Asymmetric Electrophilic Reactions in Phosphorus Chemistry


Abstract
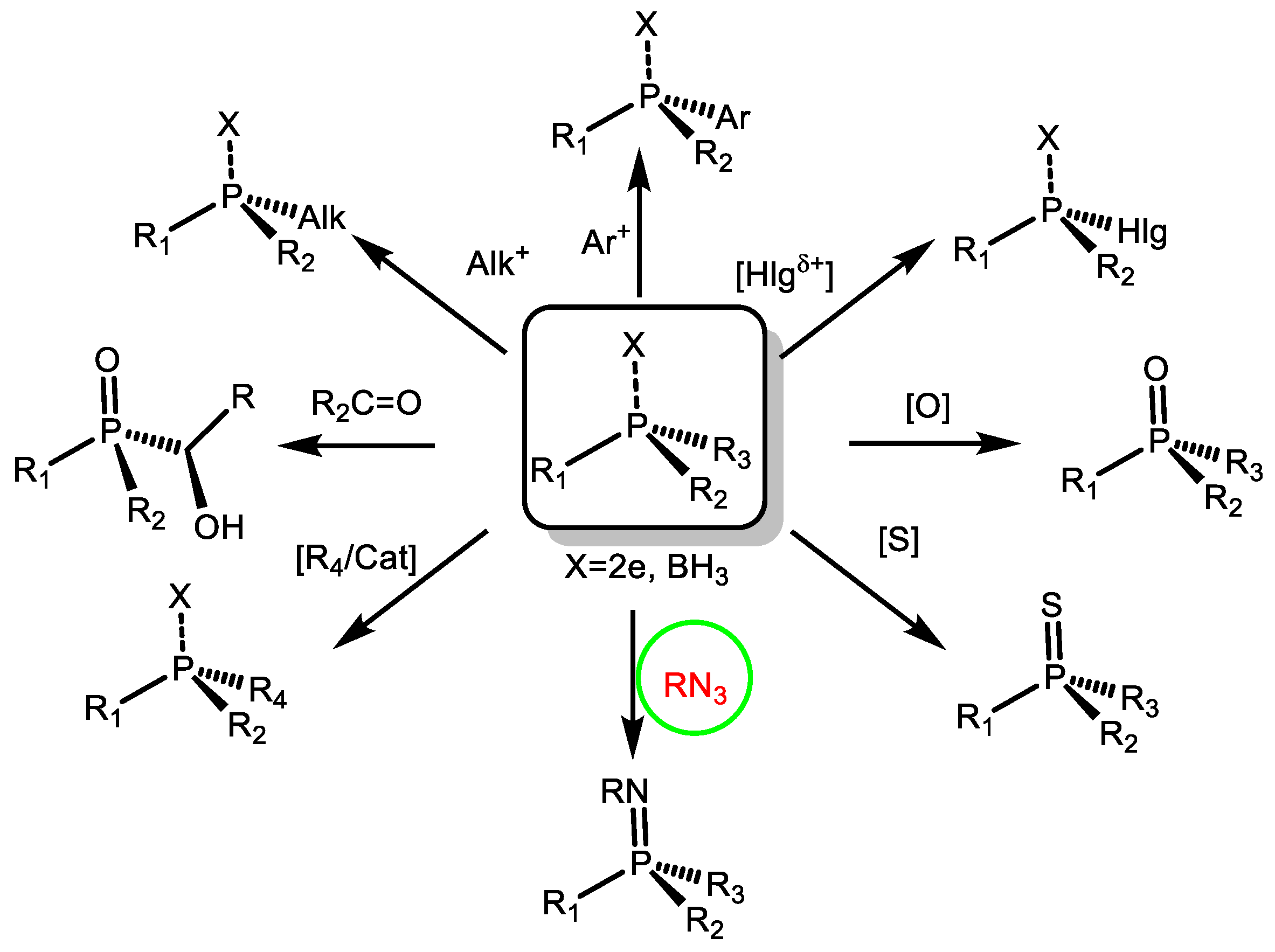
:1. Introduction
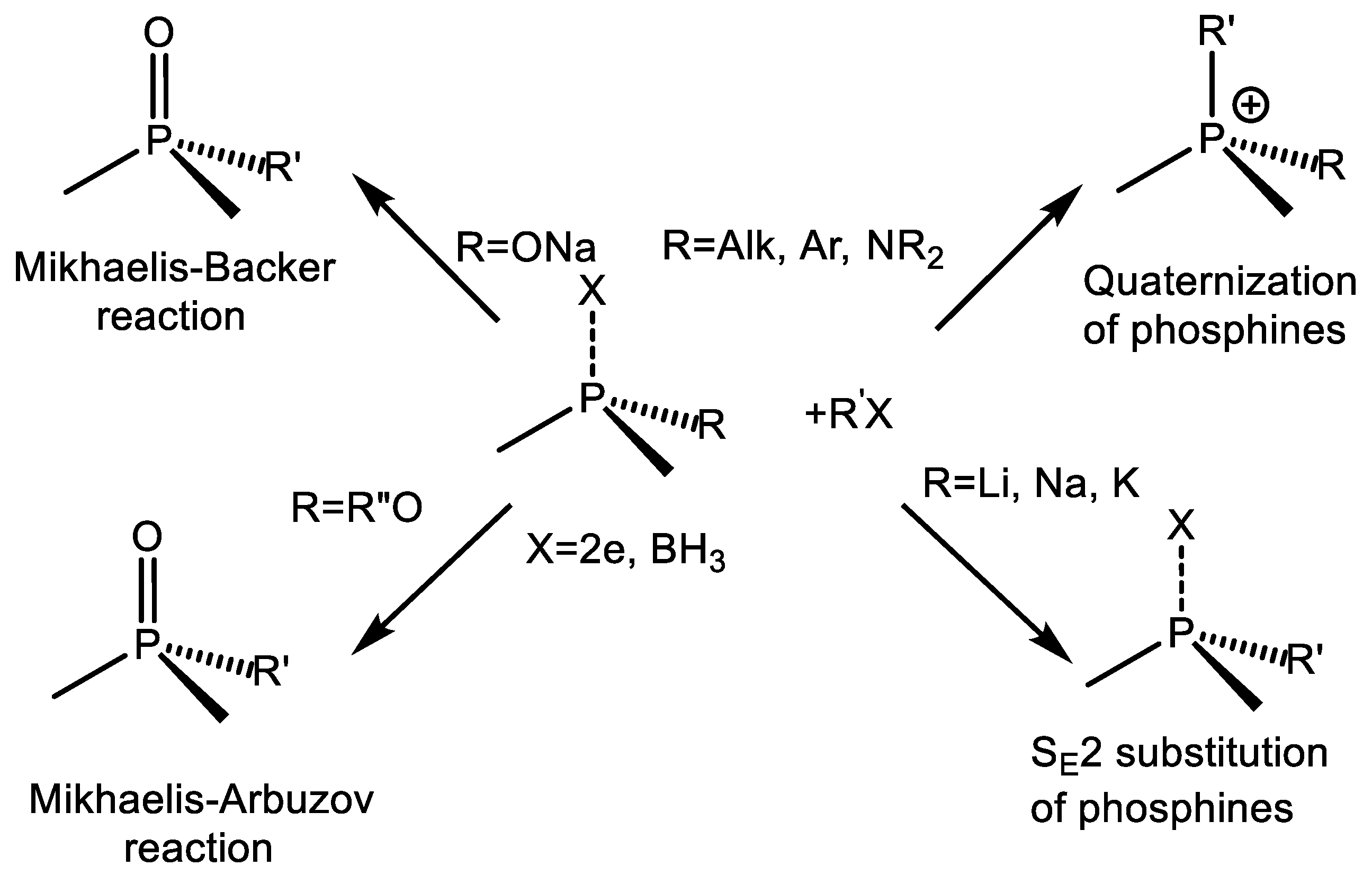
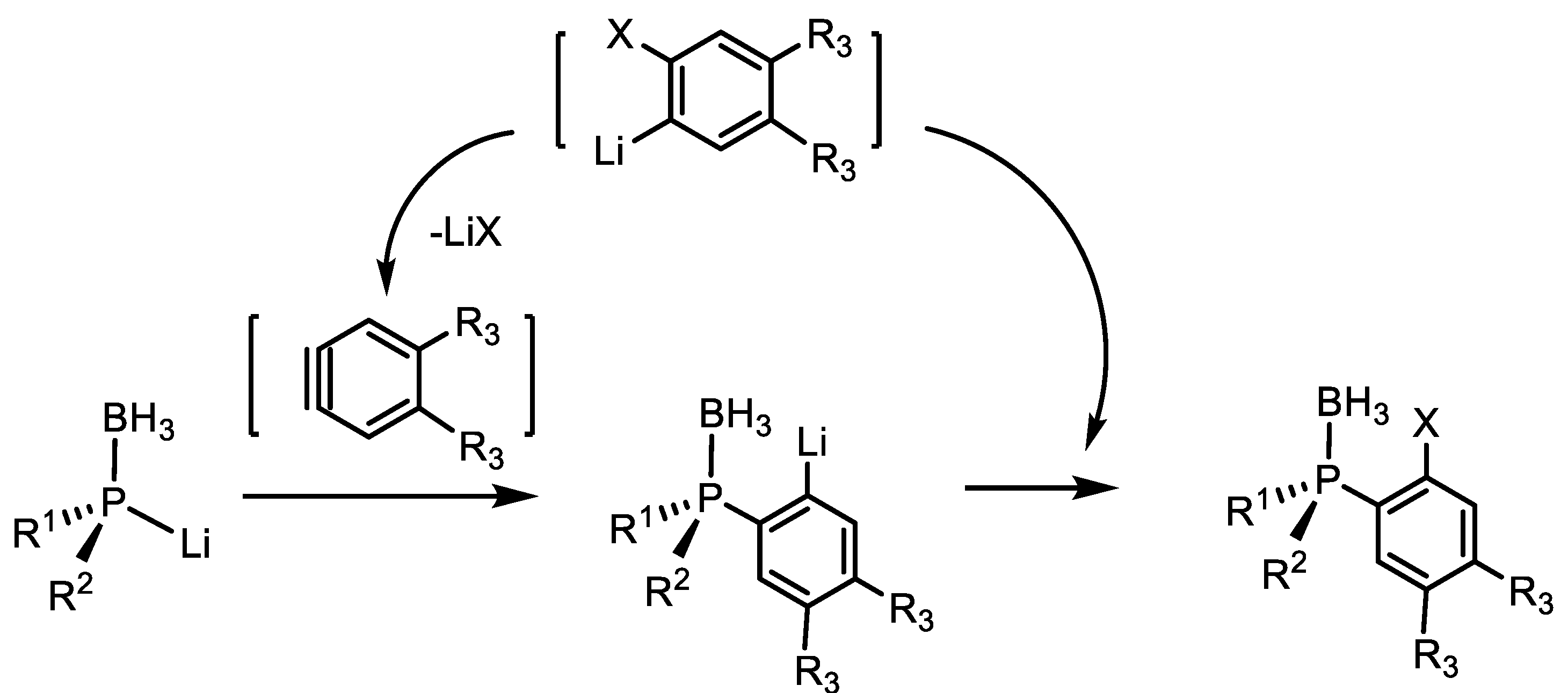
2. Alkylation and Arylation Reactions








































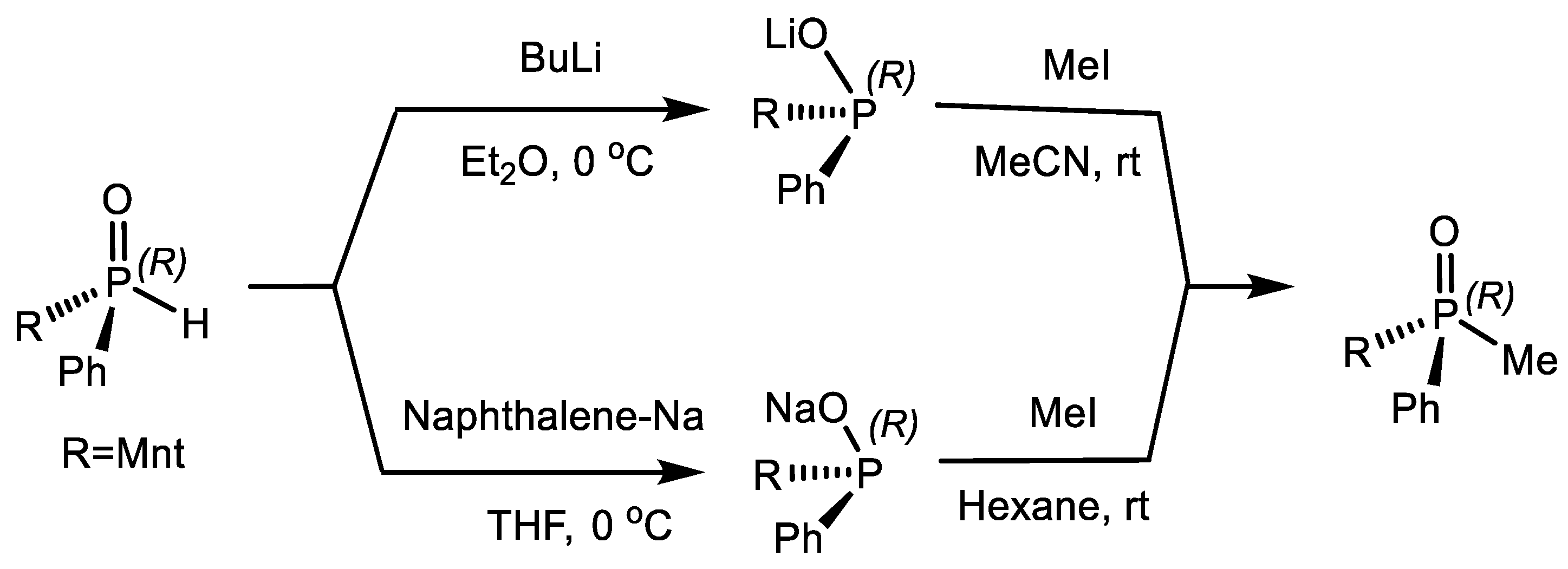
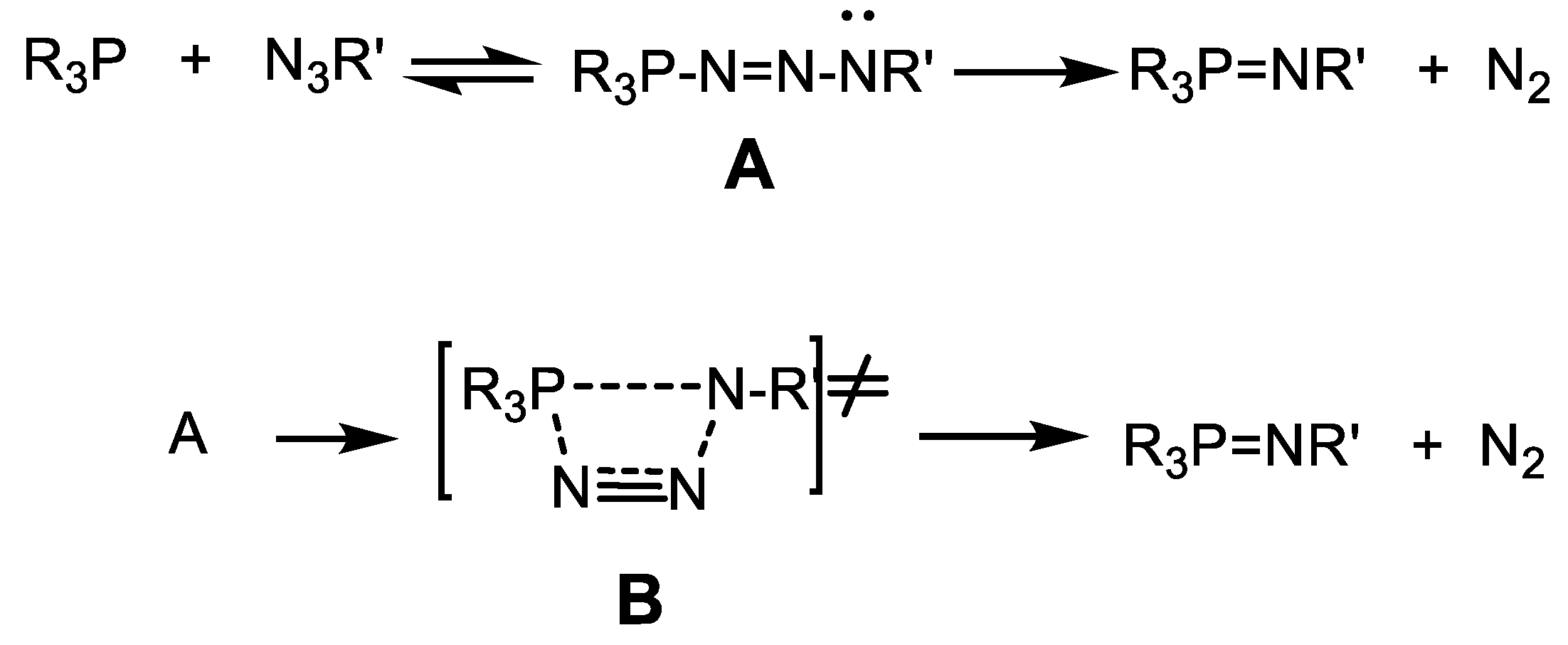
 B occurred, which then quickly turned into phosphonate. The discovery of stable intermediate A, according to the authors, indicates a monomolecular mechanism of reaction (Equation (42)) [53].
