Structural Features and PF4 Functions that Occur in Heparin-Induced Thrombocytopenia (HIT) Complicated by COVID-19


Abstract
:1. Introduction
2. Platelets and Viral Infection of the Lung
3. Role of PF4 in Infections
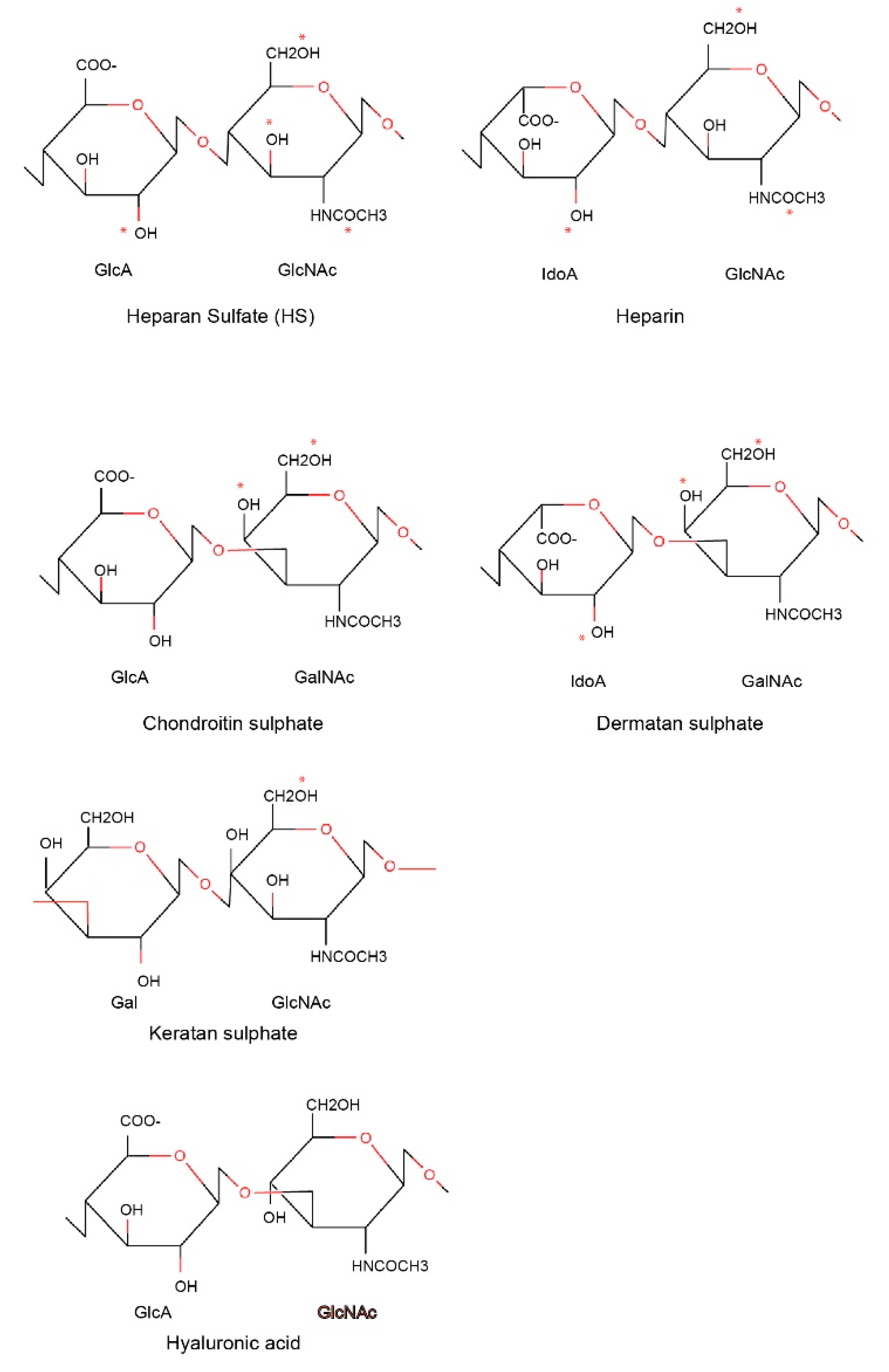
4. Structural Basis for PF4 Function: Binding Activity of PF4 to Polysaccharides
- Heparin/heparan sulfate;
- Chondroitin sulfate/dermatan sulfate (CSGAGs);
- Keratan sulfate;
- Hyaluronic acid.
