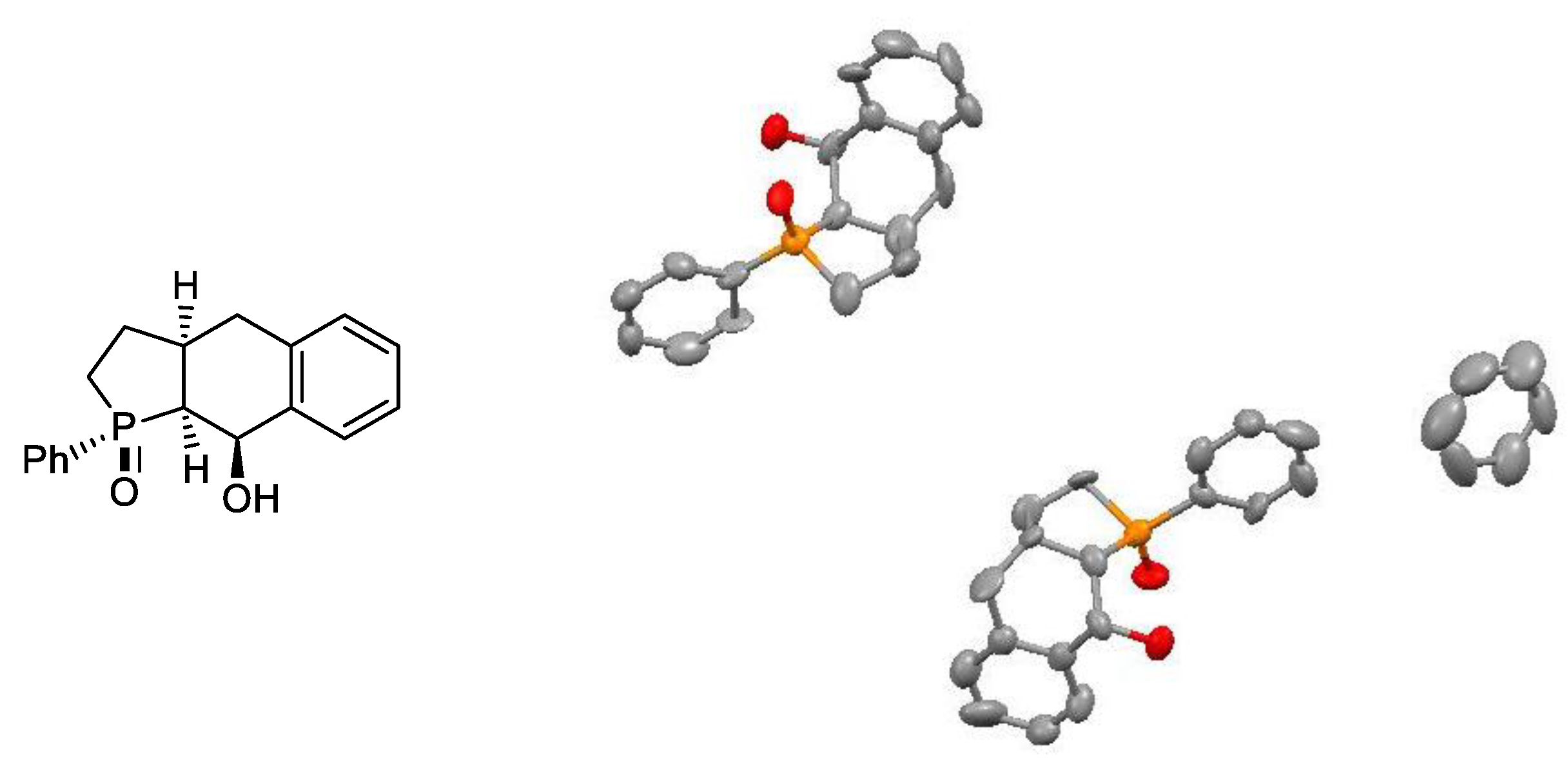
In a flask equipped with a magnetic stirrer septum were placed 15 mL of dry DMSO and 10 mL of pyridine. The stirred mixture was chilled to 0 °C and 170 µL (300 mg, 2 mmol) of triflic acid was added. Then, 300 mg (1 mmol) of rac-39 disolved in 5 mL of DMSO was slowly added, and 400 mg (1.9 mmol) DCC was added in one portion at 0 °C. The resulting reaction mixture was then stirred for 24 h at room temperature and, at the end, heated to 60 °C for additional 2 h. The end point of the reaction was detected by NMR, which indicated a nearly complete conversion of rac-39. The formed DCU was removed by filtration through a syringe filter and a small part of the product crystallized out from the filtrate during cooling to ambient temperature. Due to a minute amount of isolated hardly soluble crystalline rac-44, it was not fully characterized except for an X-ray diffraction analysis which unambiguously confirmed its molecular structure.