B occurred, which then quickly turned into phosphonate. The discovery of stable intermediate A, according to the authors, indicates a monomolecular mechanism of reaction (Equation (42)) [53].




















3. Halogenophilic Reactions of P(III) Compounds







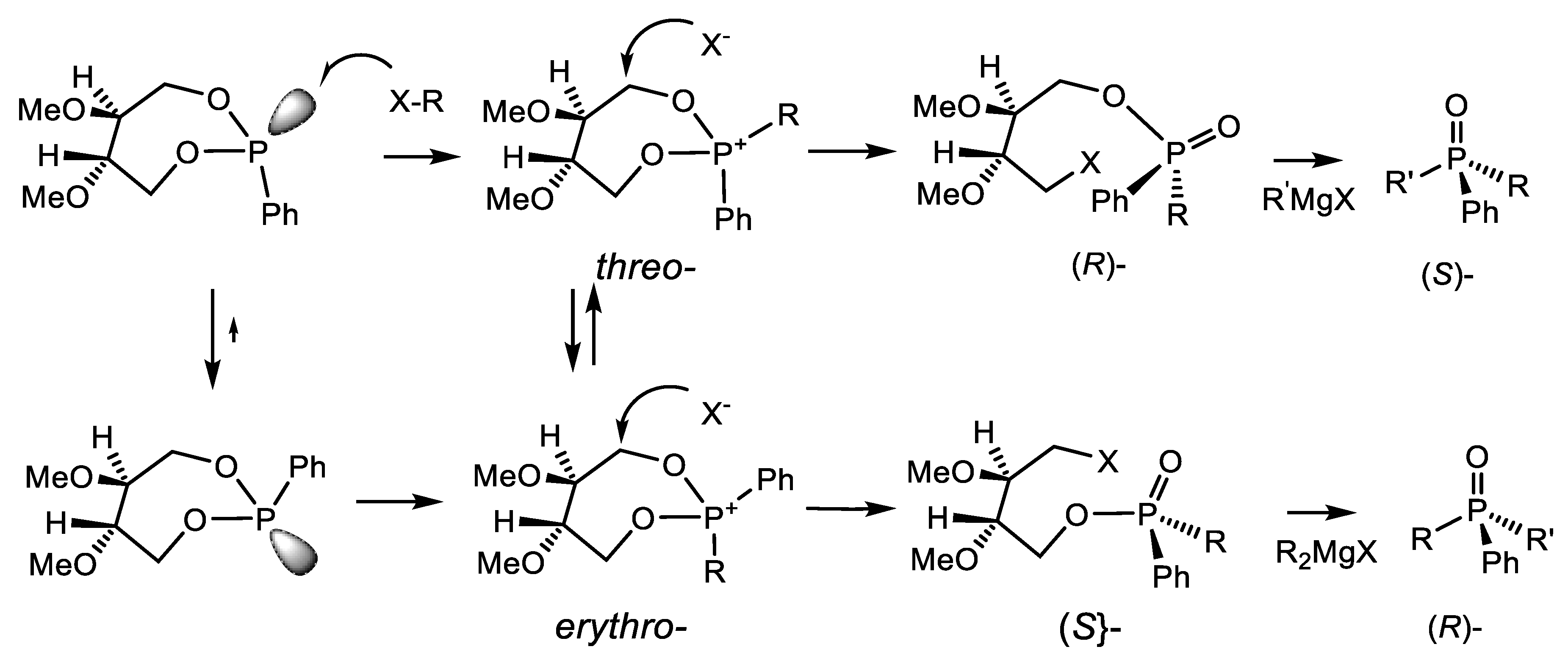
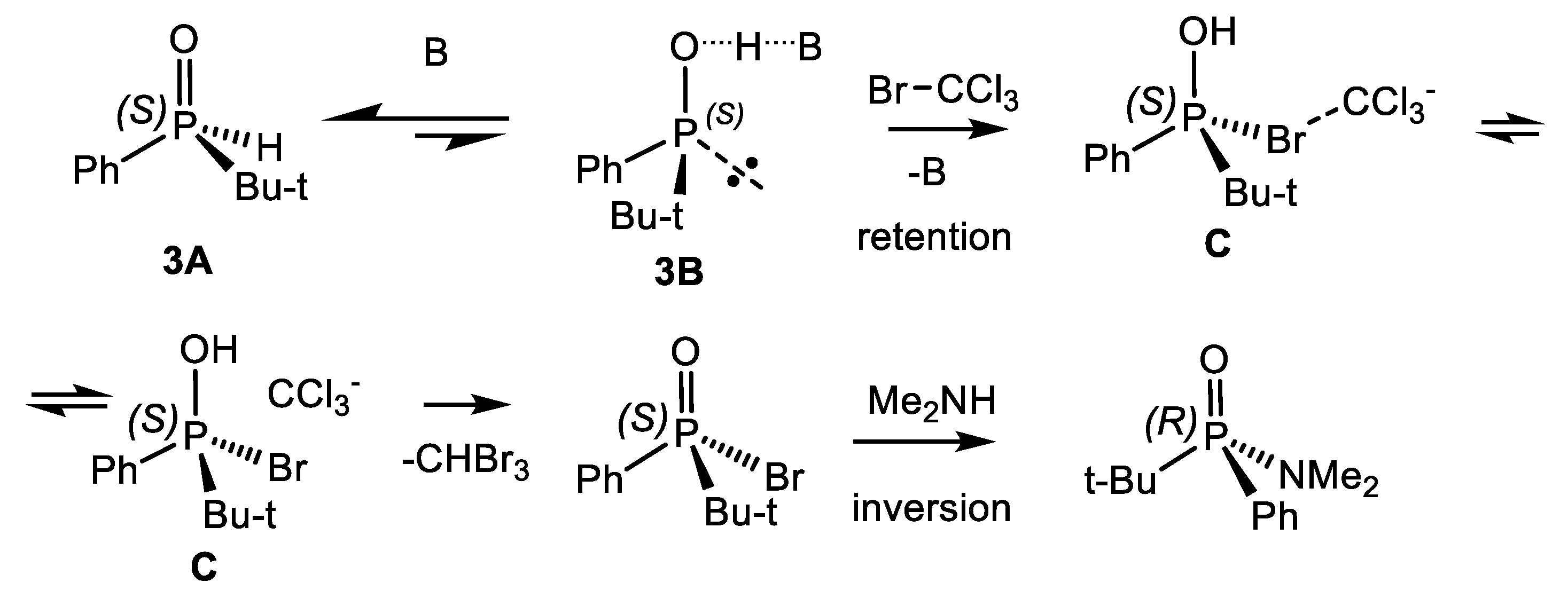
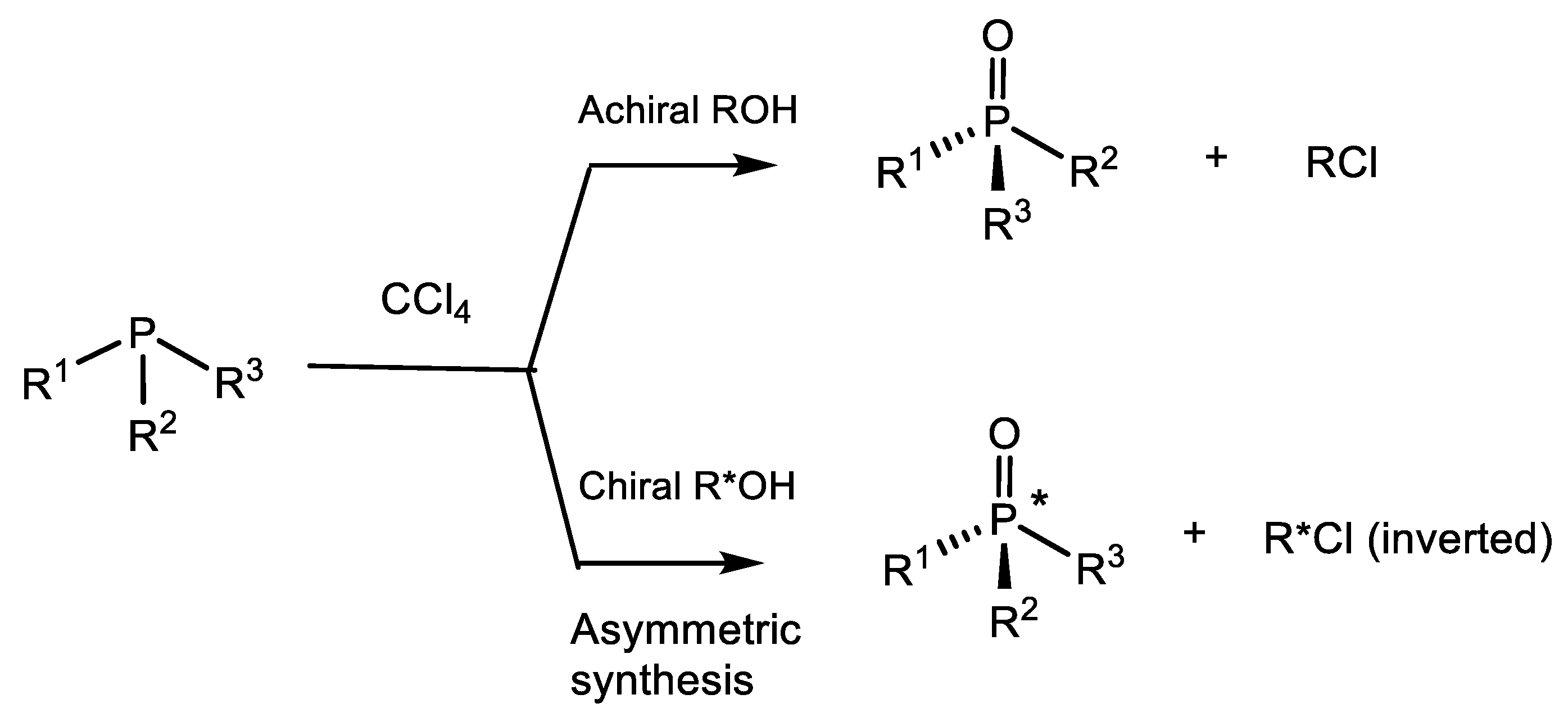
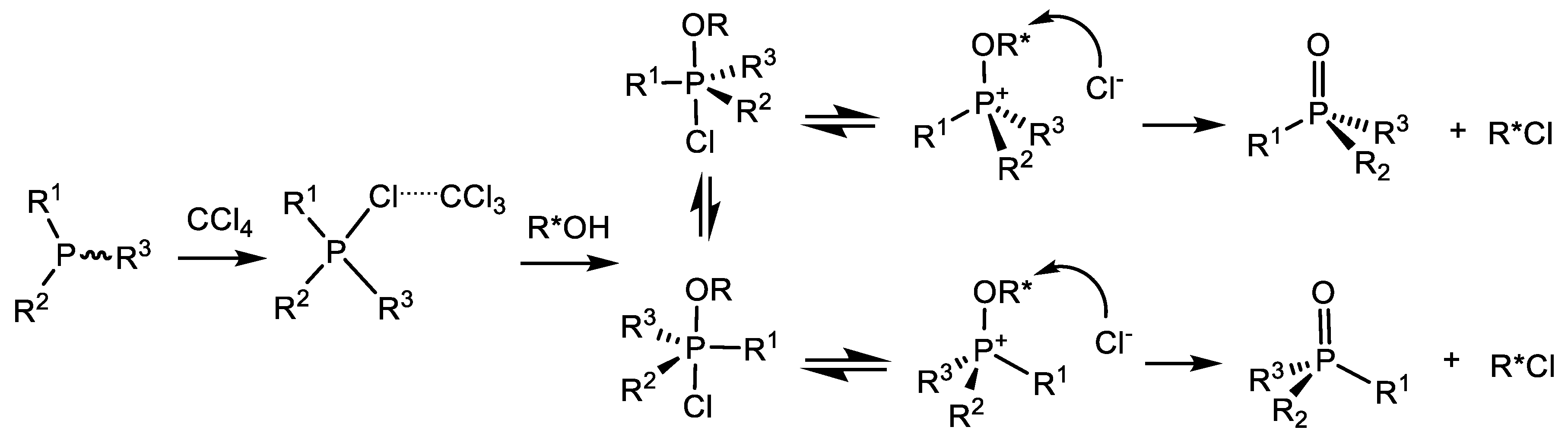
 P(IV) equilibrium of compounds towards the alkoxyphosphonium salt. The pseudo-rotation of ligands at the P(V) atom leads to the formation of the most thermodynamically stable diastereomer of the halogen-phosphonium intermediate, which then turns into one diastereomer of the final product (Equation (69)) [89].
