Understanding Inter-Individual Variability in Monoclonal Antibody Disposition


Abstract
:1. Introduction
2. Mechanisms Influencing Antibody Pharmacokinetics
2.1. Mechanisms of Antibody Absorption
2.1.1. Determinants of Convective Transport
2.1.2. Role of Anatomical Site for Subcutaneous Injection
2.1.3. Role of Neonatal Fc Receptor
2.1.4. Role of Pre-Systemic Catabolism
2.1.5. Role of Target at Subcutaneous Site
2.1.6. Role of Antibody Dose
2.1.7. Role of PEGylation and Co-Formulation Strategies
2.2. Mechanisms of Antibody Distribution
Tissue-Specific Properties Affecting mAb Distribution
2.3. Mechanisms of Antibody Elimination
3. Variability in Determinants of Antibody Disposition
3.1. FcRn Gene Polymorphism and Expression
3.2. Fc Gamma Receptor Expression and Polymorphism
3.3. Target Properties
3.3.1. Target Expression
3.3.2. Target Shedding
3.3.3. Target Turnover and Internalization
3.3.4. Target Heterogeneity
3.3.5. Target Polymorphism
3.4. Anti-Drug Antibodies
4. Common Covariates Identified in Population Pharmacokinetic Modeling
4.1. Body Size
4.2. Sex
4.3. Race
4.4. Age
4.5. Albumin
5. Additional Factors Contributing to Inter-Individual Variability
5.1. Influence of Pathophysiological Elements of Disease
5.1.1. Proteinuria and Renal Protein Catabolism
5.1.2. Protein Losing Enteropathy
5.1.3. Blood-Brain Barrier
5.2. Influence of Co-Administered Drugs
5.2.1. Saturation of Neonatal Fc Receptor
5.2.2. Alteration in Convective Transport and Tumor Uptake
5.2.3. Decrease in Anti-Drug Antibody Response
5.2.4. Alteration in Target Expression and Conformation
6. Summary and Future Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Porter, C.J.H.; Edwards, G.A.; Charman, S.A. Lymphatic transport of proteins after s.c. injection: Implications of animal model selection. Adv. Drug Deliv. Rev. 2001, 50, 157–171. [Google Scholar] [CrossRef]
- Gill, K.L.; Machavaram, K.K.; Rose, R.H.; Chetty, M. Potential Sources of Inter-Subject Variability in Monoclonal Antibody Pharmacokinetics. Clin. Pharmacokinet. 2016, 55, 789–805. [Google Scholar] [CrossRef]
- Leak, L.V. Studies on the permeability of lymphatic capillaries. J. Cell Biol. 1971, 50, 300–323. [Google Scholar] [CrossRef] [Green Version]
- Leak, L.V. Electron microscopic observations on lymphatic capillaries and the structural components of the connective tissue-lymph interface. Microvasc. Res. 1970, 2, 361–391. [Google Scholar] [CrossRef]
- Sarin, H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J. Angiogenesis Res. 2010, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Reddy, N.P. Lymph circulation: Physiology, pharmacology, and biomechanics. Crit. Rev. Biomed. Eng. 1986, 14, 45–91. [Google Scholar]
- Reth, M. Matching cellular dimensions with molecular sizes. Nat Immunol 2013, 14, 765–767. [Google Scholar] [CrossRef]
- Starling, E.H. On the Absorption of Fluids from the Connective Tissue Spaces. J. Physiol. 1896, 19, 312–326. [Google Scholar] [CrossRef]
- Ogawa, K.; Imai, M.; Ogawa, T.; Tsukamoto, Y.; Sasaki, F. Caveolar and intercellular channels provide major transport pathways of macromolecules across vascular endothelial cells. Anat. Rec. 2001, 264, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Bendayan, M. Morphological and cytochemical aspects of capillary permeability. Microsc. Res. Tech. 2002, 57, 327–349. [Google Scholar] [CrossRef]