5. Heparin-Induced Thrombocytopenia (HIT)
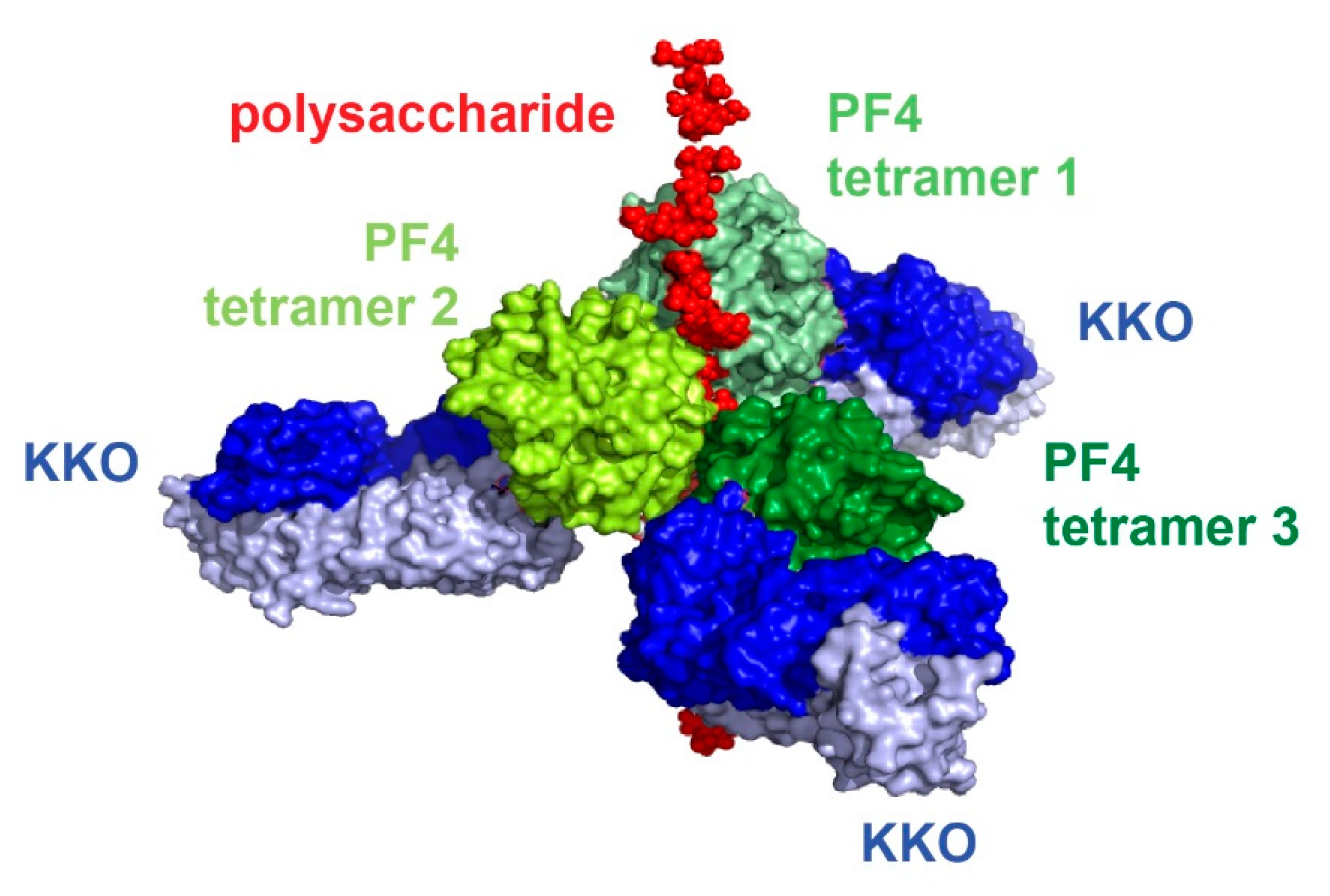
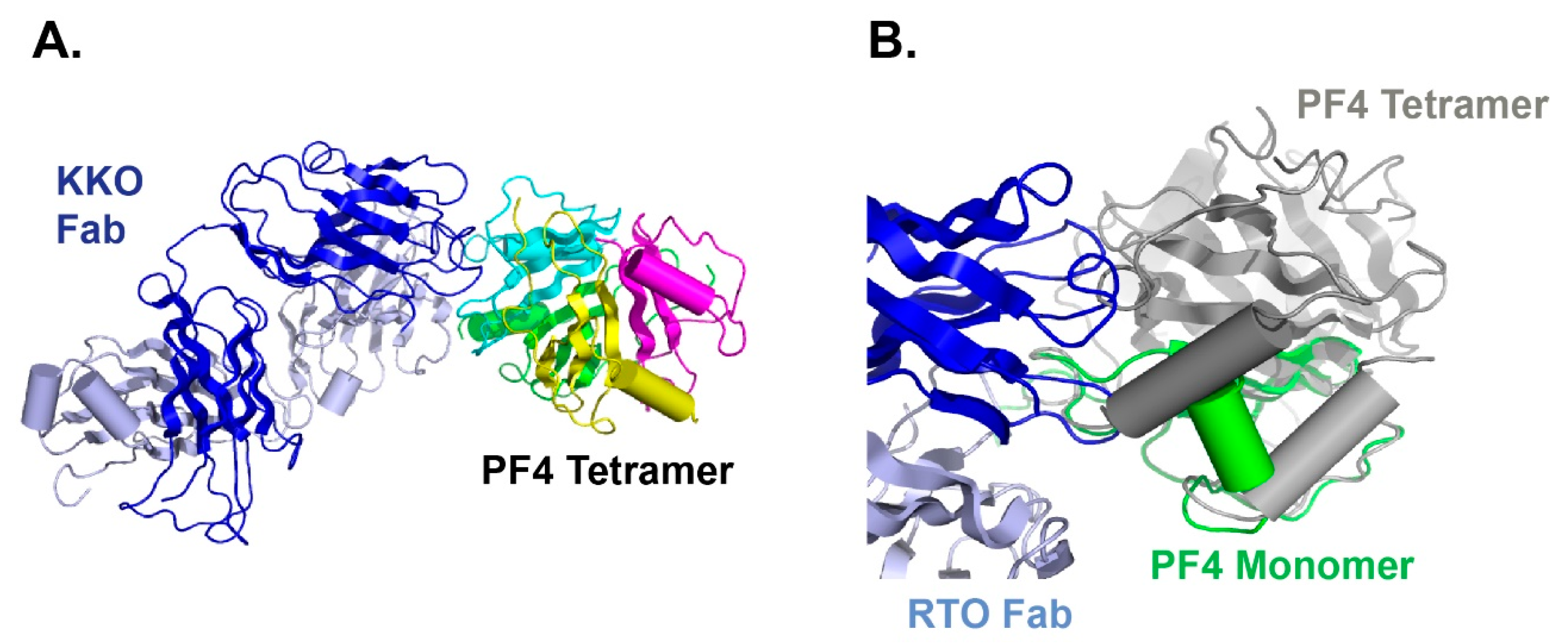
6. Structural Basis for Heparin-Induced Thrombocytopenia (HIT)
7. Coagulation Dysfnction in COVID-19 and the Risk of HIT
8. Potential Use of a Humanized PF4 Antibody to Prevent HIT
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Poncz, M.; Surrey, S.; LaRocco, P.; Weiss, M.J.; Rappaport, E.F.; Conway, T.M.; Schwartz, E. Cloning and characterization of platelet factor 4 cDNA derived from a human erythroleukemic cell line. Blood 1987, 69, 219–223. [Google Scholar] [CrossRef]
- Barber, A.J.; Kaser-Glanzmann, R.; Jakabova, M.; Luscher, E.F. Characterization of a chondroitin 4 -sulfate proteoglycan carrier for heparin neutralizing activity (platelet factor 4) released from human blood platelets. Biochim. Biophys. Acta 1972, 286, 312–329. [Google Scholar] [CrossRef]
- Holt, J.C.; Niewiarowski, S. Biochemistry of alpha granule proteins. Semin. Hematol. 1985, 22, 151–163. [Google Scholar]
- Qiu, J.; Ma, J.; Zhang, S.; Han, J.; Liu, S. Promoting platelets is a therapeutic option to combat severe viral infection of the lung. Blood Adv. 2020, 4, 1640–1642. [Google Scholar] [CrossRef]
- Salamanna, F.; Maglio, M.; Landini, M.P.; Fini, M. Platelet functions and activities as potential hematologic parameters related to Coronavirus Disease 2019 (COVID-19). Platelets 2020, 31, 627–632. [Google Scholar] [CrossRef]
- Schrottmaier, W.C.; Mussbacher, M.; Salzmann, M.; Assinger, A. Platelet-leukocyte interplay during vascular disease. Atherosclerosis 2020. [Google Scholar] [CrossRef]
- Lefrancais, E.; Ortiz-Munoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.B.; David, T.; Coughlin, S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017. [Google Scholar] [CrossRef]
- Koupenova, M.; Corkrey, H.A.; Vitseva, O.; Manni, G.; Pang, C.J.; Clancy, L.; Yao, C.; Rade, J.; Levy, D.; Wang, J.P.; et al. The role of platelets in mediating a response to human influenza infection. Nat. Commun. 2019, 10, 1780. [Google Scholar] [CrossRef] [Green Version]
- van der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; van den Heuvel, G.; Mantere, T.; Kersten, S.; van Deuren, R.C.; Steehouwer, M.; van Reijmersdal, S.V.; Jaeger, M.; et al. Presence of Genetic Variants among Young Men With Severe COVID-19. JAMA 2020. [Google Scholar] [CrossRef]
- Rosowski, E.E.; Huttenlocher, A. Motile Collectors: Platelets Promote Innate Immunity. Immunity 2018, 48, 16–18. [Google Scholar] [CrossRef] [Green Version]
- Worth, R.G.; Chien, C.D.; Chien, P.; Reilly, M.P.; McKenzie, S.E.; Schreiber, A.D. Platelet FcgammaRIIA binds and internalizes IgG-containing complexes. Exp. Hematol. 2006, 34, 1490–1495. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.J.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.J.; et al. Platelet Gene Expression and Function in COVID-19 Patients. Blood 2020. [Google Scholar] [CrossRef]
- Zuchtriegel, G.; Uhl, B.; Puhr-Westerheide, D.; Pörnbacher, M.; Lauber, K.; Krombach, F.; Reichel, C.A. Platelets Guide Leukocytes to Their Sites of Extravasation. PLoS Biol. 2016, 14, e1002459. [Google Scholar] [CrossRef] [Green Version]
- Kuckleburg, C.J.; Yates, C.M.; Kalia, N.; Zhao, Y.; Nash, G.B.; Watson, S.P.; Rainger, G.E. Endothelial cell-borne platelet bridges selectively recruit monocytes in human and mouse models of vascular inflammation. Cardiovasc. Res. 2011, 91, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Carlow, D.A.; Tra, M.C.; Ziltener, H.J. A cell-extrinsic ligand acquired by activated T cells in lymph node can bridge L-selectin and P-selectin. PLoS ONE 2018, 13, e0205685. [Google Scholar] [CrossRef] [Green Version]
- Han, P.; Hanlon, D.; Arshad, N.; Lee, J.S.; Tatsuno, K.; Robinson, E.; Filler, R.; Sobolev, O.; Cote, C.; Rivera-Molina, F.; et al. Platelet P-selectin initiates cross-presentation and dendritic cell differentiation in blood monocytes. Sci. Adv. 2020, 6, eaaz1580. [Google Scholar] [CrossRef] [Green Version]