P(IV) equilibrium of compounds towards the alkoxyphosphonium salt. The pseudo-rotation of ligands at the P(V) atom leads to the formation of the most thermodynamically stable diastereomer of the halogen-phosphonium intermediate, which then turns into one diastereomer of the final product (Equation (69)) [89].








4. Electrophilic Asymmetric Catalysis
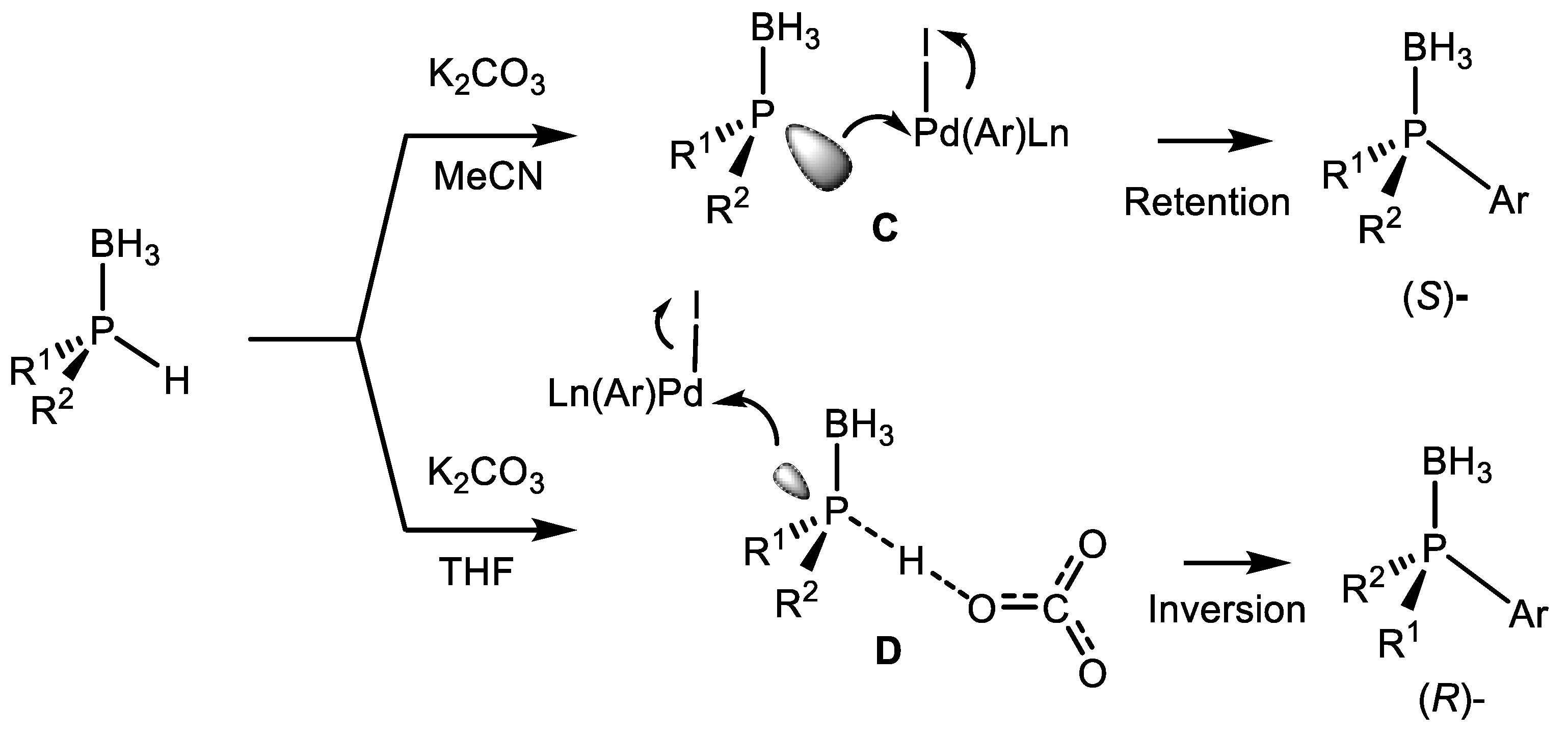
(RP)-4 (Equation (77)). Adducts were isolated and studied by low temperature NMR. X-Ray diffraction analysis showed the (RP)-absolute configuration of this compound [107]. The treatment of this adduct with benzyl bromide resulted in the formation of (RP)-tertiary phosphine with 72–78% ee and the initial catalyst. Alkylation of secondary phosphines occurred with retention of absolute configuration, that confirmed the proposed mechanism. Based on these results, it was concluded that the enantioselectivity of electrophilic substitution is determined by the thermodynamic preference of one of the diastereomers in the equilibrium between the (SP)-4 and (RP)-4 complexes.













5. Oxidation, Thionation, Imination











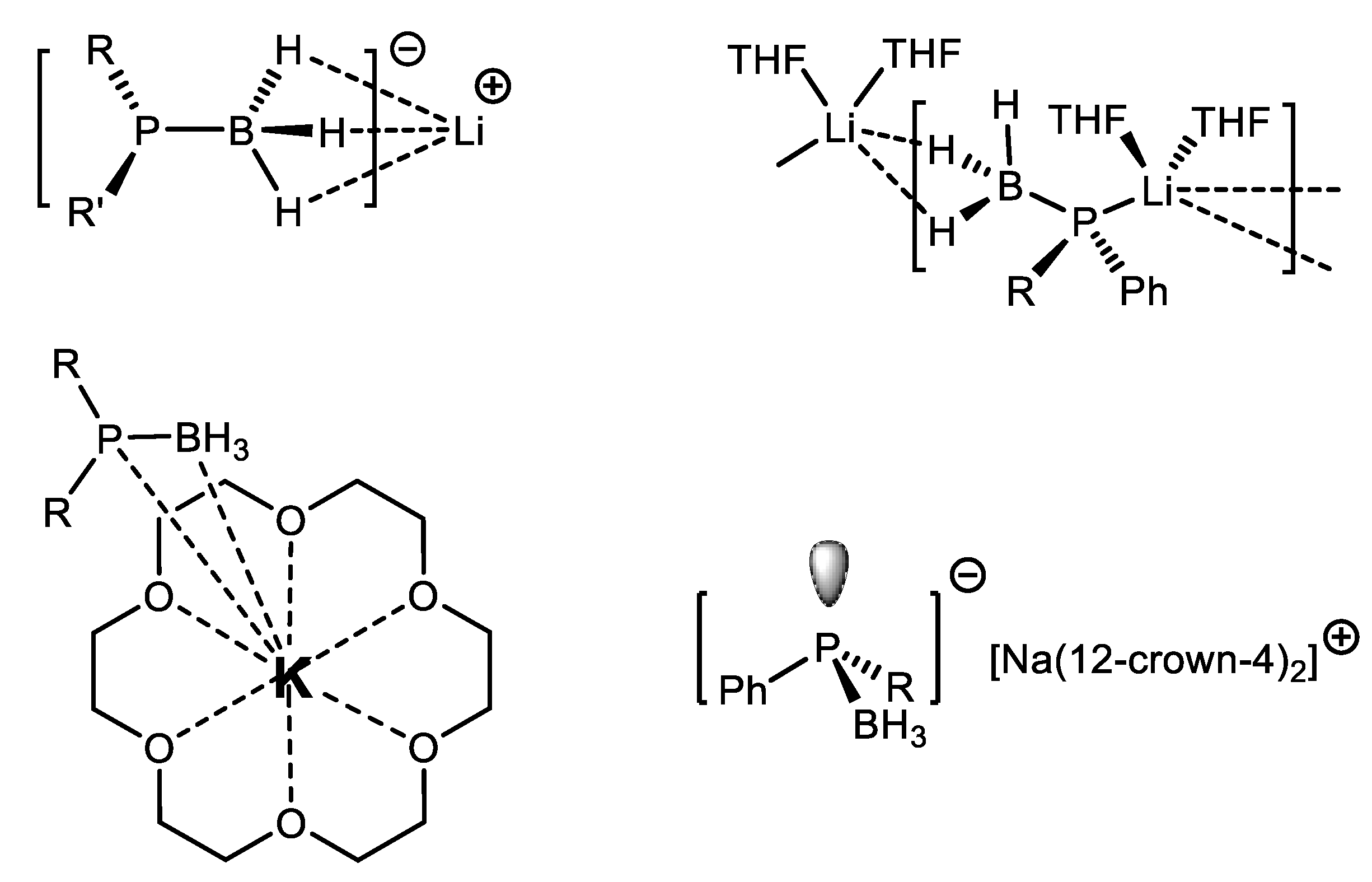
6. Reduction







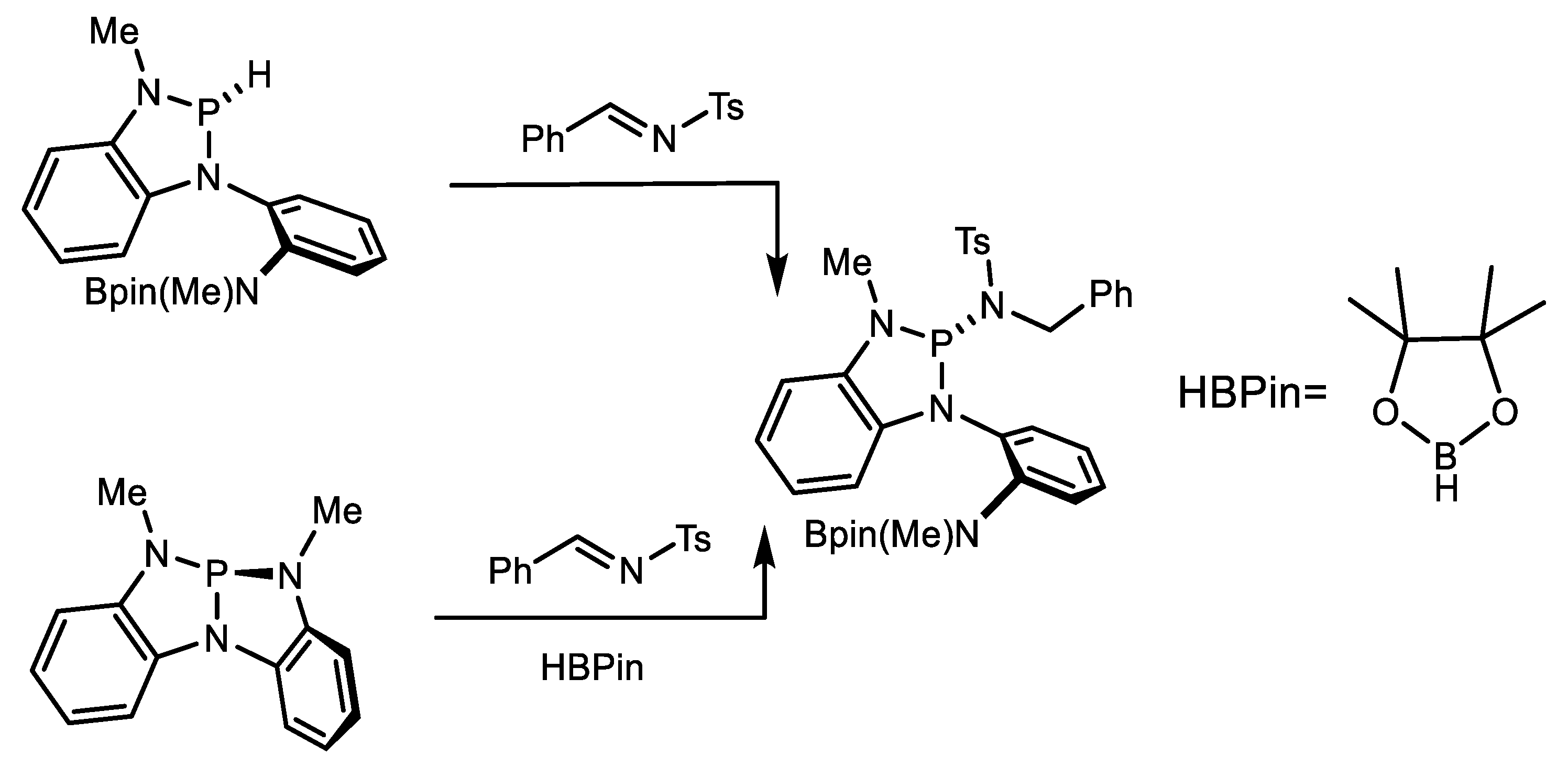
7. Imination

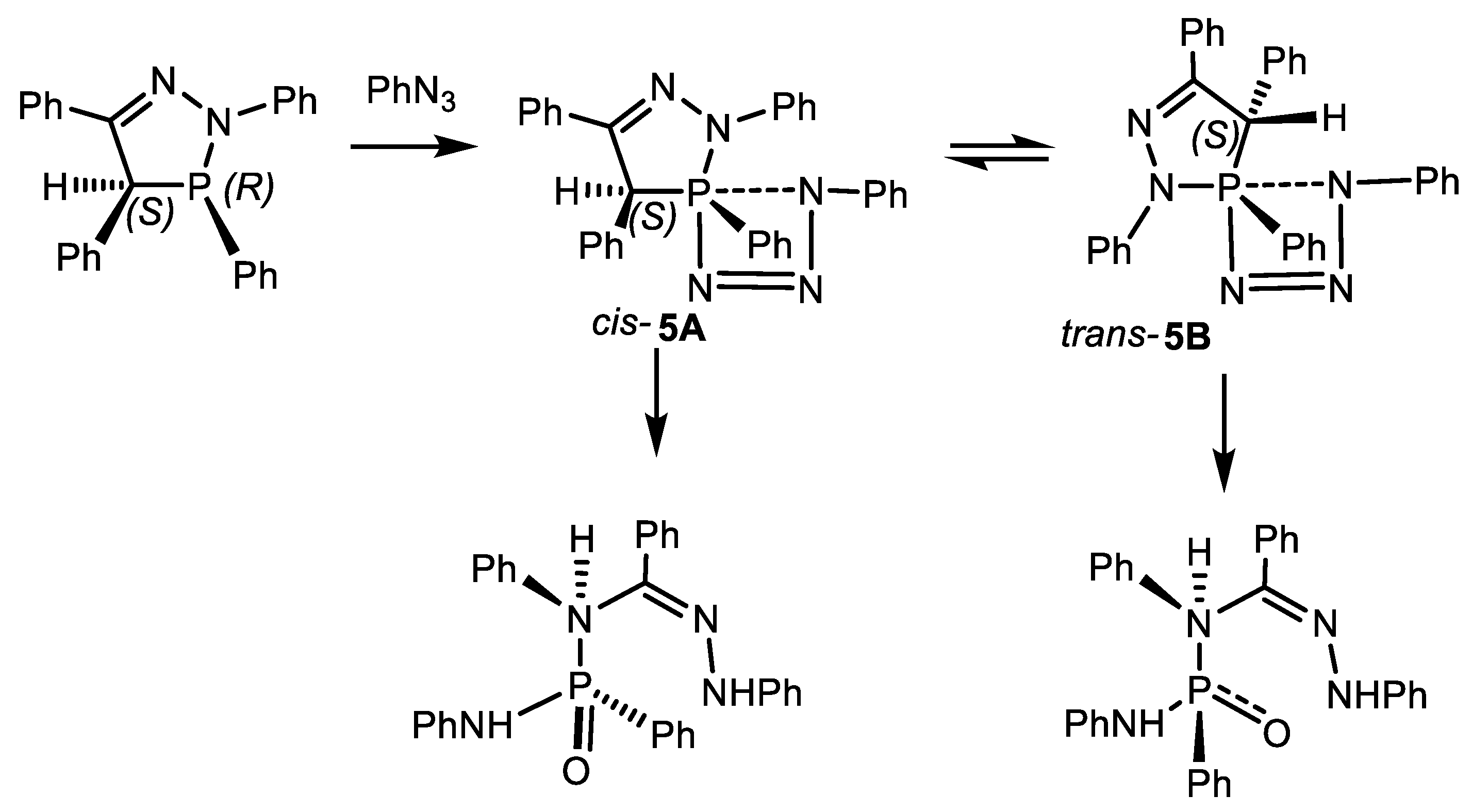
 5B [148].
5B [148].
8. Addition to Multiple Bonds



















9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Fukuto, J.; Jensen, F.R. Mechanisms of SE2 reactions: Emphasis on organotin compounds. Acc. Chem. Res. 1983, 16, 177–184. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Asymmetric Synthesis in Organophosphorus Chemistry: Synthetic Methods, Catalysis and Application; Wiley-VCH: Weinheim, Germany, 2016. [Google Scholar]
- Kolodiazhnyi, O.I.; Kolodiazhna, A.O. Nucleophilic substitution at phosphorus: Stereochemistry and mechanisms. Tetrahedron Asymmetry 2017, 28, 1651–1674. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Recent Advances in Asymmetric Synthesis of P-Stereogenic Phosphorus Compounds. In Topics in Current Chemistry; Montchamp, J.-L., Ed.; Springer Intern. Publ.: Cham, Switzerland, 2015; Volume 361, pp. 161–236. [Google Scholar] [CrossRef]
- Eliel, E.L.; Wilen, S.H.; Doyle, P. Basic Organic Stereochemistry; J. Wiley Interscience: New York, NY, USA, 2001. [Google Scholar]
- Kolodiazhnyi, O.I.; Kolodiazhna, A.O. Stereoselective Synthesis of Organophosphorus Compounds; Naukova Dumka: Kiev, Ukraine, 2017. [Google Scholar]
- Nandi, P.; Dye, J.L.; Bentley, P.; Jackson, J.E. Preparation of diphenyl phosphide and substituted phosphines using alkali metal in silica gel (M-SG). Org. Lett. 2009, 11, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Naylor, R.A.; Walker, B. New routes to optically active phosphorus compounds. Asymmetric alkylation of phosphide anions. J. Chem. Soc. Chem. Commun. 1975, 6, 45–46. [Google Scholar] [CrossRef]
- Valentine, D., Jr.; Blount, J.F.; Toth, K. Synthesis of phosphines having chiral organic groups ligated to chiral phosphorus. J. Org. Chem. 1980, 45, 3691–3698. [Google Scholar] [CrossRef]
- Fisher, C.; Mosher, H. Asymmetric homogeneous hydrogenation with phosphine-rhodium complexes chiral both at phosphorus and carbon. Tetrahedron Lett. 1977, 18, 2487–2490. [Google Scholar] [CrossRef]
- Burgess, K.; Ohimeyer, M.J.; Whitmire, K.H. Stereochemically matched (and mismatched) bisphosphine ligands: DIOP-DIPAMP hybrids. Organometallics 1992, 11, 3588–3600. [Google Scholar] [CrossRef]
- Nagel, U.; Krink, T. Neue optisch reine 3,4-Bis(phosphanyl)pyrrolidine mit Phenyl-und Anisylgruppen sowie deren Palladium-und Rhodiumkomplexe. Chem. Ber. 1993, 126, 1091–1100. [Google Scholar] [CrossRef]
- Nagel, U.; Bublewitz, A. Neue 1,2-Bisphosphanliganden mit vier stereogenen Zentren und zusatzlichen Methoxygruppen fur die asymmetrische katalytische Hydrierung. Chem. Ber. 1992, 125, 1061–1072. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Recent developments in the asymmetric synthesis of P-chiral phosphorus compounds. Tetrahedron Asymmetry 2012, 23, 1–46. [Google Scholar] [CrossRef]
- Mathey, F.; Herrmann, G.; Bellus, D.; Błażewska, K.M. Science of Synthesis: Houben-Weyl Methods of Molecular Transformations. Category 5. Compounds with One Saturated Carbon-Heteroatom Bond. Organophosphorus Compounds; Georg Thieme Verlag KG: Stuttgard, Germany; New York, NY, USA, 2009; Volume 42. [Google Scholar]
- Remond, E.; Tessier, A.; Leroux, F.R.; Bayardon, J.; Juge, S. Efficient synthesis of quaternary and p-stereogenic phosphonium triflates. Org. Lett. 2010, 12, 1568–1571. [Google Scholar] [CrossRef]
- Meddour, A.; Uziel, J.; Courtieu, J.; Juge, S. Enantiodifferentiation of acyclic phosphonium salts in chiral liquid crystalline solutions. Tetrahedron Asymmetry 2006, 17, 1424–1429. [Google Scholar] [CrossRef]
- Beaumont, A.J.; Kiely, C.; Rooney, A.D. Synthesis of novel chiral quaternary phosphonium fluorides: Reagents for simple asymmetric nucleophilic fluorination reactions. J. Fluor. Chem. 2001, 108, 47–50. [Google Scholar] [CrossRef] [Green Version]
- Coumbe, T.; Lawrence, N.J.; Muhammad, F. Titanium (IV) catalysis in the reduction of phosphine oxides. Tetrahedron Lett. 1994, 35, 625–628. [Google Scholar] [CrossRef]
- Dutartre, M.; Bayardon, J.; Jugé, S. Applications and stereoselective syntheses of P-chirogenic phosphorus compounds. Chem. Soc. Rev. 2016, 45, 5771–5794. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, S.C.; Hughes, R.P.; Glueck, D.S.; Rheingold, A.L. Synthesis, reactivity, and resolution of a C2Symmetric, PStereogenic benzodiphosphetane, a building block for chiral bis(phosphines). Org. Lett. 2012, 14, 4238–6241. [Google Scholar] [CrossRef]