- Simionescu, M.; Gafencu, A.; Antohe, F. Transcytosis of plasma macromolecules in endothelial cells: A cell biological survey. Microsc. Res. Tech. 2002, 57, 269–288. [Google Scholar] [CrossRef]
- Lewis, J.H. The route and rate of absorption of subcutaneously injected serum. JAMA 1921, 76, 1342–1345. [Google Scholar]
- Field, M.E.; Drinker, C.K. The permeability of the capillaries of the dog to protien. Am. J. Physiol. Leg. Content 1931, 97, 40–51. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, J.; Steller, M.; Keenan, A.; Covell, D.; Key, M.; Sieber, S.; Oldham, R.; Hwang, K.; Parker, R. Monoclonal antibodies in the lymphatics: Selective delivery to lymph node metastases of a solid tumor. Science 1983, 222, 423–426. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Steller, M.A.; Covell, D.G.; Holton, O.D., 3rd; Keenan, A.M.; Sieber, S.M.; Parker, R.J. Monoclonal antitumor antibodies in the lymphatics. Cancer Treat. Rep. 1984, 68, 257–264. [Google Scholar]
- Hein, W.R.; Supersaxo, A. Effect of interferon-alpha-2a on the output of recirculating lymphocytes from single lymph nodes. Immunology 1988, 64, 469–474. [Google Scholar]
- Supersaxo, A.; Hein, W.R.; Steffen, H. Effect of Molecular Weight on the Lymphatic Absorption of Water-Soluble Compounds Following Subcutaneous Administration. Pharm. Res. 1990, 7, 167–169. [Google Scholar] [CrossRef]
- Charman, S.A.; McLennan, D.N.; Edwards, G.A.; Porter, C.J. Lymphatic absorption is a significant contributor to the subcutaneous bioavailability of insulin in a sheep model. Pharm. Res. 2001, 18, 1620–1626. [Google Scholar] [CrossRef]
- McLennan, D.N.; Porter, C.J.H.; Edwards, G.A.; Brumm, M.; Martin, S.W.; Charman, S.A. Pharmacokinetic model to describe the lymphatic absorption of r-metHu-leptin after subcutaneous injection to sheep. Pharm. Res. 2003, 20, 1156–1162. [Google Scholar] [CrossRef]
- Charman, S.A.; Segrave, A.M.; Edwards, G.A.; Porter, C.J. Systemic availability and lymphatic transport of human growth hormone administered by subcutaneous injection. J. Pharm. Sci. 2000, 89, 168–177. [Google Scholar] [CrossRef]
- McLennan, D.N.; Porter, C.J.H.; Edwards, G.A.; Martin, S.W.; Heatherington, A.C.; Charman, S.A. Lymphatic Absorption Is the Primary Contributor to the Systemic Availability of Epoetin Alfa following Subcutaneous Administration to Sheep. J. Pharmacol. Exp. Ther. 2005, 313, 345–351. [Google Scholar] [CrossRef] [Green Version]
- McLennan, D.N.; Porter, C.J.; Edwards, G.A.; Heatherington, A.C.; Martin, S.W.; Charman, S.A. The absorption of darbepoetin alfa occurs predominantly via the lymphatics following subcutaneous administration to sheep. Pharm. Res. 2006, 23, 2060–2066. [Google Scholar] [CrossRef]
- Wang, W.; Chen, N.; Shen, X.; Cunningham, P.; Fauty, S.; Michel, K.; Wang, B.; Hong, X.; Adreani, C.; Nunes, C.N.; et al. Lymphatic transport and catabolism of therapeutic proteins after subcutaneous administration to rats and dogs. Drug Metab. Dispos. 2012, 40, 952–962. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Bateman, T.J.; Adreani, C.; Shen, X.; Cunningham, P.K.; Wang, B.; Trinh, T.; Christine, A.; Hong, X.; Nunes, C.N.; et al. Lymphatic absorption, metabolism, and excretion of a therapeutic peptide in dogs and rats. Drug Metab. Dispos. 2013, 41, 2206–2214. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.A.; Sawchuk, R.J.; Brundage, R.C.; Horvath, C.; Mendenhall, H.V.; Gunther, R.A.; Braeckman, R.A. Plasma and Lymph Pharmacokinetics of Recombinant Human Interleukin-2 and Polyethylene Glycol-Modified Interleukin-2 in Pigs. J. Pharmacol. Exp. Ther. 2000, 293, 248–259. [Google Scholar]