3.3. X-Ray Crystallographic Data
The X-ray data for compounds
rac-
19 and
rac-
26 were collected at 100(2) K on a Nonius Kappa CCD diffractometer [
70] using graphite monochromated MoKα radiation (λ = 0.71073 Å). The crystals were mounted in a nylon loop in a drop of silicon oil to prevent the possibility of decay of the crystal during data collection. The unit cells’ parameters were determined from ten frames and then refined on all data. The data were processed with
DENZO and
SCALEPACK (
HKL2000 package) [
71]. The structures were solved by direct methods using the SHELXS-97 [
72] program and was refined by full matrix least–squares on F
2 using the program SHELXL-97 [
73]. All non-hydrogen atoms were refined with anisotropic displacement parameters. The hydrogen atoms were introduced at geometrically idealized coordinates with a fixed isotropic displacement parameter equal to 1.5 (methyl groups) times the value of the equivalent isotropic displacement parameter of the parent carbon.
The X-ray data for complex
rac-
39a were collected at 293(2) K on an Enraf Nonius MACH3 diffractometer [
71] using graphite monochromated CuKα radiation (λ = 1.54178 Å). The unit cell parameters were determined from ten frames and then refined on all data. The data were processed with
OpenMolEN, Nonius BV. The structure was solved by direct methods using the SHELXS97 [
71] program and was refined by full matrix least–squares on F
2 using the program SHELXL 97 [
73] All of the non-hydrogen atoms were refined with anisotropic displacement parameters. The hydrogen atoms were introduced at geometrically idealized coordinates with a fixed isotropic displacement parameter equal to 1.5 (methyl groups) times the value of the equivalent isotropic displacement parameter of the parent carbon.
Crystallographic data for compounds
rac-
19, rac-
26 and
rac-
44 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication.: CCDC–1981155 (
rac-
19), CCDC–1981156 (
rac-
26) and CCDC–239150 (
rac-
44). Copies of the data can be obtained free of charge on application to CCDC via
www.ccdc.cam.ac.uk/data_request/cif, or by e-mail data_request@ccdc.cam.ac.uk.