- Vedejs, E.; Donde, Y. Stereogenic P-Trisubstituted phosphorus by crystallization-induced asymmetric transformation: A practical synthesis of phenyl(o-anisyl)methylphosphine borane. J. Am. Chem. Soc. 1997, 119, 9293–9294. [Google Scholar] [CrossRef]
- Vedejs, E.; Donde, Y. Crystallization-induced asymmetric transformation of a tertiary phosphine. J. Org. Chem. 2000, 65, 2337–2343. [Google Scholar] [CrossRef]
- Alayrac, C.; Lakhdar, S.; Abdellah, I.; Gaumont, A.-C. Recent advances in synthesis of P-BH3 compounds. Top. Curr. Chem. 2015, 361, 1–83. [Google Scholar] [CrossRef]
- Izod, K.; Watson, J.M.; Clegg, W.; Harrington, R.W. Phosphido-borane and phosphido-bis(borane) complexes of the alkali metals, a comparative study. Inorg. Chem. 2013, 52, 1466–1475. [Google Scholar] [CrossRef]
- Imamoto, T. Synthesis and reactions of new phosphine-boranes. Pure Appl. Chern. 1993, 65, 655–660. [Google Scholar] [CrossRef] [Green Version]
- Petit, C.; Favre-Reguillon, A.; Mignani, G.; Lemaire, M. A straightforward synthesis of unsymmetrical secondary phosphine-boranes. Green Chem. 2010, 12, 326–330. [Google Scholar] [CrossRef]
- Stankevic, X.M.; Pietrusiewicz, K.M. Resolution and stereochemistry of tert-butylphenylphosphinous acid-borane. J. Org. Chem. 2007, 72, 816–822. [Google Scholar] [CrossRef]
- Wauters, I.; Debrouwer, W.; Stevens, C.V. Preparation of phosphines through C–P bond formation. Beilstein J. Org. Chem. 2014, 10, 1064–1096. [Google Scholar] [CrossRef]
- Gatineau, D.; Giordano, L.; Buono, G. Optically active p-stereogenic Phosphine–boranes from pure h-menthylphosphinates. J. Am. Chem. Soc. 2011, 133, 10728–10731. [Google Scholar] [CrossRef]
- Imamoto, T.; Oshiki, T.; Onozawa, T.; Matsuo, M.; Hikosaka, T.; Yanagawa, M. Synthesis and reactions of optically active phosphine-boranes. Heteroatom Chem. 1992, 3, 563–575. [Google Scholar] [CrossRef]
- Bauduin, C.; Moulin, D.; Kaloun, E.B.; Darcel, C.; Juge, S. Highly enantiomerically enriched chlorophosphine boranes: Synthesis and applications as p-chirogenic electrophilic blocks. J. Org. Chem. 2003, 68, 4293–4301. [Google Scholar] [CrossRef]
- Rajendran, K.V.; Gilheany, D.G. Simple unprecedented conversion of phosphine oxides and sulfides to phosphine-boranes using sodium borohydrides. Chem. Commun. 2012, 48, 817–819. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.-J.; Nie, S.-Z.; Wang, J.-P.; Wen, J.-H.; Zhang, Y.; Qiu, M.-R.; Zhao, C.-Q. Nucleophilic substitution of p-stereogenic chlorophosphines: Mechanism, stereochemistry, and stereoselective conversions of diastereomeric secondary phosphine oxides to tertiary phosphines. Org. Lett. 2017, 19, 5384–5387. [Google Scholar] [CrossRef]
- Al-Masum, M.; Kumaraswamy, G.; Livinghouse, T. A New Synthetic Route to P-Chiral Phosphine-Boranes of High Enantiopurity via Stereocontrolled Pd(0)-Cu(I) Cocatalyzed Aromatic Phosphorylation. J. Org. Chem. 2000, 65, 4776–4778. [Google Scholar] [CrossRef]
- Wolfe, B.; Livinghouse, T. A Direct Synthesis of P-Chiral Phosphine-Boranes via Dynamic Resolution of Lithiated Racemic tert-Butylphenylphosphine-Borane with (-)-Sparteine. J. Am. Chem. Soc. 1998, 120, 5116–5117. [Google Scholar] [CrossRef]
- Nie, S.-Z.; Zhou, Z.-Y.; Wang, J.-P.; Yan, H.; Wen, J.-H.; Ye, J.-J.; Cui, Y.-Y.; Zhao, C.-Q. Nonepimerizing Alkylation of H–P Species to Stereospecifically Generate P-Stereogenic Phosphine Oxides: A Shortcut to Bidentate Tertiary Phosphine Ligands. J. Org. Chem. 2017, 82, 9425–9434. [Google Scholar] [CrossRef] [PubMed]
- Moraleda, D.; Gatineau, D.; Martin, D.; Giordano, L.; Buono, G. A simple route to chiral phosphinous acid–boranes. Chem. Commun. 2008, 3031–3033. [Google Scholar] [CrossRef] [PubMed]
- Juge, S. Enantioselective Synthesis of P-Chirogenic Phosphorus Compounds via the Ephedrine-Borane Complex Methodology. Phosphorus Sulfur Silicon Relat. Elem. 2008, 183, 233–248. [Google Scholar] [CrossRef]
- Bayardon, J.; Laureano, H.; Diemer, V.; Dutartre, M.; Das, U.; Rousselin, Y.; Henry, J.-C.; Colobert, F.; Leroux, F.R.; Jugeé, S. Stereoselective synthesis of o-bromo (or iodo)aryl P-chirogenic phosphines based on aryne chemistry. J. Org. Chem. 2012, 77, 5759–5769. [Google Scholar] [CrossRef]
- Kaloun, E.B.; Merdes, R.; Genet, J.P.; Uziel, J.; Jugé, S. Asymmetric synthesis of (S,S)-(+)-1,1′-bis(methyl-phenylphosphino)ferrocene. J. Organometall. Chem. 1997, 529, 455–463. [Google Scholar] [CrossRef]
- Uziel, J.; Riegel, N.; Aka, B.; Figuiere, P.; Juge, S. A Practical Synthesis of Chiral and Achiral Phosphonium Salts. Tetrahedron Lett. 1997, 38, 3405–3408. [Google Scholar] [CrossRef]
- Maienza, F.; Spindler, F.; Thommen, T.M.; Pugin, B.; Mezzetti, A. Exploring Stereogenic Phosphorus: The Search for New Chiral Diphosphines. Chimia 2001, 55, 694–698. [Google Scholar] [CrossRef]
- Minois, P.; Bayardon, J.; Meunier-Prest, R.; Jugé, S. Fullerene l-Amino Acids and Peptides: Synthesis under Phase-Transfer Catalysis Using a Phosphine–Borane Linker. Electrochemical Behavior. J. Org. Chem. 2017, 82, 11358–11369. [Google Scholar] [CrossRef]
- Imamoto, T.; Yamanoi, Y. Methylene Insertion Reactions of Samarium Carbenoids into Boron-Hydrogen and Phosphorus-Hydrogen Bonds. Chem. Lett. 1996, 8, 705–706. [Google Scholar] [CrossRef]
- Imamoto, T. Development of P-Chirogenic Phosphine Ligands Based on Chemistry of Phosphine–Boranes: Searching for Novelty and Utility in Synthetic Organic Chemistry. TCIMAIL 2017, N174, 1–18. Available online: https://www.tcichemicals.com/en/in/support-download/tcimail/backnumber/article/174drE_1.pdf (accessed on 15 November 2019).