- Bocci, V.; Muscettola, M.; Grasso, G.; Magyar, Z.; Naldini, A.; Szabo, G. The lymphatic route. 1) Albumin and hyaluronidase modify the normal distribution of interferon in lymph and plasma. Experientia 1986, 42, 432–433. [Google Scholar] [CrossRef]
- Kojima, K.; Takahashi, T.; Nakanishi, Y. Lymphatic transport of recombinant human tumor necrosis factor in rats. J. Pharm. Dyn. 1988, 11, 700–706. [Google Scholar] [CrossRef]
- Kagan, L.; Gershkovich, P.; Mendelman, A.; Amsili, S.; Ezov, N.; Hoffman, A. The role of the lymphatic system in subcutaneous absorption of macromolecules in the rat model. Eur. J. Pharm. Biopharm. 2007, 67, 759–765. [Google Scholar] [CrossRef]
- Kaminskas, L.M.; Kota, J.; McLeod, V.M.; Kelly, B.D.; Karellas, P.; Porter, C.J. PEGylation of polylysine dendrimers improves absorption and lymphatic targeting following SC administration in rats. J. Control. Release 2009, 140, 108–116. [Google Scholar] [CrossRef]
- Dahlberg, A.M.; Kaminskas, L.M.; Smith, A.; Nicolazzo, J.A.; Porter, C.J.; Bulitta, J.B.; McIntosh, M.P. The lymphatic system plays a major role in the intravenous and subcutaneous pharmacokinetics of trastuzumab in rats. Mol. Pharm. 2014, 11, 496–504. [Google Scholar] [CrossRef]
- Wu, F.; Bhansali, S.G.; Law, W.C.; Bergey, E.J.; Prasad, P.N.; Morris, M.E. Fluorescence imaging of the lymph node uptake of proteins in mice after subcutaneous injection: Molecular weight dependence. Pharm. Res. 2012, 29, 1843–1853. [Google Scholar] [CrossRef] [PubMed]
- Bhansali, S.G.; Balu-Iyer, S.V.; Morris, M.E. Influence of route of administration and liposomal encapsulation on blood and lymph node exposure to the protein VEGF-C156S. J. Pharm. Sci. 2012, 101, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Bhansali, S.G.; Tamhane, M.; Kumar, R.; Vathy, L.A.; Ding, H.; Yong, K.T.; Bergey, E.J.; Prasad, P.N.; Morris, M.E. Noninvasive real-time fluorescence imaging of the lymphatic uptake of BSA-IRDye 680 conjugate administered subcutaneously in mice. J. Pharm. Sci. 2012, 101, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Tamhane, M.; Morris, M.E. Pharmacokinetics, lymph node uptake, and mechanistic PK model of near-infrared dye-labeled bevacizumab after IV and SC administration in mice. AAPS J. 2012, 14, 252–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilney, N.L. Patterns of lymphatic drainage in the adult laboratory rat. J. Anat. 1971, 109, 369–383. [Google Scholar] [PubMed]
- Xie DD, H.V. Factors affecting the lymphatic absorption of macromolecules following extravascular administration. Pharm. Res. 1996, 13, S396. [Google Scholar]
- Bagby, T.R.; Cai, S.; Duan, S.; Thati, S.; Aires, D.J.; Forrest, L. Impact of molecular weight on lymphatic drainage of a biopolymer-based imaging agent. Pharmaceutics 2012, 4, 276–295. [Google Scholar] [CrossRef] [Green Version]
- Richter, W.F.; Bhansali, S.G.; Morris, M.E. Mechanistic determinants of biotherapeutics absorption following SC administration. AAPS J. 2012, 14, 559–570. [Google Scholar] [CrossRef] [Green Version]
- Olszewski, W.L. Surgical pathophysiology of the lymphatic system. Z. Fur Exp. Chir. 1981, 14, 9–20. [Google Scholar]