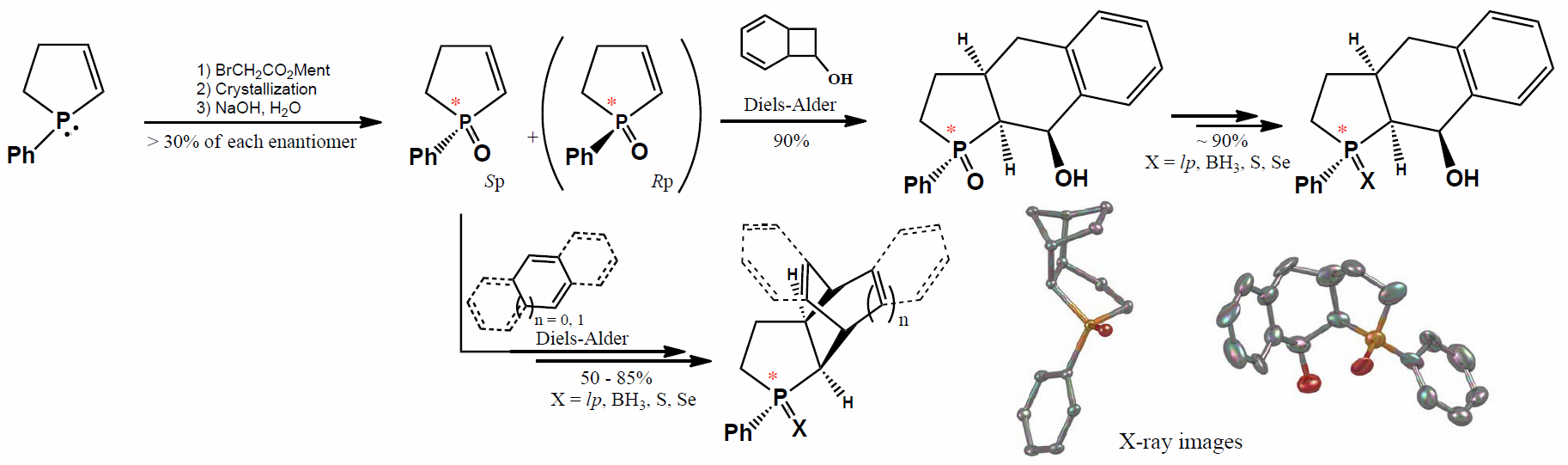
Crystal data for rac-19: C15H19P1O1: M = 246.27, orthorhombic, space group P 212,121 (no. 19), a = 9.4360(2) Å, b = 11.3710(2) Å, c = 11.5660(2) Å, U = 1240.99(4) Å3, Z = 4, F(000) = 528, Dc = 1.318 g cm−3, T = 100(2)K, μ(Mo-Kα) = 0.202 mm−1, θmax = 27.505°, 2833 unique reflections. Refinement converged at R1 = 0.0346, wR2 = 0.0816 for all data and 154 parameters (R1 = 0.0317, wR2 = 0.0803 for 2716 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal 1.052. A weighting scheme w = [σ2(Fo)2 + (0.0418P)2 + 3.1964P]−1 where P = (Fo2 + 2Fc2)/3 was used in the final stage of refinement. The residual electron density = +0.24/−0.24 eÅ−3.
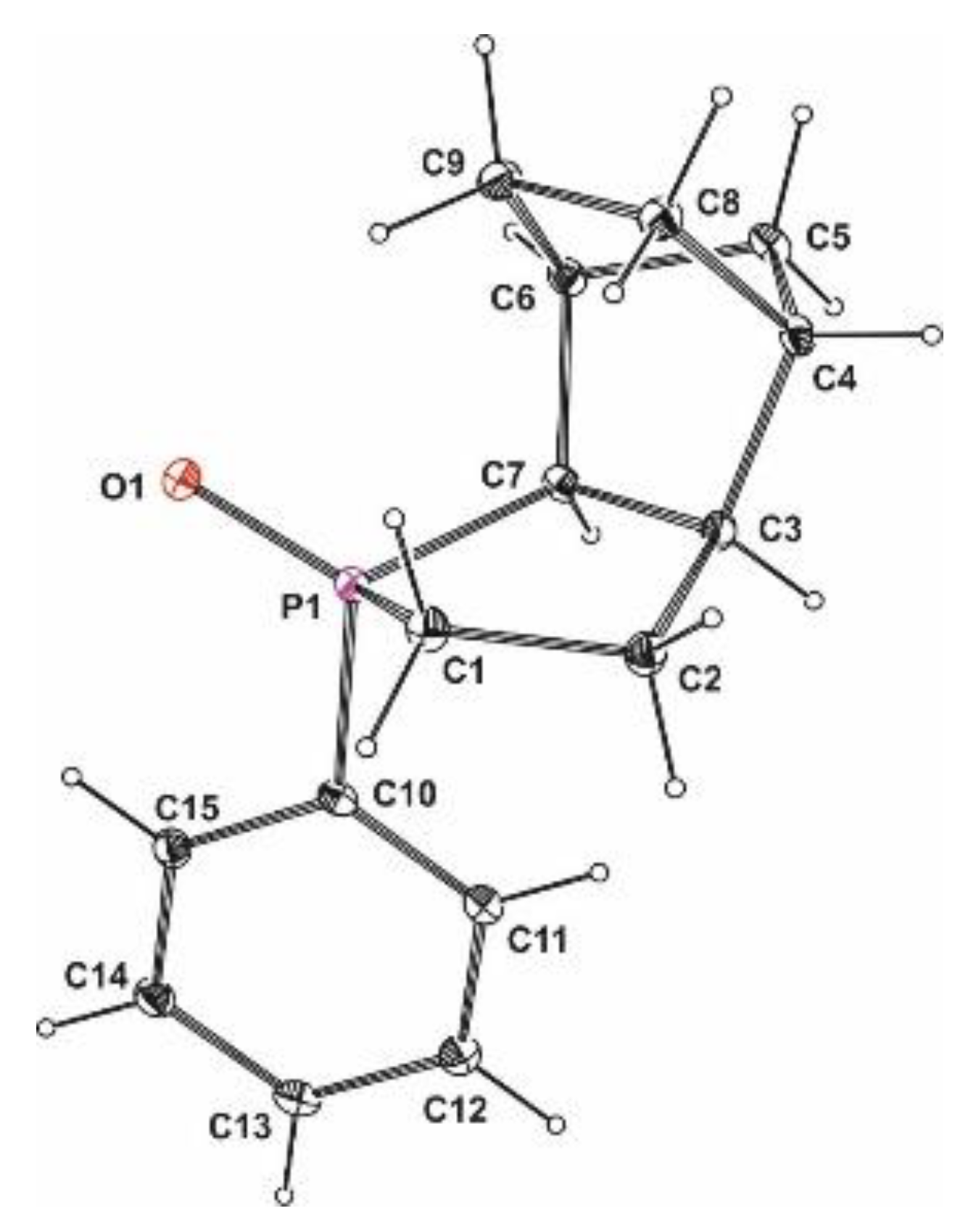
Crystal data for rac-26: C16H22P1B1: M = 256.11, monoclinic, space group P 21 (no. 4), a = 6.37100(10) Å, b = 13.5760(3) Å, c = 8.4160(2) Å, β = 101.4930(10)°, U = 713.33(3) Å3, Z = 2, F(000) = 276, Dc = 1.192 g cm−3, T = 100(2)K, μ(Mo-Kα) = 0.172 mm−1, θmax = 27.471°, 2639 