- Katagiri, K.; Danjo, H.; Yamaguchi, K.; Imamoto, T. Nucleophilic aromatic substitution reactions of fluorobenzenechromium complexes with P-chiral secondary phosphine–boranes: Synthesis of optically pure P-chiral (dialkyl) arylphosphine-boranes. Tetrahedron 2005, 61, 4701–4707. [Google Scholar] [CrossRef]
- Jayaraman, A.; Nilewar, S.; Jacob, T.V.; Sterenberg, B.T. Sequential Electrophilic Substitution Reactions of Tungsten-Coordinated Phosphenium Ions and Phosphine Triflates. ACS Omega 2017, 2, 7849–7861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, T.; Kobayashi, K.; Masuda, S.; Segi, M.; Nakajima, T.; Suga, S. Asymmetric Synthesis of Phosphine Oxides with the Arbuzov Reaction. Chem. Lett. 1987, 1915–1918. [Google Scholar] [CrossRef] [Green Version]
- Bhatacharya, A.K.; Thyagarman, G. The Michaelis-Arbuzov Rearrangement. Chem. Rev. 1981, 81, 415–430. [Google Scholar] [CrossRef]
- Berlin, K.D.; Hellwege1, D.M.; Nagabhushanam, M.; Gaudy, E.T. Evidence for a stereospecific Michaelis-Arbuzov rearrangement in 4-t-butylcyclohexyl diphenylphosphinite, a conformationally homogeneous system—III. Tetrahedron 1966, 22, 2191–2201. [Google Scholar] [CrossRef]
- Segi, M.; Nakamura, Y.; Nakajima, T.; Suga, S. Preparation of optically active phosphine oxides by regioselective cleavage of cyclic phenylphosphonite with alkyl halides. Chem. Lett. 1983, 913–916. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Valle, M.E.; Martínez-Álvarez, R.; Molero-Vílchez, D.Z. 2D Ultrafast HMBC 1H,31P: Obtaining Mechanistic Details on the Michaelis–Arbuzov Reaction. J. Org. Chem. 2015, 80, 799–805. [Google Scholar] [CrossRef]
- Michalski, J.; Mikołajczak, J.; Pakulski, M.; Skowronska, A. The stereochemistry of dealkylation step in the Arbuzov reaction involving fivecoordinate intermediate. Evidence for equilibrium between phosphorane and phosphonium species. Phosphorus Sulfur Silicon Relat. Elem. 1978, 4, 233–234. [Google Scholar] [CrossRef]
- van den Berg, G.R.; Platenburg, D.H.J.M.; Benschop, H.P. Stereochemistry of a Michaelis–Arbusov reaction: Alkylation of optically active ethyl trimethylsilyl phenylphosphonite with retention of configuration. Chem. Commun. 1971, 606–607. [Google Scholar] [CrossRef]
- Bodalski, R.; Rutkowska-Olma, E.; Pietrusiewicz, K.M. Optically active Phosphine oxides: Synthesis and absolute configuration of (menthoxycarbonylmethyl) phenylvinyl phosphine oxide. Tetrahedron 1980, 36, 2353–2355. [Google Scholar] [CrossRef]
- Pietrusiewicz, K.M. Stereoselective Synthesis and Resolution of P-Chiral Phosphine Chalcogenides. Phosphorus Sulfur Silicon Relat. Elem. 1996, 109, 573–576. [Google Scholar] [CrossRef]
- Johnson, C.R.; Imamoto, T. Synthesis of Polydentate Ligands with Homochiral Phosphine Centers. J. Org. Chem. 1987, 52, 2170–2174. [Google Scholar] [CrossRef]
- Faure, B.; Archavlis, A.; Buono, G. Stereoselective synthesis of (Rp)-benzylphenyl-[2-(S)-bromomethylpyrrolidine-1-yl]phosphine oxide from (S)-(+)-prolinol by the Michaelis–Arbuzov reaction: Application in the synthesis of a chiral hybrid phosphine–phosphine oxide ligand. Chem. Commun. 1989, 805–807. [Google Scholar] [CrossRef]
- Savignac, P.; Iorga, B. Modern Phosphonate Chemistry; CRC Press: Boca Raton, FL, USA, 2003; p. 552. [Google Scholar]
- Zhang, J.; Xu, Y.; Huang, G.; Guo, H. Palladium-catalyzed synthesis of chiral, nonracemic isopropyl arylmethylphosphinates. Tetrahedron Lett. 1988, 29, 1955–1958. [Google Scholar] [CrossRef]
- Kato, T.; Tejlma, M.; Ebiike, H.; Achiwa, K. Asymmetric synthesis of (S)-(+)-and (R)-(-)-NZ-105 through the modified Michaelis-Arbuzov rearrangement as a key step. Chem. Pharm. Bull. 1996, 44, 1132–1134. [Google Scholar] [CrossRef] [Green Version]
- Mikolajczyk, M.; Krzywanski, J.; Ziemnicka, B.J. Stereochemistry of organophosphorus cyclic compounds. 6. Stereochemistry of the reaction between sulfenyl chlorides and trivalent phosphorus compounds. J. Org. Chem. 1977, 42, 190–199. [Google Scholar] [CrossRef]
- Berger, O.A.; Montchamp, J.L. A General Strategy for the Synthesis of P-Stereogenic Compounds. Angew. Chem. Int. Ed. Engl. 2013, 52, 11377–11380. [Google Scholar] [CrossRef]
- Montchamp, J.L. Organophosphorus synthesis without phosphorus trichloride: The case for the hypophosphorous pathway. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 66–75. [Google Scholar] [CrossRef]
- Haynes, R.K.; Freeman, R.N.; Mitchell, C.R.; Vonwiller, S.C. Preparation of Enantiomerically Pure Tertiary Phosphine Oxides from, and Assay of Enantiomeric Purity with, (Rp)- and (Sp)-tert-Butylphenylphosphinothioic Acids. J. Org. Chem. 1994, 59, 2919–2921. [Google Scholar] [CrossRef]
- Hirao, T.; Masunaga, T.; Ohshiro, Y.; Agawa, T. Stereoselective synthesis of vinylphosphonate. Tetrahedron Lett. 1980, 21, 3595–3596. [Google Scholar] [CrossRef]
- Keglevich, G.; Jablonkai, E.; Balázsa, L.B. A “green” variation of the Hirao reaction: The P–C coupling of diethyl phosphite, alkyl phenyl-Hphosphinates and secondary phosphine oxides with bromoarenes using a P-ligand-free Pd(OAc)2 catalyst under microwave and solvent-free conditions†. RSC Adv. 2014, 4, 22808–22816. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wei, H.; Zhang, J.; Huang, G. An efficient synthesis of chiral, nonracemic isopropyl alkenylmethylpbosphinates via palladium route. Tetrahedron Lett. 1989, 30, 949–952. [Google Scholar] [CrossRef]
- Kalek, M.; Jezowska, M.; Stawinskia, J. Preparation of Arylphosphonates by Palladium(0)-Catalyzed Cross-Coupling in the Presence of Acetate Additives: Synthetic and Mechanistic Studies. Adv. Synth. Catal. 2009, 351, 3207–3216. [Google Scholar] [CrossRef]
- Zhang, Y.; He, H.; Wang, Q.; Cai, Q. Asymmetric synthesis of chiral P-stereogenic triaryl phosphine oxides via Pd-catalyzed kinetic arylation of diaryl phosphine oxides. Tetrahedron Lett. 2016, 57, 5308–5311. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, Y. Stereochemistry at the Phosphorus Atom during Palladium-catalysed Formation of Carbon-Phosphorus Bonds and Mechanistic Implications. Chem. Commun. 1986, 1606. [Google Scholar] [CrossRef]
- Anderson, B.J.; Glueck, D.S.; DiPasquale, A.G.; Rheingold, A.L. Substrate and Catalyst Screening in Platinum-Catalyzed Asymmetric Alkylation of Bis(secondary) Phosphines. Synthesis of an Enantiomerically Pure C2-Symmetric Diphosphine. Organometallics 2008, 27, 4992–5001. [Google Scholar] [CrossRef]
- Whittaker, B.; de Lera Ruiz, M.; Hayes, C.J. Stereoselective synthesis of highly functionalised P-stereogenic nucleosides via palladium-catalysed P–C cross-coupling reactions. Tetrahedron Lett. 2008, 49, 6984–6987. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Reaction of sterically hindered phosphines with tetrahalo methanes. P-Halogenylides. Russ. J. Gen. Chem. 1981, 51, 2466–2480. [Google Scholar]
- Zhou, Y.; Wang, G.; Saga, Y.; Shen, R.; Goto, M.; Zhao, Y.; Han, L.-B. Stereospecific Halogenation of P(O)-H Bonds with Copper(II) Chloride Affording Optically Active Z1Z2P(O)Cl. J. Org. Chem. 2010, 75, 7924–7927. [Google Scholar] [CrossRef]
- Appel, R. Tertiary Phosphane/Tetrachloromethane, a Versatile Reagent for Chlorination, Dehydration, and P-N Linkage. Angew. Chem. Int. Ed. Engl. 1975, 14, 801–811. [Google Scholar] [CrossRef]
- Sazonov, P.K.; Artamkina, G.A.; Beletskaya, I.P. Nucleophilic substitution at the halogen atom (halogenophilic reactions). Russ. Chem. Rev. 2012, 81, 317–335. [Google Scholar] [CrossRef]
- Atherton, F.R.; Openshaw, H.T.; Todd, A.R. Studies on Phosphorylation. Part 11. The Reaction of Diallcyl Phosphites with Polyhalogen Compounds in Presence of Bases. A New Method for the Phosphorylation of Amines. J. Chem. Soc. 1945, 660–664. [Google Scholar] [CrossRef]
- Steinberg, G.M. Reactions of dialkyl phosphites. Synthesis of dialkyl chlorophosphates, tetraalkyl pyrophosphates, and mixed orthophosphate esters. J. Org. Chem. 1950, 15, 637–647. [Google Scholar] [CrossRef]
- Wang, G.; Shen, R.; Xu, Q.; Goto, M.; Zhao, Y.; Han, L.-B. Stereospecific Coupling of H-Phosphinates and Secondary Phosphine Oxides with Amines and Alcohols: A General Method for the Preparation of Optically Active Organophosphorus Acid Derivatives. J. Org. Chem. 2010, 75, 3890–3892. [Google Scholar] [CrossRef]
- Xiong, B.; Zhou, Y.; Zhao, C.; Goto, M.; Yin, S.-F.; Han, L.-B. Systematic study for the stereochemistry of the Athertone-Todd reaction. Tetrahedron 2013, 69, 9373–9380. [Google Scholar] [CrossRef]