- Olszewski, W.; Engeset, A.; Jaeger, P.M.; Sokolowski, J.; Theodorsen, L. Flow and composition of leg lymph in normal men during venous stasis, muscular activity and local hyperthermia. Acta Physiol. Scand. 1977, 99, 149–155. [Google Scholar] [CrossRef]
- Olszewski, W.L. Contractility patterns of normal and pathologically changed human lymphatics. Ann. N. Y. Acad. Sci. 2002, 979, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Unno, N.; Tanaka, H.; Suzuki, M.; Yamamoto, N.; Mano, Y.; Sano, M.; Saito, T.; Konno, H. Influence of age and gender on human lymphatic pumping pressure in the leg. Lymphology 2011, 44, 113–120. [Google Scholar] [PubMed]
- Gashev, A.A.; Chatterjee, V. Aged Lymphatic Contractility: Recent Answers and New Questions. Lymphat. Res. Biol. 2013, 11, 2–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, A.; Tannenbaum, S.; Rordorf, C.; Lowe, P.J.; Floch, D.; Gram, H.; Roy, S. Pharmacokinetic and Pharmacodynamic Properties of Canakinumab, a Human Anti-Interleukin-1β Monoclonal Antibody. Clin. Pharmacokinet. 2012, 51, e1–e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, A.; Van, L.M.; Skerjanec, A.; Floch, D.; Klein, U.R.; Krammer, G.; Sunkara, G.; Howard, D. Pharmacokinetic and pharmacodynamic properties of canakinumab in patients with gouty arthritis. J. Clin. Pharmacol. 2013, 53, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Sutjandra, L.; Rodriguez, R.D.; Doshi, S.; Ma, M.; Peterson, M.C.; Jang, G.R.; Chow, A.T.; Perez-Ruixo, J.J. Population pharmacokinetic meta-analysis of denosumab in healthy subjects and postmenopausal women with osteopenia or osteoporosis. Clin. Pharmacokinet. 2011, 50, 793–807. [Google Scholar] [CrossRef]
- Gibiansky, L.; Sutjandra, L.; Doshi, S.; Zheng, J.; Sohn, W.; Peterson, M.C.; Jang, G.R.; Chow, A.T.; Perez-Ruixo, J.J. Population pharmacokinetic analysis of denosumab in patients with bone metastases from solid tumours. Clin. Pharmacokinet. 2012, 51, 247–260. [Google Scholar] [CrossRef]
- Kakkar, T.; Sung, C.; Gibiansky, L.; Vu, T.; Narayanan, A.; Lin, S.L.; Vincent, M.; Banfield, C.; Colbert, A.; Hoofring, S.; et al. Population PK and IgE pharmacodynamic analysis of a fully human monoclonal antibody against IL4 receptor. Pharm. Res. 2011, 28, 2530–2542. [Google Scholar] [CrossRef]
- Cromer, W.E.; Zawieja, S.D.; Tharakan, B.; Childs, E.W.; Newell, M.K.; Zawieja, D.C. The effects of inflammatory cytokines on lymphatic endothelial barrier function. Angiogenesis 2014, 17, 395–406. [Google Scholar] [CrossRef] [Green Version]
- Aldrich, M.B.; Sevick-Muraca, E.M. Cytokines are systemic effectors of lymphatic function in acute inflammation. Cytokine 2013, 64, 362–369. [Google Scholar] [CrossRef] [Green Version]
- Hollander, W.; Reilly, P.; Burrows, B.A. Lymphatic flow in human subjects as indicated by the disappearance of i (131)-labeled albumin from the subcutaneous tissue. J. Clin. Investig. 1961, 40, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.F.; Jacobsen, B. Subcutaneous absorption of biotherapeutics: Knowns and unknowns. Drug Metab. Dispos. 2014, 42, 1881–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modi, S.; Stanton, A.W.; Svensson, W.E.; Peters, A.M.; Mortimer, P.S.; Levick, J.R. Human lymphatic pumping measured in healthy and lymphoedematous arms by lymphatic congestion lymphoscintigraphy. J. Physiol. 2007, 583, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Levakov, A.; Vuckovic, N.; Dolai, M.; Kacanski, M.M.; Bozanic, S. Age-related skin changes. Med. Pregl. 