unique reflections. Refinement converged at R1 = 0.0367, wR2 = 0.0946 for all data and 180 parameters (R1 = 0.0352, wR2 = 0.0924 for 2566 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal 1.063. A weighting scheme w = [σ2(Fo)2 + (0.0418P)2 + 3.1964P]−1 where P = (Fo2 + 2Fc2)/3 was used in the final stage of refinement. The residual electron density = +0.25/−0.30 eÅ−3.
Crystal data for rac-44: C24H25P1S1F3N1O5: M = 246.27, triclinic, space group P -1 (no. 2), a = 9.6444(6) Å, b = 11.0887(7) Å, c = 11.7985(7), Å, α = 95.281(5)°, β = 105.384(5)°, γ = 95.097(5)°, U = 1202.94(13) Å3, Z = 2, F(000) = 548, Dc = 1.456 g cm−3, μ(Mo-Kα) = 2.363 mm−1, θmax = 74.24°, 3948 unique reflections. Refinement converged at R1 = 0.0858, wR2 = 0.1772 for all data and 337 parameters. The goodness-of-fit on F2 was equal 1.430. A weighting scheme w = [σ2(Fo)2 + (0.0418P)2 + 3.1964P]−1, where P = (Fo2 + 2Fc2)/3 was used in the final stage of refinement. The residual electron density = +0.67/−0.31 eÅ−3.