- Denton, R.M.; An, J.; Adeniran, B.; Blake, A.J.; Lewis, W.; Poulton, A.M. Catalytic Phosphorus(V)-Mediated Nucleophilic Substitution Reactions: Development of a Catalytic Appel Reaction. J. Org. Chem. 2011, 76, 6749–6767. [Google Scholar] [CrossRef]
- Le Corre, S.S.; Berchel, M.; Couthon-Gourvès, H.; Haelters, J.-P.; Jaffrès, P.-A. Atherton–Todd reaction: Mechanism, scope and applications. Beilstein J. Org. Chem. 2014, 10, 1166–1196. [Google Scholar] [CrossRef] [Green Version]
- Reiff, L.P.; Aaron, H.S. Stereospecific Synthesis and Reactions of Optically Active Isopropyl Methylphosphinate. J. Am. Chem. Soc. 1970, 92, 5275–5276. [Google Scholar] [CrossRef]
- Stec, W.; Mikolajczyk, M. Stereochemistry of organophosphorus cyclic compounds-ii stereospecific synthesis of cis- and trans-2-halogeno-2-oxo-4-methyl-l,3,2-dioxaphosphorinans and their chemical transformations. Tetrahedron 1973, 29, 539–546. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. (1R,2S,5R)-Menthyl Phosphinate and Its Propertie. Russ. J. Gen. Chem. 2005, 75, 656–657. [Google Scholar] [CrossRef]
- Cao, S.X.; Gao, P.; Guo, Y.C.; Zhao, H.M.; Wang, J.; Liu, Y.F.; Zhao, Y.F. Unexpected Insertion of CO2 into the Pentacoordinate P–N Bond: Atherton–Todd-Type Reaction of Hydrospirophosphorane with Amines. J. Org. Chem. 2013, 78, 11283–11293. [Google Scholar] [CrossRef] [PubMed]
- Kolodiazhnyi, O.I. Stereoselective Oxidation of N-Phosphor (Ill) Substituted Amino Acids. Tetrahedron Lett. 1995, 36, 3921–3924. [Google Scholar] [CrossRef]
- Bergin, E.; O’Connor, C.T.; Robinson, S.B.; McGarrigle, E.M.; O’Mahony, C.P.; Gilheany, D.G. Synthesis of P-stereogenic phosphorus compounds. asymmetric oxidation of phosphines under Appel conditions. J. Am. Chem. Soc. 2007, 129, 9566–9567. [Google Scholar] [CrossRef]
- Rajendran, K.V.; Gilheany, D.G. Identification of a key intermediate in the asymmetric Appel process: One pot stereoselective synthesis of P-stereogenic phosphines and phosphine-boranes from racemic phosphine oxides. Chem. Commun. 2012, 48, 10040–10042. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, K.V.; Kennedy, L.; Gilheany, D.G. P-Stereogenic phosphorus compounds: Effect of aryl substituents on the oxidation of arylmethylphenylphosphanes under asymmetric Appel conditions. Eur. J. Org. Chem. 2010, N 29, 5642–5649. [Google Scholar] [CrossRef]
- Rajendran, K.V.; Kudavalli, J.S.; Dunne, K.S.; Gilheany, D.G. A U-turn in the asymmetric appel reaction: Stereospecific reduction of diastereomerically enriched alkoxyphosphonium salts allows the asymmetric synthesis of P-stereogenic phosphanes and phosphane boranes. Eur. J. Org. Chem. 2012, N 14, 2720–2723. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, K.V.; Kennedy, L.; O’Connor, C.T. Systematic survey of positive chlorine sources in the asymmetric Appel reaction: Oxalyl chloride as a new phosphine activator. Tetrahedron Lett. 2013, 54, 7009–7012. [Google Scholar] [CrossRef]
- Kolodiazhna, A.O.; Kolodiazhnyi, O.I. Synthesis, Properties and Stereochemistry of 2-Halo-1,2λ5-oxaphosphetanes. Molecules 2016, 21, 1371. [Google Scholar] [CrossRef] [Green Version]
- Kolodiazhnyi, O.I. Thiocetenes phosphores. Tetrahedron Lett. 1987, 28, 881–884. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Chemistry of P-F Ylides. Russ. J. Gen. Chem. 2005, 75, 1017–1039. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Phosphorus Ylides. Chemistry and Application in Organic Synthesis; J. Wiley-VCH: Weinheim, Germany; New York, NY, USA; Chichester, UK, 1999. [Google Scholar]
- Kolodyazhnyi, O.I.; Kolodyazhnaya, A.O. A New Approach Towards Synthesis of Phosphorylated Alkenes. Russ. J. Gen. Chem. 2015, 85, 359–365. [Google Scholar] [CrossRef]
- Kolodiazhna, A.O.; Kolodiazhnyi, O.I. Synthesis and Properties of Four-Membered Phosphorus Heterocycles-2-Fluoro-1,2λ5-Oxaphosphetanes. Phosphorus Sulfur Silicon Relat. Elem. 2015, 190, 2232–2245. [Google Scholar] [CrossRef]
- Kolodyazhna, O.O.; Grishkun, E.V.; Sheiko, S.Y.; Kolodyazhna, A.O.; Kolodyazhnyi, O.I. Synthesis and Properties of tert-Butylphenylmethylene(chloro)phosphorane. Russ. J. Gen. Chem. 2015, 85, 1639–1643. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Phosphorus Ylide Chemistry; Naukova Dumka: Kiev, Ukraine, 1994. [Google Scholar]
- Corey, E.J.; Fuchs, P.L. A synthetic method for formyl→ethynyl conversion (RCHO→RCCH or RCCR′). Tetrahedron Lett. 1972, 1972, 3769–3772. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. P-Halogen-substituted phosphorus ylids. Russ. Chem. Rev. 1991, 60, 391–409. [Google Scholar] [CrossRef]
- Anderson, B.J.; Reynolds, S.C.; Guino-o, M.A.; Xu, Z.; Glueck, D.S. Effect of Linker Length on Selectivity and Cooperative Reactivity in Platinum-Catalyzed Asymmetric Alkylation of Bis(phenylphosphino)alkanes. ACS Catal. 2016, 6, 8106–8108. [Google Scholar] [CrossRef]
- Scriban, C.; Glueck, D.S. Platinum-Catalyzed Asymmetric Alkylation of Secondary Phosphines: Enantioselective Synthesis of P-Stereogenic Phosphines. J. Am. Chem. Soc. 2006, 128, 2788–2789. [Google Scholar] [CrossRef]
- Scriban, C.; Glueck, D.S.; Golen, J.A.; Rheingold, A.L. Platinum-Catalyzed Asymmetric Alkylation of a Secondary Phosphine: Mechanism and Origin of Enantioselectivity. Organometallics 2007, 26, 1788–1800. [Google Scholar] [CrossRef]
- Chan, V.S.; Chiu, M.; Bergman, R.G.; Toste, F.D. Development of Ruthenium Catalysts for the Enantioselective Synthesis of P-Stereogenic Phosphines via Nucleophilic Phosphido Intermediates. J. Am. Chem. Soc. 2009, 131, 6021–6032. [Google Scholar] [CrossRef]
- Chan, V.S.; Stewart, I.C.; Bergman, R.G.; Toste, F.D. Asymmetric Catalytic Synthesis of P-Stereogenic Phosphines via a Nucleophilic Ruthenium Phosphido Complex. J. Am. Chem. Soc. 2006, 128, 2786–2787. [Google Scholar] [CrossRef] [PubMed]
- Scriban, C.; Glueck, D.S.; DiPasquale, A.G.; Rheingold, A.L.; Scriban, C. Chiral Platinum Duphos Terminal Phosphido Complexes: Synthesis, Structure, Phosphido Transfer, and Ligand Behavior. Organometallics 2006, 25, 5435–5448. [Google Scholar] [CrossRef]
- Chapp, T.W.; Schoenfeld, A.J.; Glueck, D.S. Effects of Linker Length on the Rate and Selectivity of Platinum-Catalyzed Asymmetric Alkylation of the Bis(isitylphosphino)alkanes IsHP(CH2)nPHIs (Is = 2,4,6-(i-Pr)3C6H2, n = 1-5). Organometallics 2010, 29, 2465–2473. [Google Scholar] [CrossRef]
- Moncarz, J.R.; Brunker, T.J.; Glueck, D.S.; Sommer, R.D.; Rheingold, A.L. Stereochemistry of Palladium-Mediated Synthesis of PAMP–BH3:Retention of Configuration at P in Formation of Pd–P and P–C Bonds. J. Am. Chem. Soc. 2003, 125, 1180–1181. [Google Scholar] [CrossRef] [PubMed]
- Glueck, D.S. Recent Advances in Metal-Catalyzed C–P Bond Formation. Top. Organomet. Chem. 2010, 31, 65–100. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I.; Kukhar, V.P.; Kolodiazhna, A.O. Asymmetric catalysis as a method for the synthesis of chiral organophosphorus compounds. Tetrahedron Asymmetry 2014, 25, 865–922. [Google Scholar] [CrossRef]
- Chapp, T.W.; Glueck, D.S.; Golen, J.A.; Moore, C.E.; Rheingold, A.L. Platinum-Catalyzed Asymmetric Alkylation of Bis(isitylphosphino)ethane: Stereoselectivity Reversal in Successive Formation of Two P–C Bonds. Organometallics 2010, 29, 378–388. [Google Scholar] [CrossRef]
- Imamoto, T.; Yashio, K.; Crépy, K.V.L.; Katagiri, K.; Takahashi, H.; Kouchi, M.; Gridnev, I.D. P-Chiral tetraphosphine dirhodium complex as a catalyst for asymmetric hydrogenation: Synthesis, structure, enantioselectivity, and mechanism. stereoselective formation of a dirhodium tetrahydride complex and its reaction with methyl (Z)-α-acetamidocinnamate. Organometallics 2006, 25, 908–914. [Google Scholar] [CrossRef]
- Guino’o, M.A.; Zureick, A.H.; Blank, N.F.; Anderson, B.J.; Chapp, T.W.; Kim, Y.; Glueck, D.S.; Rheingold, A.L. Synthesis and Structure of Platinum Bis(phospholane) Complexes Pt(diphos*)(R)(X), Catalyst Precursors for Asymmetric Phosphine Alkylation. Organometallics 2012, 31, 6900–6910. [Google Scholar] [CrossRef]
- Moncarz, J.R.; Brunker, T.J.; Jewett, J.C.; Orchowski, M.; Glueck, D.S.; Sommer, R.D.; Lam, K.C.; Incarvito, C.D.; Concolino, T.E.; Ceccarelli, C.; et al. Palladium Catalyzed Asymmetric Phosphination Enantioselective Synthesis of PAMP–BH3, Ligand Effects on Catalysis, and Direct Observation of the Stereochemistry of Transmetalation and Reductive Elimination. Organometallics 2003, 22, 3205–3221. [Google Scholar] [CrossRef]
- Chan, V.S.; Bergman, R.G.; Toste, F.D. Catalyzed Dynamic Kinetic Enantioselective Arylation of Silylphosphines. J. Am. Chem. Soc. 2007, 129, 15122–15123. [Google Scholar] [CrossRef] [PubMed]