2012, 65, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Akkus, O.; Oguz, A.; Uzunlulu, M.; Kizilgul, M. Evaluation of Skin and Subcutaneous Adipose Tissue Thickness for Optimal Insulin Injection. J. Diabetes Metab. 2012, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Mariman, E.C.M.; Wang, P. Adipocyte extracellular matrix composition, dynamics and role in obesity. Cell. Mol. Life Sci. 2010, 67, 1277–1292. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.C.; Ng, E.H.; Chan, M.M.; Tang, O.S.; Lau, E.Y.; Yeung, W.S.; Ho, P.C. Bioavailability of hCG after intramuscular or subcutaneous injection in obese and non-obese women. Hum. Reprod. 2003, 18, 2294–2297. [Google Scholar] [CrossRef] [Green Version]
- Olsson-Gisleskog, P.; Jacqmin, P.; Perez-Ruixo, J.J. Population pharmacokinetics meta-analysis of recombinant human erythropoietin in healthy subjects. Clin. Pharmacokinet. 2007, 46, 159–173. [Google Scholar] [CrossRef]
- Koivisto, V.A.; Felig, P. Alterations in insulin absorption and in blood glucose control associated with varying insulin injection sites in diabetic patients. Ann. Intern. Med. 1980, 92, 59–61. [Google Scholar] [CrossRef]
- Ter Braak, E.W.; Woodworth, J.R.; Bianchi, R.; Cerimele, B.; Erkelens, D.W.; Thijssen, J.H.; Kurtz, D. Injection site effects on the pharmacokinetics and glucodynamics of insulin lispro and regular insulin. Diabetes Care 1996, 19, 1437–1440. [Google Scholar] [CrossRef]
- Beshyah, S.A.; Anyaoku, V.; Niththyananthan, R.; Sharp, P.; Johnston, D.G. The effect of subcutaneous injection site on absorption of human growth hormone: Abdomen versus thigh. Clin. Endocrinol. 1991, 35, 409–412. [Google Scholar] [CrossRef]
- Macdougall, I.C.; Jones, J.M.; Robinson, M.I.; Miles, J.B.; Coles, G.A.; Williams, J.D. Subcutaneous erythropoietin therapy: Comparison of three different sites of injection. Contrib. Nephrol. 1991, 88, 152–156; discussion 157–158. [Google Scholar] [PubMed]
- Jensen, J.D.; Jensen, L.W.; Madsen, J.K. The pharmacokinetics of recombinant human erythropoietin after subcutaneous injection at different sites. Eur. J. Clin. Pharmacol. 1994, 46, 333–337. [Google Scholar] [CrossRef]
- Kota, J.; Machavaram, K.K.; McLennan, D.N.; Edwards, G.A.; Porter, C.J.; Charman, S.A. Lymphatic absorption of subcutaneously administered proteins: Influence of different injection sites on the absorption of darbepoetin alfa using a sheep model. Drug Metab. Dispos. 2007, 35, 2211–2217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, L.; Turner, M.R.; Balu-Iyer, S.V.; Mager, D.E. Subcutaneous absorption of monoclonal antibodies: Role of dose, site of injection, and injection volume on rituximab pharmacokinetics in rats. Pharm. Res. 2012, 29, 490–499. [Google Scholar] [CrossRef]
- Kagan, L.; Mager, D.E. Mechanisms of subcutaneous absorption of rituximab in rats. Drug Metab. Dispos. 2013, 41, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Kagan, L.; Zhao, J.; Mager, D.E. Interspecies pharmacokinetic modeling of subcutaneous absorption of rituximab in mice and rats. Pharm. Res. 2014, 31, 3265–3273. [Google Scholar] [CrossRef] [Green Version]
- Sandby-Moller, J.; Poulsen, T.; Wulf, H.C. Epidermal thickness at different body sites: Relationship to age, gender, pigmentation, blood content, skin type and smoking habits. Acta Derm. Venereol. 2003, 83, 410–413. [Google Scholar] [CrossRef]