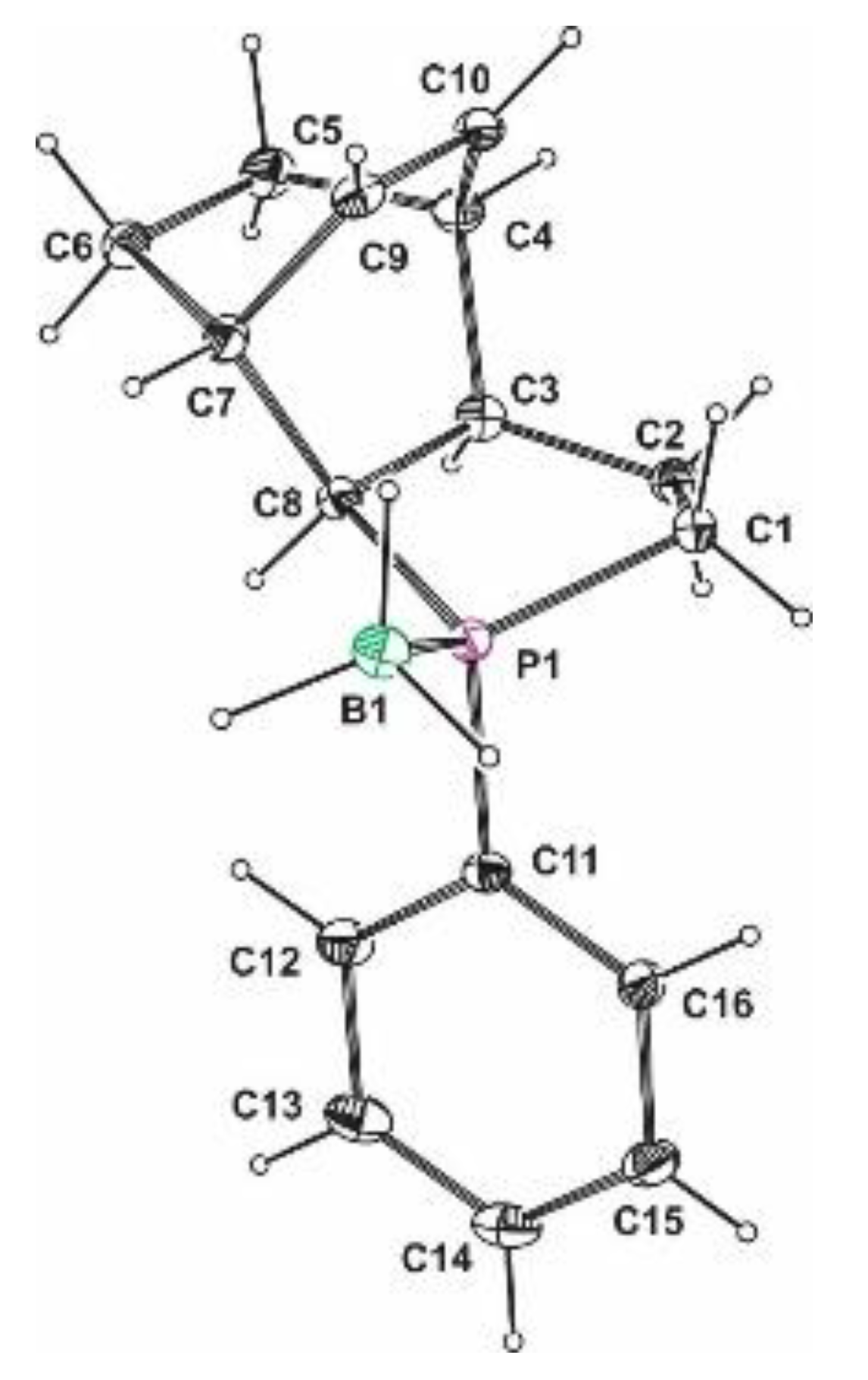
Crystal data for rac-39: There are no complete crystallographic data for rac-39. The overview picture was built based on the RES file whose quality was sufficient to determine the relative configuration of the substituents.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}