- Julienne, D.; Lohier, J.F.; Delacroix, O.; Gaumont, A.C. Palladium-Catalyzed C–P Coupling Reactions between Vinyl Triflates and Phosphine–Boranes: Efficient Access to Vinylphosphine–Boranes. J. Org. Chem. 2007, 72, 2247–2250. [Google Scholar] [CrossRef] [PubMed]
- Korff, C.; Helmchen, G. Preparation of chiral triarylphosphines by Pd-catalysed asymmetric P–C cross-coupling. Chem. Commun. 2004, 530–531. [Google Scholar] [CrossRef] [PubMed]
- Blank, N.F.; Moncarz, J.R.; Brunker, T.J.; Scriban, C.; Anderson, B.J.; Amir, O.; Glueck, D.S.; Zakharov, L.N.; Golen, J.A.; Incarvito, C.D.; et al. Palladium-Catalyzed Asymmetric Phosphination. Scope, Mechanism, and Origin of Enantioselectivity. J. Am. Chem. Soc. 2007, 129, 6847–6858. [Google Scholar] [CrossRef] [PubMed]
- Julienne, D.; Delacroix, O.; Gaumont, A.C. First study on the enantioselective palladium-catalyzed C-P crosscoupling reaction between an alkenyltriflate and a phosphine-borane. C. R. Chim. 2010, 13, 1099–1103. [Google Scholar] [CrossRef]
- Liu, L.-J.; Wang, W.-M.; Yao, L.; Meng, F.J.; Sun, Y.-M.; Xu, H.; Xu, Z.-Y.; Li, Q.; Zhao, C.-Q.; Han, L.-B. Reinvestigation of the Substitutions Reaction of Stereogenic Phosphoryl Compounds: Stereochemistry, Mechanism, and Applications. J. Org. Chem. 2017, 82, 11990–12002. [Google Scholar] [CrossRef]
- Skrzypczynski, Z.; Michalski, J. Stereoselective Synthesis and Stereochemistry of Optically Active tert-Butylphenylphosphine Sulfide. J. Org. Chem. 1988, 53, 4549–4551. [Google Scholar] [CrossRef]
- Omelahczuk, J.; Mikolajczyk, M. Optically Active Trivalent Phosphorus Compounds. 2. Reactivity of Alkylthio- and Alkylselenophosphonium Salts. The First Stereospecific Synthesis of a Chiral Phosphinite. J. Am. Chem. Soc. 1979, 101, 7292–7295. [Google Scholar] [CrossRef]
- Le Maux, P.; Bahri, H.; Simonneaux, G.; Toupet, L. Enantioselective Oxidation of Racemic Phosphines with Chiral Oxoruthenium Porphyrins and Crystal Structure of [5,10,15,20-Tetrakis[o-((2-methoxy-2-phenyl-3,3,3-trifluoropropanoyl)amino)phenyl] porphyrinato] (carbonyl) (tetrahydrofuran)ruthenium (II) (alpha, beta, alpha, beta. Isomer). Inorg. Chem. 1995, 34, 4691–4697. [Google Scholar] [CrossRef]
- Van den Berg, G.R.; Platenburg, D.H.J.M.; Benschop, H.P. Organophosphorus compounds XI Stereochemistry of Grignard displacement reactions at phosphorus in isopropyl methylphosphonohalogenates. Recueil Pays-Bas 1972, 91, 929–934. [Google Scholar] [CrossRef]
- Perlikowska, W.; Gouygou, M.; Daran, J.C.; Balavoine, G.; Mikołajczyk, M. Kinetic resolution of P-chiral tertiary phosphines and chlorophosphines: A new approach to optically active phosphoryl and thiophosphoryl compou.nds. Tetrahedron Lett. 2001, 42, 7841–7845. [Google Scholar] [CrossRef]
- Michalski, J.; Skrzypczynski, Z. Novel Reaction of Phosphinothioic Methanesulphonic Anhydride with Aluminium Halides. Stereoselective Synthesis of Phosphinothioic Halides. Chem. Commun. 1977, 66–67. [Google Scholar] [CrossRef]
- Gryshkun, E.V.; Andrushko, N.V.; Kolodiazhnyi, O.I. Stereoselective reactions of chiral amines with racemic chlorophosphines. Phosphorus Sulfur Silicon Relat. Elem. 2004, 179, 1027–1046. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I.; Andrushko, N.V.; Gryshkun, E.V. Stereoselective reactions of optically active derivatives of α−methylbenzylaminophosphine. J. Russ. Gen. Chem. 2004, 74, 515–522. [Google Scholar] [CrossRef]
- Herault, D.; Nguyen, D.H.; Nuel, D.; Buono, G. Reduction of secondary and tertiary phosphine oxides to phosphines. Chem. Soc. Rev. 2015, 44, 2508–2528. [Google Scholar] [CrossRef] [PubMed]
- Imamoto, S.; Kikuchi, S.-I.; Miura, T.; Wada, Y. Stereospecific Reduction of Phosphine Oxides to Phosphines by the Use of a Methylation Reagent and Lithium Aluminum Hydride. Org. Lett. 2001, 3, 87–90. [Google Scholar] [CrossRef]
- Cernia, E.; Giongo, G.M.; Marcati, F.; Marconi, W.; Palladino, N. Optically Active Phosphines by Asymmetric Reduction of Racemic Phosphine Oxides. Inorg. Chim. Acta 1974, 11, 195–200. [Google Scholar] [CrossRef]
- Wu, H.-C.; Yu, J.-Q.; Spencer, J.B. Stereospecific Deoxygenation of Phosphine Oxides with Retention of Configuration Using Triphenylphosphine or Triethyl Phosphite as an Oxygen Acceptor. Org. Lett. 2004, 6, 4675–4678. [Google Scholar] [CrossRef]
- Kerrigan, N.J.; Dunne, E.C.; Cunningham, D.; McArdle, P.; Gilligan, K.; Gilheany, D.G. Studies in the preparation of novel P-chirogenic binaphthyl monophosphanes (MOPs). Tetrahedron Lett. 2003, 44, 8461–8465. [Google Scholar] [CrossRef]
- Keglevich, G.; Fekete, M.; Chuluunbaatar, T.; Dobo, A.; Harmat, V.; Toke, L. Studies in the preparation of novel P-chirogenic binaphthyl monophosphanes (MOPs). J. Chem. Soc. Perkin Trans. 2000, 1, 4451–4455. [Google Scholar] [CrossRef]
- Marsi, K.L. Phenylsilane Reduction of Phosphine Oxides with Complete Stereospecificity. J. Org. Chem. 1974, 39, 265–267. [Google Scholar] [CrossRef]
- Krenske, E.H. Theoretical Investigation of the Mechanisms and Stereoselectivities of Reductions of Acyclic Phosphine Oxides and Sulfides by Chlorosilanes. J. Org. Chem. 2012, 77, 3969–3977. [Google Scholar] [CrossRef] [PubMed]
- Krenske, E.H. Reductions of Phosphine Oxides and Sulfides by Perchlorosilanes: Evidence for the Involvement of Donor-Stabilized Dichlorosilylene. J. Org. Chem. 2012, 77, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kenny, N.P.; Rajendran, K.V.; Gilheany, D.G. Chemoselective reduction of the phosphoryl bond of O-alkyl phosphinates and related compounds: An apparently impossible transformation. Chem. Commun. 2015, 51, 16561–16566. [Google Scholar] [CrossRef] [PubMed]
- Gololobov, Y.G.; Kasukhin, L.F. Recent advances in the Staudinger reaction. Trahedron 1992, 48, 1353–1406. [Google Scholar] [CrossRef]
- Gololobov, Y.G.; Zhmurova, I.N.; Kasukhin, L.F. Sixty years of Staudinger reaction. Tetrahedron 1981, 37, 437–472. [Google Scholar] [CrossRef]
- Heesing, A.; Steinkamp, H. Mechanismen bei elektrophilen Reaktionen von reaktiven Stickstoffverbindungen mit Phosphanen. Chem. Ber. 1982, 115, 2854–2864. [Google Scholar] [CrossRef]
- Horner, L.; Jordan, M. Phosphororganische verbindungen 92. Zur Stereochemie und zum chemischen Verhalten optisch aktiver Amidophosphoniumsalze und optisch aktiver Phosphinigsaureamide. Phosphorus Sulfur 1980, 8, 225–234. [Google Scholar] [CrossRef]
- Dimukhametov, M.N.; Nuretdinov, I.A. Optically active silylphosphonites in the Staudinger reaction. Russ. Chem. Bull. 1983, 1103–1104. [Google Scholar] [CrossRef]
- Baccolini, G.; Todesco, P.E.; Bartoli, G. The Staudinger reaction between 2-h-1,2,3-diazaphospholenes and aromatic azides. Phosphorus Sulfur 1981, 10, 387–394. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Asymmetric synthesis of hydroxyphosphonates. Tetrahedron Asymmetry 2005, 16, 3295–3340. [Google Scholar] [CrossRef]
- Andersen, N.G.; Ramsden, P.D.; Che, D.; Parvez, M.; Keay, B.A. A Novel Resolution Procedure for the Preparation of P-Stereogenic Phosphine Oxides. Org. Lett. 1999, 1, 2009–2011. [Google Scholar] [CrossRef]
- Andersen, N.G.; Ramsden, P.D.; Che, D.; Parvez, M.; Keay, B.A. A Simple Resolution Procedure Using the Staudinger Reaction for the Preparation of P-Stereogenic Phosphine Oxides. J. Org. Chem. 2001, 66, 7478–7486. [Google Scholar] [CrossRef] [PubMed]
- Headley, C.E.; Marsden, S.P. Synthesis and Application of P-Stereogenic Phosphines as Superior Reagents in the Asymmetric Aza-Wittig Reaction. J. Org. Chem. 2007, 72, 7185–7189. [Google Scholar] [CrossRef] [PubMed]
- Scriven, E.F.V.; Turnbull, K. Azides: Their Preparation and Synthetic Uses. Chem. Rev. 1988, 88, 297–368. [Google Scholar] [CrossRef]
- Keglevich, G.; Bálint, E. The Kabachnik–Fields Reaction: Mechanism and Synthetic Use. Molecules 2012, 17, 12821–12835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherkasov, R.A.; Galkin, V.I. The Kabachnik–Fields reaction: Synthetic potential and the problem of the mechanism. Russ. Chem. Rev. 1998, 67, 857–882. [Google Scholar] [CrossRef]
- Enders, D.; Saint-Dizier, A.; Lannou, M.-I.; Lenzen, A.; Enders, D. The Phospha-Michael Addition in Organic Synthesis. Eur. J. Org. Chem. 2006, 2006, 29–49. [Google Scholar] [CrossRef]
- Gancarz, R. Nucleophilic Addition to Carbonyl Compounds. Competition Between Hard (Amine) and Soft (Phosphite) Nucleophile. Tetrahedron 1995, 51, 10627–10632. [Google Scholar] [CrossRef]