- Mrsny, R.J. Metabolic Processes at Injection Sites Affecting Pharmacokinetics, Pharmacodynamics and Metabolism of Protein and Peptide Therapeutics. In Proteins and Peptides; Mrsny, R.J., Daugherty, A., Eds.; CRC Press: Boca Raton, FL, USA, 2009; pp. 80–105. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Q.; Zhuang, Y.; Frederick, B.; Yan, H.; Bouman-Thio, E.; Marini, J.C.; Keen, M.; Snead, D.; Davis, H.M.; et al. Subcutaneous bioavailability of golimumab at 3 different injection sites in healthy subjects. J. Clin. Pharmacol. 2010, 50, 276–284. [Google Scholar] [CrossRef]
- Ling, J.; Lyn, S.; Xu, Z.; Achira, M.; Bouman-Thio, E.; Shishido, A.; Ford, J.; Shankar, G.; Wagner, C.; Kim, K.T.; et al. Lack of racial differences in the pharmacokinetics of subcutaneous golimumab in healthy Japanese and Caucasian male subjects. J. Clin. Pharmacol. 2010, 50, 792–802. [Google Scholar] [CrossRef]
- Zhuang, Y.; Xu, Z.; de Vries, D.E.; Wang, Q.; Shishido, A.; Comisar, C.; Ford, J.A.; Keen, M.; Achira, M.; Tsukamoto, Y.; et al. Pharmacokinetics and safety of sirukumab following a single subcutaneous administration to healthy Japanese and Caucasian subjects. Int. J. Clin. Pharmacol. Ther. 2013, 51, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Garg, A. Investigation of the role of FcRn in the absorption, distribution, and elimination of monoclonal antibodies (Order No. 3301453). Available from Dissertations & Theses @ SUNY Buffalo 2007; ProQuest Dissertations & Theses Global. (304779017). Available online: https://search-proquest-com.gate.lib.buffalo.edu/docview/304779017?accountid=14169 (accessed on 2 December 2019).
- Deng, R.; Loyet, K.M.; Lien, S.; Iyer, S.; DeForge, L.E.; Theil, F.P.; Lowman, H.B.; Fielder, P.J.; Prabhu, S. Pharmacokinetics of humanized monoclonal anti-tumor necrosis factor-{alpha} antibody and its neonatal Fc receptor variants in mice and cynomolgus monkeys. Drug Metab. Dispos. 2010, 38, 600–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, R.; Meng, Y.G.; Hoyte, K.; Lutman, J.; Lu, Y.; Iyer, S.; DeForge, L.E.; Theil, F.P.; Fielder, P.J.; Prabhu, S. Subcutaneous bioavailability of therapeutic antibodies as a function of FcRn binding affinity in mice. mAbs 2012, 4, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta-Mannan, A.; Witcher, D.R.; Lu, J.; Wroblewski, V.J. Influence of improved FcRn binding on the subcutaneous bioavailability of monoclonal antibodies in cynomolgus monkeys. mAbs 2012, 4, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, M.; Halban, P.A.; Girardier, L.; Seydoux, J.; Offord, R.E.; Renold, A.E. Absorption kinetics of subcutaneously injected insulin. Evidence for degradation at the injection site. Diabetologia 1979, 17, 97–99. [Google Scholar] [CrossRef] [Green Version]
- Hori, R.; Komada, F.; Iwakawa, S.; Seino, Y.; Okumura, K. Enhanced bioavailability of subcutaneously injected insulin coadministered with collagen in rats and humans. Pharm. Res. 1989, 6, 813–816. [Google Scholar] [CrossRef]
- Takeyama, M.; Ishida, T.; Kokubu, N.; Komada, F.; Iwakawa, S.; Okumura, K.; Hori, R. Enhanced bioavailability of subcutaneously injected insulin by pretreatment with ointment containing protease inhibitors. Pharm. Res. 1991, 8, 60–64. [Google Scholar] [CrossRef]