- Di Micco, P.; Russo, V.; Carannante, N.; Imparato, M.; Rodolfi, S.; Cardillo, G.; Lodigiani, C. Clotting Factors in COVID-19: Epidemiological Association and Prognostic Values in Different Clinical Presentations in an Italian Cohort. J. Clin. Med. 2020, 9, 1371. [Google Scholar] [CrossRef]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; El Haj, R.B. Platelets can contain SARS-CoV-2 RNA and are hyperactivated in COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Kullaya, V.; van der Ven, A.; Mpagama, S.; Mmbaga, B.T.; de Groot, P.; Kibiki, G.; de Mast, Q. Platelet-monocyte interaction in Mycobacterium tuberculosis infection. Tuberculosis (Edinb.) 2018, 111, 86–93. [Google Scholar] [CrossRef]
- Wilkinson, J.M.; Ladinig, A.; Bao, H.; Kommadath, A.; Stothard, P.; Lunney, J.K.; Harding, J.C.; Plastow, G.S. Differences in Whole Blood Gene Expression Associated with Infection Time-Course and Extent of Fetal Mortality in a Reproductive Model of Type 2 Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Infection. PLoS ONE 2016, 11, e0153615. [Google Scholar] [CrossRef]
- Guo, L.; Feng, K.; Wang, Y.C.; Mei, J.J.; Ning, R.T.; Zheng, H.W.; Wang, J.J.; Worthen, G.S.; Wang, X.; Song, J.; et al. Critical role of CXCL4 in the lung pathogenesis of influenza (H1N1) respiratory infection. Mucosal Immunol. 2017, 10, 1529–1541. [Google Scholar] [CrossRef]
- McMorran, B.J.; Wieczorski, L.; Drysdale, K.E.; Chan, J.A.; Huang, H.M.; Smith, C.; Mitiku, C.; Beeson, J.G.; Burgio, G.; Foote, S.J. Platelet factor 4 and Duffy antigen required for platelet killing of Plasmodium falciparum. Science 2012, 338, 1348–1351. [Google Scholar] [CrossRef]
- Auerbach, D.J.; Lin, Y.; Miao, H.; Cimbro, R.; Difiore, M.J.; Gianolini, M.E.; Furci, L.; Biswas, P.; Fauci, A.S.; Lusso, P. Identification of the platelet-derived chemokine CXCL4/PF-4 as a broad-spectrum HIV-1 inhibitor. Proc. Natl. Acad. Sci. USA 2012, 109, 9569–9574. [Google Scholar] [CrossRef] [Green Version]
- Parker, Z.F.; Rux, A.H.; Riblett, A.M.; Lee, F.H.; Rauova, L.; Cines, D.B.; Poncz, M.; Sachais, B.S.; Doms, R.W. Platelet Factor 4 Inhibits and Enhances HIV-1 Infection in a Concentration-Dependent Manner by Modulating Viral Attachment. AIDS Res. Hum. Retrovir. 2016, 32, 705–717. [Google Scholar] [CrossRef] [Green Version]
- Messina, F.; Giombini, E.; Agrati, C.; Vairo, F.; Bartoli, T.A.; Moghazi, S.A.; Piacentini, M.; Locatelli, F.; Kobinger, G.; Maeurer, M.; et al. COVID-19: Viral-host interactome analyzed by network based-approach model to study pathogenesis of SARS-CoV-2 infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wilson, N.O.; Jain, V.; Roberts, C.E.; Lucchi, N.; Joel, P.K.; Singh, M.P.; Nagpal, A.C.; Dash, A.P.; Udhayakumar, V.; Singh, N.; et al. CXCL4 and CXCL10 predict risk of fatal cerebral malaria. Dis. Markers 2011, 30, 39–49. [Google Scholar] [CrossRef]
- Trinh, M.; Chang, D.R.; Govindarajulu, U.S.; Kane, E.; Fuster, V.; Kohli-Seth, R.; Ahmed, S.; Levin, M.A.; Chen, M.D. Therapeutic Anticoagulation Is Associated with Decreased Mortality in Mechanically Ventilated COVID-19 Patients. medRxiv 2020. [Google Scholar] [CrossRef]
- Kolset, S.O.; Mann, D.M.; Uhlin-Hansen, L.; Winberg, J.O.; Ruoslahti, E. Serglycin-binding proteins in activated macrophages and platelets. J. Leukoc. Biol. 1996, 59, 545–554. [Google Scholar] [CrossRef]
- Chong, B.H. Evolving concepts of pathogenesis of heparin-induced thrombocytopenia: Diagnostic and therapeutic implications. Int. J. Lab. Hematol. 2020, 42 (Suppl. S1), 25–32. [Google Scholar] [CrossRef]
- Arepally, G.M.; Cines, D.B. Pathogenesis of heparin-induced thrombocytopenia. Transl. Res. J. Lab. Clin. Med. 2020. [Google Scholar] [CrossRef]
- Rauova, L.; Hirsch, J.D.; Greene, T.K.; Zhai, L.; Hayes, V.M.; Kowalska, M.A.; Cines, D.B.; Poncz, M. Monocyte-bound PF4 in the pathogenesis of heparin-induced thrombocytopenia. Blood 2010, 116, 5021–5031. [Google Scholar] [CrossRef] [Green Version]
- Joseph, J.; Rabbolini, D.; Enjeti, A.K.; Favaloro, E.; Kopp, M.C.; McRae, S.; Pasalic, L.; Tan, C.W.; Ward, C.M.; Chong, B.H. Diagnosis and management of heparin-induced thrombocytopenia: A consensus statement from the Thrombosis and Haemostasis Society of Australia and New Zealand HIT Writing Group. Med. J. Aust. 2019, 210, 509–516. [Google Scholar] [CrossRef]