- Wroblewski, A.E.; Konieczko, V.T. Stereochemistry of 1,2-oxaphospholanes, III Evidence for the retro-Abramov pathway in methoxide-catalysed equilibration of substituted 2-methoxy-2-oxo-1,2-oxaphospholan-3-ols. Monatsh. Chem. 1984, 115, 785–791. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Chiral hydroxy phosphonates: Synthesis, configuration and biological properties. Russ. Chem. Rev. 2006, 75, 227–253. [Google Scholar] [CrossRef]
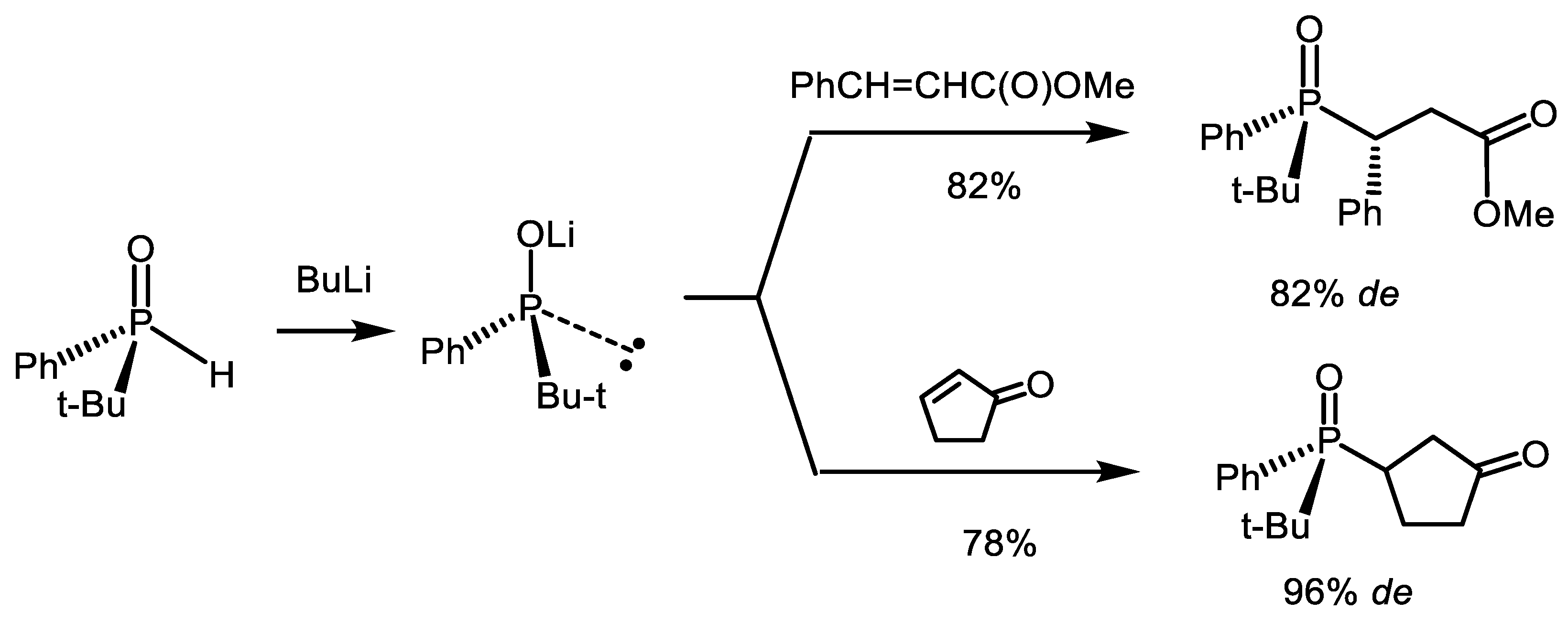
- Haynes, R.K.; Lam, W.W.L.; Yeung, L.L. Stereoselective Preparation of Functionalized Tertiary P-Chiral Phosphine Oxides by Nucleophilic Addition of Lithiated tert-Butylpbenylphospbine Oxide to Carbonyl Compounds. Tetrahedron Lett. 1996, 37, 4729–4732. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, Y.-M.; Zhao, Y.; Zhou, Z.-Y.; Wang, J.-P.; Xin, N.; Nie, S.-Z.; Zhao, C.-Q.; Han, L.-B. One-Pot Process That Efficiently Generates Single Stereoisomers of 1,3-Bisphosphinylpropanes Having Five Chiral Centers. Org. Lett. 2015, 17, 142–145. [Google Scholar] [CrossRef] [PubMed]
- Kolodiazhnyi, O.I.; Guliaiko, I.V.; Kolodiazhna, A.O. Highly stereoselective addition of silylphosphines to chiral aldehydes. Tetrahedron Lett. 2004, 45, 6955–6957. [Google Scholar] [CrossRef]
- Kortmann, F.A.; Chang, M.-C.; Otten, E.; Couzijn, E.P.A.; Lutz, M.; Minnaard, A. Consecutive dynamic resolutions of phosphine oxides. J. Chem. Sci. 2014, 5, 1322–1327. [Google Scholar] [CrossRef] [Green Version]
- Holt, J.; Maj, A.M.; Schudde, E.P.; Pietrusiewicz, K.M.; Sieron, L.; Wieczorek, W.; Jerphagnon, T.; Arends, I.W.C.E.; Hanefeld, U.; Minnaard, A.J. On the Resolution of Secondary Phosphine Oxides via Diastereomeric Complex Formation: The Case of tert-Butylphenylphosphine Oxide. Synthesis 2009, 2061–2065. [Google Scholar] [CrossRef]
- Sowa, S.; Stankevic, M.; Szmigielska, A.; Maluszynska, H.; Koziol, A.E.; Pietrusiewicz, K.M. Reduction of Functionalized Tertiary Phosphine Oxides with BH3. J. Org. Chem. 2015, 80, 1672–1688. [Google Scholar] [CrossRef]
- Gatineau, D.; Nguyen, D.H.; Hrault, D.; Vanthuyne, N.; Leclaire, J.; Giordano, L.; Buono, G. H-Adamantylphosphinates as Universal Precursors of P-Stereogenic Compounds. J. Org. Chem. 2015, 80, 4132–4141. [Google Scholar] [CrossRef]
- Lemouzy, S.; Nguyen, D.H.; Camy, V.; Jean, M.; Gatineau, D.; Giordano, L.; Naubron, J.-V.; Vanthuyne, N.; Hrault, D.; Buono, G. Stereospecific Synthesis of a- and b-Hydroxyalkyl P-Stereogenic Phosphine–Boranes and Functionalized Derivatives: Evidence of the P=O Activation in the BH3-Mediated Reduction. Chem. Eur. J. 2015, 21, 15607–15621. [Google Scholar] [CrossRef]
- Lemouzy, S.; Jean, M.; Giordano, L.; Hérault, D.; Buono, G. The Hydroxyalkyl Moiety as a Protecting Group for the Stereospecific Alkylation of Masked Secondary Phosphine-Boranes. Org. Lett. 2016, 18, 140–143. [Google Scholar] [CrossRef]
- Kumaraswamy, G.; Rao, G.V.; RamaKrishna, G. Stereocontrolled Copper Iodide Catalyzed Phosphorus–Carbon Bond Formation: An Efficient Synthesis of Scalemic Tertiary Phosphineboranes. Synlett 2006, 1122–1124. [Google Scholar] [CrossRef]
- Fu, X.; Loh, W.-T.; Zhang, Y.; Chen, T.; Ma, T.; Liu, H.; Wang, J.; Tan, C.-H. Chiral Guanidinium Salt Catalyzed Enantioselective Phospha-Mannich Reactions. Angew. Chem. Int. Ed. 2009, 48, 7387–7390. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Hatzakis, E.; McCarthy, S.M.; Reichl, K.D.; Lai, T.-Y.; Yennawar, H.P.; Radosevich, A.T. P–N Cooperative Borane Activation and Catalytic Hydroboration by a Distorted Phosphorous Triamide Platform. J. Am. Chem. Soc. 2017, 139, 6008–6016. [Google Scholar] [CrossRef] [PubMed]
- Keglevich, G.; Fehervari, A.; Csontos, I. A study on the Kabachnik–Fields reaction of benzaldehyde, propylamine, and diethyl phosphite by in situ Fourier transform IR spectroscopy. Heteroat. Chem. 2011, 22, 599–604. [Google Scholar] [CrossRef]
- Kolodiazhna, A.O.; Guliaiko, I.V.; Kolodiazhnyi, O.I. Diasteroselective Addition of Mono and Bis-Silylphosphines to Chiral Aldehydes. Phosphorus Sulfur Silicon Relat. Elem. 2005, 180, 2335–2346. [Google Scholar] [CrossRef]
- Tedeschi, L.; Enders, D. Asymmetric synthesis of beta-phosphono malonates via Fe2O3-mediated phospha-Michael addition to Knoevenagel acceptors. Org. Lett. 2001, 3, 3515–3517. [Google Scholar] [CrossRef]
- Wang, J.-P.; Nie, S.-Z.; Zhou, Z.-Y.; Ye, J.-J.; Wen, J.-H.; Zhao, C.-Q. Preparation of optically pure tertiary phosphine oxides via the addition of P-Stereogenic secondary phosphine oxide to activated alkenes. J. Org. Chem. 2016, 81, 7644–7653. [Google Scholar] [CrossRef]
- Kolodiazhna, A.O.; Kolodiazhnyi, O.I. Stereochemistry of nucleophilic substitution at trivalent phosphorus. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 621–633. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Stereochemistry of electrophilic and nucleophilic substitutions at phosphorus. Pure Appl. Chem. 2019, 91, 43–57. [Google Scholar] [CrossRef]
- Chmielewska, E.; Kafarski, P. Synthetic procedures leading towards aminobisphosphonates. Molecules 2016, 21, 1474. [Google Scholar] [CrossRef] [Green Version]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
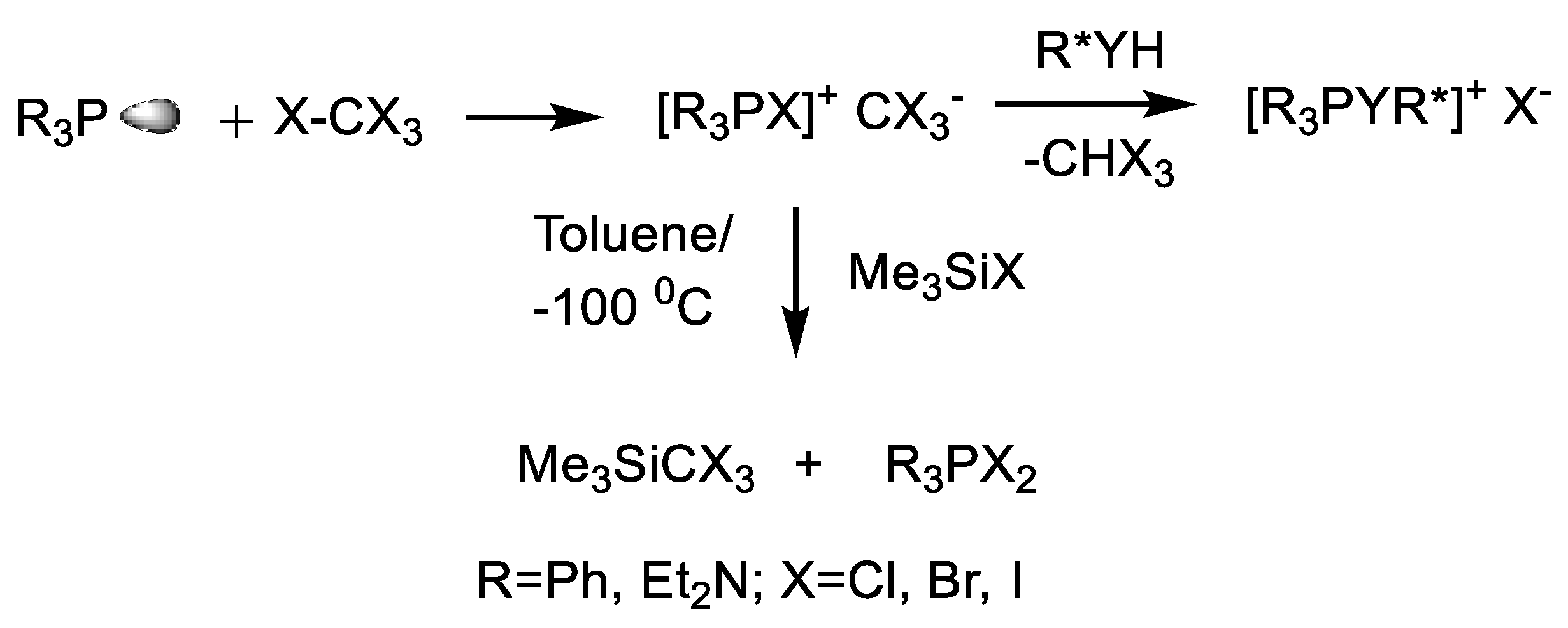{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | RX | Product | Yield, % | ee, % |
|---|---|---|---|---|
| (SP)-1a | MeI | (RP)-2a | 85 | 99 |
| (SP)-1a | BnBr | (RP)-2b | 91 | 98 |
| (SP)-1a | CH2=CHCH2Br | (RP)-2c | 83 | 89 |
| (SP)-1a | CH≡CCH2Br | (RP)-2d | 65 | 89 |
| (SP)-1a | Me3SiCH2I | (RP)-2e | 93 | 86 |
| (SP)-1a | o-PyCH2Cl | (RP)-2f | 68 | 99 |
| (SP)-1b | MeI | (RP)-2g | 77 | 98 |
| (SP)-1b | BnBr | (RP)-2h | 90 | 99 |
| (SP)-1b | CH2=CHCH2Br | (RP)-2i | 90 | 78 |
| (SP)-1b | CH≡CCH2Br | (RP)-2j | 68 | 77 |
| (SP)-1c | MeI | (RP)-2k | 89 | 91 |
| R | X | Base | LiX | Yield, % | ee, %/(Config) |
|---|---|---|---|---|---|
| CO2Bu-t | 2-Ph | NEt3 | - | 63 | 71 (S) |
| CO2Bu-t | 2-Ph | NBnMe2 | - | 45 | 77 (S) |
| CO2Bu-t | 2-Ph | N-Me-piperidine | - | 76 | 84 (S) |
| CO2Bu-t | 2-Ph | NEt3 | LiF | 76 | 66 (S) |
| CO2Bu-t | 2-Ph | NEt3 | LiCl | 66 | 86 (S) |
| CO2Bu-t | 2-Ph | NEt3 | LiBr | 76 | 90 (S) |
| CO2Bu-t | 2-Ph | NEt3 | LiI | 58 | 87 (S) |
| CO2Bu-t | 2-MeO | N-Me-piperidine | LiBr | 43 | 86 (S) |
| CO2Bu-t | 2-CF3 | NEt3 | LiBr | 39 | 93 (R) |
| CO2Bu-t | 2-Ph | NEt3 | LiBr | 69 | 85 (S) |
| CHO | 2-Ph | NEt3 | LiBr | 71 | 63 (S) |
| R1 | R2 | Yield, (%) | % ee | Configuration |
|---|---|---|---|---|
| Np | Ph | 92 | 97 | (S) |
| Np | Ph | 90 | 98 | (S) |
| Ph | o-An | 85 | 98 | (R) |
| c-C6H11 | o-An | 74 | 95 | (R) |
| c-C6H11 | Np | 81 | 88 | (R) |
| Ph | o-(Pr-i)C6H4 | 97 | 98 | (R) |
| (CH2)2Ph | Bu-t | 55 | 97 | (R) |
| (CH2)2Ph | c-C6H11 | 96 | 92 | (R) |
| R1 | R2 | Yield, (%) 6 | Yield, (%) 7 | Configuration |
|---|---|---|---|---|
| Me | C6H11 | 94 | 93 | SP |
| Me | C5H9 | 90 | 93 | - |
| Me | CH(CH3)2 | 87 | 94 | RP |
| Me | 1-Np | 94 | 96 | SP |
| Me | 9-Phnt | 89 | >99 | - |
| 1-Np | p-PhC6H4 | 91 | 93 | RP |
| RCH=O | Yield, (%) | dr |
|---|---|---|
| PhCH=CHCHO | 77 | 80:20 |
| PhCH=CHCHO | 83 | 80:20 |
| CH2=CHCHO | 65 | 78:22 |
| MeCH(=CH2)CHO | 77.2 | 82:18 |
| c-C6H11CHO | 80 | 85:15 |
| PhCHO | 77 | 98:2 |
| MeCHO | 80 | 70:30 |
| 2-PyCHO | 76 | 80:20 |
| Me2CHCH2CHO | 78 | 67:33 |
| 1-NpCHO | 71 | 98:8 |
| 2-NpCHO | 71 | 85:15 |
| 2-ThienylCHO | 68 | 82:18 |
| 2-FurylCHO | 76 | 90:10 |
| R | dr | Configuration | Yield, % |
|---|---|---|---|
| Ph | 20:1 | RP,R | 89 |
| p-Tl | 1:20 | RP,S | 94 |
| o-Tl | 1:20 | RP,S | 94 |
| p-(i-Pr)C6H4 | 20:1 | RP,R | 96 |
| p-An | 20:1 | RP,S | 96 |
| m-HOC6H4 | 1:20 | RP,S | 90 |
| p-ClC6H4 | 1:3.5 | - | 90 |
| p-O2NC6H4 | 1:4.4 | - | 93 |
| Pr-i | 1:20 | RP,S | 95 |
| Bu-t | 1:20 | RP,S | 89 |
| H | - | - | 100 |
| R1 | R2 | Yield (%) | dr | ee (%) |
|---|---|---|---|---|
| Bn | Ph | 83 | 6:1 | 94 |
| Bn | 4-FC6H4 | 90 | 6:1 | 90 |
| Bn | 4-ClC6H4 | 90 | 4:1 | 92 |
| Bn | 4-Tol | 85 | 4:1 | 90 |
| Bn | 2-naphthyl | 93 | 6:1 | 91 |
| Bn | 2-furyl | 71 | 7:1 | 94 |
| Bn | E-CH=CHPh | 92 | 3:1 | 90 |
| 2-Naphthylmethyl | Ph | 92 | 6.5:1 | 94 |
| 4-F3CC6H4CH2 | Ph | 92 | 16:1 | 94 |
| 4-TolCH2 | Ph | 83 | 5:1 | 88 |
| (E)-H2CCH=CHPh | Ph | 82 | 7:1 | 82 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolodiazhna, A.O.; Kolodiazhnyi, O.I. Asymmetric Electrophilic Reactions in Phosphorus Chemistry. Symmetry 2020, 12, 108. https://doi.org/10.3390/sym12010108
Kolodiazhna AO, Kolodiazhnyi OI. Asymmetric Electrophilic Reactions in Phosphorus Chemistry. Symmetry. 2020; 12(1):108. https://doi.org/10.3390/sym12010108
Chicago/Turabian StyleKolodiazhna, Anastasy O., and Oleg I. Kolodiazhnyi. 2020. "Asymmetric Electrophilic Reactions in Phosphorus Chemistry" Symmetry 12, no. 1: 108. https://doi.org/10.3390/sym12010108