- Hua, Y.; Nair, S. Proteases in cardiometabolic diseases: Pathophysiology, molecular mechanisms and clinical applications. Biochim. Biophys. Acta 2015, 1852, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Davis, C.B.; Bugelski, P.J. Subcutaneous bioavailability of a PRIMATIZED IgG1 anti-human CD4 monoclonal antibody is dose dependent in transgenic mice bearing human CD4. Drug Deliv. 1998, 5, 95–100. [Google Scholar] [CrossRef]
- Mao, C.P.; Brovarney, M.R.; Dabbagh, K.; Birnbock, H.F.; Richter, W.F.; Del Nagro, C.J. Subcutaneous versus intravenous administration of rituximab: Pharmacokinetics, CD20 target coverage and B-cell depletion in cynomolgus monkeys. PLoS ONE 2013, 8, e80533. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.K. Pharmacokinetic strategies to improve the safety and efficacy of intraperitoneal chemotherapy (Order No. 3423520). Available from Dissertations & Theses @ SUNY Buffalo 2010; ProQuest Dissertations & Theses Global. (759395016). Available online: https://search-proquest-com.gate.lib.buffalo.edu/docview/759395016?accountid=14169 (accessed on 2 December 2019).
- Kaminskas, L.M.; Ascher, D.B.; McLeod, V.M.; Herold, M.J.; Le, C.P.; Sloan, E.K.; Porter, C.J. PEGylation of interferon alpha2 improves lymphatic exposure after subcutaneous and intravenous administration and improves antitumour efficacy against lymphatic breast cancer metastases. J. Control. Release 2013, 168, 200–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, L.J.; Bulitta, J.B.; Ascher, D.B.; Haynes, J.M.; McLeod, V.M.; Porter, C.J.; Williams, C.C.; Kaminskas, L.M. PEGylation does not significantly change the initial intravenous or subcutaneous pharmacokinetics or lymphatic exposure of trastuzumab in rats but increases plasma clearance after subcutaneous administration. Mol. Pharm. 2015, 12, 794–809. [Google Scholar] [CrossRef] [PubMed]
- Bittner, B.; Richter, W.F.; Hourcade-Potelleret, F.; McIntyre, C.; Herting, F.; Zepeda, M.L.; Schmidt, J. Development of a subcutaneous formulation for Trastuzumab—Nonclinical and clinical bridging approach to the approved intravenous dosing regimen. Arzneimittelforschung 2012, 62, 401–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bittner, B.; Richter, W.F.; Hourcade-Potelleret, F.; Herting, F.; Schmidt, J. Non-clinical pharmacokinetic/pharmacodynamic and early clinical studies supporting development of a novel subcutaneous formulation for the monoclonal antibody rituximab. Drug Res. 2014, 64, 569–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shpilberg, O.; Jackisch, C. Subcutaneous administration of rituximab (MabThera) and trastuzumab (Herceptin) using hyaluronidase. Br. J. Cancer 2013, 109, 1556–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosengren, S.; Dychter, S.S.; Printz, M.A.; Huang, L.; Schiff, R.I.; Schwarz, H.-P.; McVey, J.K.; Drake, F.H.; Maneval, D.C.; Kennard, D.A.; et al. Clinical Immunogenicity of rHuPH20, a Hyaluronidase Enabling Subcutaneous Drug Administration. AAPS J. 2015, 17, 1144–1156. [Google Scholar] [CrossRef] [Green Version]
- Fathallah, A.M.; Turner, M.R.; Mager, D.E.; Balu-Iyer, S.V. Effects of hypertonic buffer composition on lymph node uptake and bioavailability of rituximab, after subcutaneous administration. Biopharm. Drug Dispos. 2015, 36, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Capillaries. Available online: https://www1.udel.edu/biology/Wags/histopage/vascularmodelingpage/circsystempage/capillaries/capillaries.html (accessed on 18 July 2019).