- Althaus, K.; Straub, A.; Haberle, H.; Rosenberger, P.; Hidiatov, O.; Hammer, S.; Nowak-Harnau, S.; Enkel, S.; Riessen, R.; Bakchoul, T. Heparin-induced thrombocytopenia: Diagnostic challenges in intensive care patients especially with extracorporeal circulation. Thromb. Res. 2020, 188, 52–60. [Google Scholar] [CrossRef]
- Warkentin, T.E. Clinical picture of heparin-induced thrombocytopenia (HIT) and its differentiation from non-HIT thrombocytopenia. Thromb. Haemost. 2016, 116, 813–822. [Google Scholar] [CrossRef]
- Barcellona, D.; Melis, M.; Floris, G.; Mameli, A.; Muroni, A.; Defazio, G.; Marongiu, F. A “Catastrophic” Heparin-Induced Thrombocytopenia. Case Rep. Med. 2020, 2020. [Google Scholar] [CrossRef] [Green Version]
- Greinacher, A.; Gopinadhan, M.; Gunther, J.U.; Omer-Adam, M.A.; Strobel, U.; Warkentin, T.E.; Papastavrou, G.; Weitschies, W.; Helm, C.A. Close approximation of two platelet factor 4 tetramers by charge neutralization forms the antigens recognized by HIT antibodies. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2386–2393. [Google Scholar] [CrossRef] [Green Version]
- Warkentin, T.E.; Cook, R.J.; Marder, V.J.; Sheppard, J.A.; Moore, J.C.; Eriksson, B.I.; Greinacher, A.; Kelton, J.G. Anti-platelet factor 4/heparin antibodies in orthopedic surgery patients receiving antithrombotic prophylaxis with fondaparinux or enoxaparin. Blood 2005, 106, 3791–3796. [Google Scholar] [CrossRef] [Green Version]
- Grouzi, E.; Kyriakou, E.; Panagou, I.; Spiliotopoulou, I. Fondaparinux for the treatment of acute heparin-induced thrombocytopenia: A single-center experience. Clin. Appl. Thromb. Hemost. 2010, 16, 663–667. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Pai, M.; Sheppard, J.I.; Schulman, S.; Spyropoulos, A.C.; Eikelboom, J.W. Fondaparinux treatment of acute heparin-induced thrombocytopenia confirmed by the serotonin-release assay: A 30-month, 16-patient case series. J. Thromb. Haemost. 2011, 9, 2389–2396. [Google Scholar] [CrossRef]
- Dulicek, P.; Ivanova, E.; Kostal, M.; Fiedlerova, Z.; Sadilek, P.; Hirmerova, J. Heparin-induced thrombocytopenia treated with fondaparinux: Single center experience. Int. Angiol. 2020, 39, 76–81. [Google Scholar] [CrossRef]
- D’Angelo, A.; Valle, P.D.; Fattorini, A.; Luciano, C. Disappearance of anti-PF4/heparin antibodies under prolonged fondaparinux administration in a patient with DVT associated with LMWH-induced thrombocytopenia. Thromb. Haemost. 2006, 95, 573–575. [Google Scholar] [CrossRef]
- Alsaleh, K.A.; Al-Nasser, S.M.; Bates, S.M.; Patel, A.; Warkentin, T.E.; Arnold, D.M. Delayed-onset HIT caused by low-molecular-weight heparin manifesting during fondaparinux prophylaxis. Am. J. Hematol. 2008, 83, 876–878. [Google Scholar] [CrossRef] [PubMed]
- Vadi, S.; Peshattiwar, V. The Fondaparinux Paradox: Fondaparinux-Related Heparin-induced Thrombocytopenia. Indian J. Crit. Care Med. 2018, 22, 116–118. [Google Scholar] [CrossRef] [PubMed]
- Krecak, I.; Tomac, G.; Skugor, J.; Gveric-Krecak, V.; Pulanic, D. In vivo and in vitro cross-reactivity to fondaparinux in a stroke patient with IgG-PF4/heparin antibody-negative delayed-onset heparin-induced thrombocytopenia. Blood Transfus. 2020. [Google Scholar] [CrossRef]
- Manji, F.; Warkentin, T.E.; Sheppard, J.I.; Lee, A. Fondaparinux cross-reactivity in heparin-induced thrombocytopenia successfully treated with high-dose intravenous immunoglobulin and rivaroxaban. Platelets 2020, 31, 124–127. [Google Scholar] [CrossRef]
- Farasatinasab, M.; Zarei, B.; Moghtadaei, M.; Nasiripour, S.; Ansarinejad, N.; Zarei, M. Rivaroxaban as an Alternative Agent for Heparin-Induced Thrombocytopenia. J. Clin. Pharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kerendi, F.; Thourani, V.H.; Puskas, J.D.; Kilgo, P.D.; Osgood, M.; Guyton, R.A.; Lattouf, O.M. Impact of Heparin-Induced Thrombocytopenia on Postoperative Outcomes After Cardiac Surgery. Ann. Thorac. Surg. 2007, 84, 1548–1555. [Google Scholar] [CrossRef]
- Haile, L.A.; Rao, R.; Polumuri, S.K.; Arepally, G.M.; Keire, D.A.; Verthelyi, D.; Sommers, C.D. PF4-HIT antibody (KKO) complexes activate broad innate immune and inflammatory responses. Thromb. Res. 2017, 159, 39–47. [Google Scholar] [CrossRef]
- Kasthuri, R.S.; Glover, S.L.; Jonas, W.; McEachron, T.; Pawlinski, R.; Arepally, G.M.; Key, N.S.; Mackman, N. PF4/heparin-antibody complex induces monocyte tissue factor expression and release of tissue factor positive microparticles by activation of FcγRI. Blood 2012, 119, 5285–5293. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef] [Green Version]
- Tutwiler, V.; Madeeva, D.; Ahn, H.S.; Andrianova, I.; Hayes, V.; Zheng, X.L.; Cines, D.B.; McKenzie, S.E.; Poncz, M.; Rauova, L. Platelet transactivation by monocytes promotes thrombosis in heparin-induced thrombocytopenia. Blood 2016, 127, 464–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cines, D.B.; Zaitsev, S.; Rauova, L.; Rux, A.H.; Stepanova, V.; Krishnaswamy, S.; Sarkar, A.; Kowalska, M.A.; Zhao, G.; Mast, A.E.; et al. FcRn augments induction of tissue factor activity by IgG-containing immune complexes. Blood 2020, 135, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Nakajima, S.; Ando, T.; Oda, K.; Sugita, M.; Maeda, K.; Nakiri, Y.; Takasaki, Y. Heparin-Related Thrombocytopenia Triggered by Severe Status of Systemic Lupus Erythematosus and Bacterial Infection. Case Rep. Rheumatol. 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Chen, L.; Bancroft, D.P.; Lai, C.K.; Maione, T.E. Crystal structure of recombinant human platelet factor 4. Biochemistry 1994, 33, 8361–8366. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Rauova, L.; Arepally, G.; Reilly, M.P.; McKenzie, S.E.; Sachais, B.S.; Poncz, M. Heparin-induced thrombocytopenia: An autoimmune disorder regulated through dynamic autoantigen assembly/disassembly. J. Clin. Apher. 2007, 22, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Rauova, L.; Poncz, M.; McKenzie, S.E.; Reilly, M.P.; Arepally, G.; Weisel, J.W.; Nagaswami, C.; Cines, D.B.; Sachais, B.S. Ultralarge complexes of PF4 and heparin are central to the pathogenesis of heparin-induced thrombocytopenia. Blood 2005, 105, 131–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.M.; Arepally, G.M. Heparin-induced thrombocytopenia. Hematol. Am. Soc. Hematol. Educ. Program 2013, 2013, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Poncz, M.; Rauova, L.; Cines, D.B. The role of surface PF4: Glycosaminoglycan complexes in the pathogenesis of heparin-induced thrombocytopenia (HIT). Pathophysiol. Haemost. Thromb. 2006, 35, 46–49. [Google Scholar] [CrossRef]
- Arepally, G.M.; Kamei, S.; Park, K.S.; Kamei, K.; Li, Z.Q.; Liu, W.; Siegel, D.L.; Kisiel, W.; Cines, D.B.; Poncz, M. Characterization of a murine monoclonal antibody that mimics heparin-induced thrombocytopenia antibodies. Blood 2000, 95, 1533–1540. [Google Scholar] [CrossRef]
- Kizlik-Masson, C.; Vayne, C.; McKenzie, S.E.; Poupon, A.; Zhou, Y.; Champier, G.; Pouplard, C.; Gruel, Y.; Rollin, J. 5B9, a monoclonal antiplatelet factor 4/heparin IgG with a human Fc fragment that mimics heparin-induced thrombocytopenia antibodies. J. Thromb. Haemost. 2017, 15, 2065–2075. [Google Scholar] [CrossRef] [Green Version]
- Huynh, A.; Arnold, D.M.; Kelton, J.G.; Smith, J.W.; Horsewood, P.; Clare, R.; Guarne, A.; Nazy, I. Characterization of platelet factor 4 amino acids that bind pathogenic antibodies in heparin-induced thrombocytopenia. J. Thromb. Haemost. 2019, 17, 389–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusuf, A.M.; Warkentin, T.E.; Arsenault, K.A.; Whitlock, R.; Eikelboom, J.W. Prognostic importance of preoperative anti-PF4/heparin antibodies in patients undergoing cardiac surgery. A systematic review. Thromb. Haemost. 2012, 107, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Sachais, B.S.; Litvinov, R.I.; Yarovoi, S.V.; Rauova, L.; Hinds, J.L.; Rux, A.H.; Arepally, G.M.; Poncz, M.; Cuker, A.; Weisel, J.W.; et al. Dynamic antibody-binding properties in the pathogenesis of HIT. Blood 2012, 120, 1137–1142. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Yarovoi, S.V.; Zhu, Z.; Rauova, L.; Hayes, V.; Lebedeva, T.; Liu, Q.; Poncz, M.; Arepally, G.; Cines, D.B.; et al. Atomic description of the immune complex involved in heparin-induced thrombocytopenia. Nat. Commun. 2015, 6, 8277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niyas, V.K.M.; Keri, V.C.; Ahuja, J.; Anand, A.; Nischal, N.; Wig, N. Severe Immune Thrombocytopenia in a Patient with HIV-HCV Co-infection: Challenges in Management. J. Assoc. Phys. India 2020, 68, 77–79. [Google Scholar]
- Abeysuriya, V.; Choong, C.S.H.; Thilakawardana, B.U.; de Mel, P.; Shalindi, M.; de Mel, C.; Chandrasena, L.; Seneviratne, S.L.; Yip, C.; Yap, E.S.; et al. The atypical lymphocyte count: A novel predictive factor for severe thrombocytopenia related to dengue. Trans. R. Soc. Trop. Med. Hyg. 2020, 114, 424–432. [Google Scholar] [CrossRef]
- Metcalf Pate, K.A.; Lyons, C.E.; Dorsey, J.L.; Shirk, E.N.; Queen, S.E.; Adams, R.J.; Gama, L.; Morrell, C.N.; Mankowski, J.L. Platelet activation and platelet-monocyte aggregate formation contribute to decreased platelet count during acute simian immunodeficiency virus infection in pig-tailed macaques. J. Infect. Dis. 2013, 208, 874–883. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Wei, J.; Zou, L.; Jiang, T.; Wang, G.; Chen, L.; Huang, L.; Meng, F.; Huang, L.; Wang, N.; et al. Ruxolitinib in treatment of severe coronavirus disease 2019 (COVID-19): A multicenter, single-blind, randomized controlled trial. J. Allergy Clin. Immunol. 2020. [Google Scholar] [CrossRef]
- Sinha, A.; Ma, Y.; Scherzer, R.; Hur, S.; Li, D.; Ganz, P.; Deeks, S.G.; Hsue, P.Y. Role of T-Cell Dysfunction, Inflammation, and Coagulation in Microvascular Disease in HIV. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-up: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Xu, S.; Yu, M.; Wang, K.; Tao, Y.; Zhou, Y.; Shi, J.; Zhou, M.; Wu, B.; Yang, Z.; et al. Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J. Allergy Clin. Immunol. 2020, 146, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fu, B.; Zheng, X.; Wang, D.; Zhao, C.; Qi, Y.; Sun, R.; Tian, Z.; Xu, X.; Wei, H. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl. Sci. Rev. 2020, 7, 998–1002. [Google Scholar] [CrossRef] [Green Version]
- Lax, S.F.; Skok, K.; Zechner, P.; Kessler, H.H.; Kaufmann, N.; Koelblinger, C.; Vander, K.; Bargfrieder, U.; Trauner, M. Pulmonary Arterial Thrombosis in COVID-19 With Fatal Outcome: Results From a Prospective, Single-Center, Clinicopathologic Case Series. Ann. Intern. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Levi, M.; Thachil, J.; Iba, T.; Levy, J.H. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020, 7, e438–e440. [Google Scholar] [CrossRef]
- McFadyen James, D.; Stevens, H.; Peter, K. The Emerging Threat of (Micro)Thrombosis in COVID-19 and Its Therapeutic Implications. Circ. Res. 2020. [Google Scholar] [CrossRef]
- Leonard-Lorant, I.; Delabranche, X.; Severac, F.; Helms, J.; Pauzet, C.; Collange, O.; Schneider, F.; Labani, A.; Bilbault, P.; Moliere, S.; et al. Acute Pulmonary Embolism in COVID-19 Patients on CT Angiography and Relationship to D-Dimer Levels. Radiology 2020. [Google Scholar] [CrossRef] [Green Version]
- Grillet, F.; Behr, J.; Calame, P.; Aubry, S.; Delabrousse, E. Acute Pulmonary Embolism Associated with COVID-19 Pneumonia Detected by Pulmonary CT Angiography. Radiology 2020. [Google Scholar] [CrossRef] [Green Version]
- Demelo-Rodríguez, P.; Cervilla-Muñoz, E.; Ordieres-Ortega, L.; Parra-Virto, A.; Toledano-Macías, M.; Toledo-Samaniego, N.; García-García, A.; García-Fernández-Bravo, I.; Ji, Z.; de-Miguel-Diez, J.; et al. Incidence of asymptomatic deep vein thrombosis in patients with COVID-19 pneumonia and elevated D-dimer levels. Thromb. Res. 2020, 192, 23–26. [Google Scholar] [CrossRef]
- Wichmann, D.; Sperhake, J.P.; Lutgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A.; Heinrich, F.; Mushumba, H.; Kniep, I.; Schroder, A.S.; et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19. Ann. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Song, J.-C.; Wang, G.; Zhang, W.; Zhang, Y.; Li, W.-Q.; Zhou, Z.; People’s Liberation Army Professional Committee of Critical Care Medicine, Chinese Society on Thrombosis and Haemostasis. Chinese expert consensus on diagnosis and treatment of coagulation dysfunction in COVID-19. Mil. Med. Res. 2020, 7, 19. [Google Scholar]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020, 18, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Paranjpe, I.; Fuster, V.; Lala, A.; Russak, A.J.; Glicksberg, B.S.; Levin, M.A.; Charney, A.W.; Narula, J.; Fayad, Z.A.; Bagiella, E. Association of treatment dose anticoagulation with in-hospital survival among hospitalized patients with COVID-19. J. Am. Coll. Cardiol. 2020, 76, 122–124. [Google Scholar] [CrossRef]
- Ayerbe, L.; Risco, C.; Ayis, S. The association between treatment with heparin and survival in patients with Covid-19. J. Thromb. Thrombolysis 2020, 50, 298–301. [Google Scholar] [CrossRef]
- Rossi, R.; Coppi, F.; Talarico, M.; Boriani, G. Protective role of chronic treatment with direct oral anticoagulants in elderly patients affected by interstitial pneumonia in COVID-19 era. Eur. J. Intern. Med. 2020, 77, 158–160. [Google Scholar] [CrossRef]
- Secco, E.; Pasqualetto, M.C.; Bombardini, T.; Picano, E.; Rigo, F. A possible benefit from therapeutic anticoagulation in COVID-19: The Dolo hospital experience in Veneto, Italy. Kardiol. Pol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Poggiali, E.; Bastoni, D.; Ioannilli, E.; Vercelli, A.; Magnacavallo, A. Deep Vein Thrombosis and Pulmonary Embolism: Two Complications of COVID-19 Pneumonia? Eur. J. Case Rep. Intern. Med. 2020, 7, 001646. [Google Scholar] [CrossRef]
- Brouns, S.H.; Brüggemann, R.; Linkens, A.E.; Magdelijns, F.J.; Joosten, H.; Heijnen, R.; ten Cate-Hoek, A.J.; Schols, J.M.G.A.; Ten Cate, H.; Spaetgens, B. Mortality and the use of Antithrombotic Therapies among Nursing Home Residents with COVID-19. J. Am. Geriatr. Soc. 2020, 68, 1647–1652. [Google Scholar] [CrossRef]
- Motta, J.K.; Ogunnaike, R.O.; Shah, R.; Stroever, S.; Cedeno, H.V.; Thapa, S.K.; Chronakos, J.J.; Jimenez, E.J.; Petrini, J.; Hegde, A. Clinical Outcomes With the Use of Prophylactic Versus Therapeutic Anticoagulation in COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Patel, N.G.; Bhasin, A.; Feinglass, J.M.; Belknap, S.M.; Angarone, M.P.; Cohen, E.R.; Barsuk, J.H. Clinical Outcomes of Hospitalized Patients with COVID-19 on Therapeutic Anticoagulants. medRxiv 2020. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.; Van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.; Tao, X.; Cui, W.; Yi, B.; Pan, T.; Young, K.H.; Qian, W. SARS-CoV-2 induced thrombocytopenia as an important biomarker significantly correlated with abnormal coagulation function, increased intravascular blood clot risk and mortality in COVID-19 patients. Exp. Hematol. Oncol. 2020, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, X.; Xiao, Y.; Gao, T.; Wang, G.; Wang, Z.; Zhang, Z.; Hu, Y.; Dong, Q.; Zhao, S. Heparin-induced thrombocytopenia is associated with a high risk of mortality in critical COVID-19 patients receiving heparin-involved treatment. medRxiv 2020. [Google Scholar] [CrossRef]
- Patell, R.; Khan, A.; Bogue, T.; Merrill, M.; Koshy, A.; Bindal, P.; Joyce, R.; Aird, W.C.; Neuberg, D.; Bauer, K.A. Heparin induced thrombocytopenia antibodies in COVID-19. Am. J. Hematol. 2020. [Google Scholar] [CrossRef]
- Riker, R.R.; May, T.L.; Fraser, G.L.; Gagnon, D.J.; Bandara, M.; Zemrak, W.R.; Seder, D.B. Heparin-induced thrombocytopenia with thrombosis in COVID-19 adult respiratory distress syndrome. Res. Pract. Thromb. Haemost. 2020, 4, 936–941. [Google Scholar] [CrossRef]
- Vayne, C.; May, M.A.; Bourguignon, T.; Lemoine, E.; Guery, E.A.; Rollin, J.; Gruel, Y.; Pouplard, C. Frequency and Clinical Impact of Platelet Factor 4-Specific Antibodies in Patients Undergoing Extracorporeal Membrane Oxygenation. Thromb. Haemost. 2019, 119, 1138–1146. [Google Scholar] [CrossRef]
- Dickie, H.; Tovey, L.; Berry, W.; Ostermann, M. Revised algorithm for heparin anticoagulation during continuous renal replacement therapy. Crit. Care 2015, 19, 376. [Google Scholar] [CrossRef] [Green Version]
- Khandelwal, S.; Ravi, J.; Rauova, L.; Johnson, A.; Lee, G.M.; Gilner, J.B.; Gunti, S.; Notkins, A.L.; Kuchibhatla, M.; Frank, M.; et al. Polyreactive IgM initiates complement activation by PF4/heparin complexes through the classical pathway. Blood 2018, 132, 2431–2440. [Google Scholar] [CrossRef] [Green Version]
- Cuker, A.; Rux, A.H.; Hinds, J.L.; Dela Cruz, M.; Yarovoi, S.V.; Brown, I.A.; Yang, W.; Konkle, B.A.; Arepally, G.M.; Watson, S.P.; et al. Novel diagnostic assays for heparin-induced thrombocytopenia. Blood 2013, 121, 3727–3732. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.Q.; Liu, W.; Park, K.S.; Sachais, B.S.; Arepally, G.M.; Cines, D.B.; Poncz, M. Defining a second epitope for heparin-induced thrombocytopenia/thrombosis antibodies using KKO, a murine HIT-like monoclonal antibody. Blood 2002, 99, 1230–1236. [Google Scholar] [CrossRef] [Green Version]
- Rauova, L.; Zhai, L.; Kowalska, M.A.; Arepally, G.M.; Cines, D.B.; Poncz, M. Role of platelet surface PF4 antigenic complexes in heparin-induced thrombocytopenia pathogenesis: Diagnostic and therapeutic implications. Blood 2006, 107, 2346–2353. [Google Scholar] [CrossRef]
- Falati, S.; Gross, P.; Merrill-Skoloff, G.; Furie, B.C.; Furie, B. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat. Med. 2002, 8, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Kuramochi, T.; Igawa, T.; Tsunoda, H.; Hattori, K. Humanization and Simultaneous Optimization of Monoclonal Antibody. Methods Mol. Biol. 2019, 1904, 213–230. [Google Scholar] [CrossRef] [PubMed]
- Ministro, J.; Manuel, A.M.; Goncalves, J. Therapeutic Antibody Engineering and Selection Strategies. Adv. Biochem. Eng. Biotechnol. 2020, 171, 55–86. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 117, 10970–10975. [Google Scholar] [CrossRef]
- Thomas, S.; Makris, M. The reversal of anticoagulation in clinical practice. Clin. Med. (Lond.) 2018, 18, 314–319. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Number | Patient Condition | Anti-Coagulation | Outcome | Ref. |
|---|---|---|---|---|
| 2773 | hospitalized | therapeutic anticoagulation | For patients who required mechanical ventilation (n = 395), in-hospital mortality was lower in those treated with anticoagulation (29.1% vs. 62.7%); no difference in general population | [84] |
| 2075 | hospitalized | Heparin | Lower mortality in patients who used Heparin (OR 0.55, 95% CI 0.37–0.82, p = 0.003) | [85] |
| 1716 | therapeutic anticoagulation | Subjects receiving new therapeutic anticoagulation, especially for those in the absence of other indications, were more likely to die (OR 5.93; 95% CI 3.71–9.47). Continuation of outpatient prescribed anticoagulant was not associated with improved clinical outcomes. | [91] | |
| 449 | severe | mainly LMWH | In patients with D-dimer >6-fold of upper limit of normal, 28-day mortality was lower in heparin users than nonusers (32.8% vs. 52.4%, p = 0.017). | [83] |
| 374 | hospitalized | therapeutic vs. prophylatic | higher in-hospital mortality in patients receiving preemptive therapeutic anticoagulation (aRR: 2.3, 95% CI = 1.0, 4.9; p = 0.04) | [90] |
| 245 | ICU | therapeutic vs. prophylatic | 79% reduction in death with therapeutic dose | [27] |
| 184 | ICU | thromboprophylaxis | thrombosis rate: 31% | [92] |
| 115 | hospitalized | therapeutic anticoagulation | Lower mortality in patients with anticoagulation (OR 0.055, 95% Cl 0.008–0.386, p = 0.03) | [87] |
| 101 | Nursing Home Residents | Only a trend of lower mortality in patients with anticoagulation (OR 0.89, 95% Cl 0.41–1.95) | [89] | |
| 70 | elderly patients with interstitial pneumonia and CHD | direct oral anticoagulants | Lower mortality in patients with anticoagulation (HR 0.38, 95% Cl 0.17–0.58, p = 0.01) | [86] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, Z.; Greene, M.I.; Zhu, Z.; Zhang, H. Structural Features and PF4 Functions that Occur in Heparin-Induced Thrombocytopenia (HIT) Complicated by COVID-19. Antibodies 2020, 9, 52. https://doi.org/10.3390/antib9040052
Cai Z, Greene MI, Zhu Z, Zhang H. Structural Features and PF4 Functions that Occur in Heparin-Induced Thrombocytopenia (HIT) Complicated by COVID-19. Antibodies. 2020; 9(4):52. https://doi.org/10.3390/antib9040052
Chicago/Turabian StyleCai, Zheng, Mark I. Greene, Zhiqiang Zhu, and Hongtao Zhang. 2020. "Structural Features and PF4 Functions that Occur in Heparin-Induced Thrombocytopenia (HIT) Complicated by COVID-19" Antibodies 9, no. 4: 52. https://doi.org/10.3390/antib9040052