- Klabunde, R. Mechanisms of Capillary Exchange. 2015. Available online: http://www.cvphysiology.com/Microcirculation/M016.htm (accessed on 18 July 2019).
- Feng, Y.; Gong, R.; Dimitrov, D.S. Design, expression and characterization of a soluble single-chain functional human neonatal Fc receptor. Protein Expr. Purif. 2011, 79, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.; Balthasar, J.P. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J. Pharmacokinet. Pharmacodyn. 2007, 34, 687–709. [Google Scholar] [CrossRef]
- Chen, N.; Wang, W.; Fauty, S.; Fang, Y.; Hamuro, L.; Hussain, A.; Prueksaritanont, T. The effect of the neonatal Fc receptor on human IgG biodistribution in mice. mAbs 2014, 6, 502–508. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, E.Q.; Balthasar, J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2008, 84, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Robinson, S.B.; Csaky, K.G. FcRn receptor-mediated pharmacokinetics of therapeutic IgG in the eye. Mol. Vis. 2009, 15, 2803–2812. [Google Scholar] [PubMed]
- Jain, R.K. Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer Res. 1990, 50, 814s–819s. [Google Scholar]
- Jain, R.K. Delivery of molecular and cellular medicine to solid tumors. Adv. Drug Deliv. Rev. 2012, 64, 353–365. [Google Scholar] [CrossRef] [Green Version]
- Hansen, R.J.; Balthasar, J.P. Effects of intravenous immunoglobulin on platelet count and antiplatelet antibody disposition in a rat model of immune thrombocytopenia. Blood 2002, 100, 2087–2093. [Google Scholar] [CrossRef] [Green Version]
- Getman, K.E.; Balthasar, J.P. Pharmacokinetic effects of 4C9, an anti-FcRn antibody, in rats: Implications for the use of FcRn inhibitors for the treatment of humoral autoimmune and alloimmune conditions. J. Pharm. Sci. 2005, 94, 718–729. [Google Scholar] [CrossRef]
- Junghans, R.P.; Anderson, C.L. The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proc. Natl. Acad. Sci. USA 1996, 93, 5512–5516. [Google Scholar] [CrossRef] [Green Version]
- Ghetie, V.; Hubbard, J.G.; Kim, J.K.; Tsen, M.F.; Lee, Y.; Ward, E.S. Abnormally short serum half-lives of IgG in beta 2-microglobulin-deficient mice. Eur. J. Immunol. 1996, 26, 690–696. [Google Scholar] [CrossRef]
- Israel, E.J.; Wilsker, D.F.; Hayes, K.C.; Schoenfeld, D.; Simister, N.E. Increased clearance of IgG in mice that lack beta 2-microglobulin: Possible protective role of FcRn. Immunology 1996, 89, 573–578. [Google Scholar] [CrossRef]
- Yeung, Y.A.; Leabman, M.K.; Marvin, J.S.; Qiu, J.; Adams, C.W.; Lien, S.; Starovasnik, M.A.; Lowman, H.B. Engineering human IgG1 affinity to human neonatal Fc receptor: Impact of affinity improvement on pharmacokinetics in primates. J. Immunol. 2009, 182, 7663–7671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta-Mannan, A.; Witcher, D.R.; Tang, Y.; Watkins, J.; Wroblewski, V.J. Monoclonal antibody clearance. Impact of modulating the interaction of IgG with the neonatal Fc receptor. J. Biol. Chem. 2007, 282, 1709–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, V.; Palma, E.; Tesar, D.B.; Mundo, E.E.; Bumbaca, D.; Torres, E.K.; Reyes, N.A.; Shen, B.Q.; Fielder, P.J.; Prabhu, S.; et al. Quantitative cumulative biodistribution of antibodies in mice: Effect of modulating binding affinity to the neonatal Fc receptor. mAbs 2014, 6, 689–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haymann, J.-P.; Levraud, J.-P.; Bouet, S.; Kappes, V.; Hagège, J.; Nguyen, G.; Xu, Y.; Rondeau, E.; Sraer, J.-D. Characterization and Localization of the Neonatal Fc Receptor in Adult Human Kidney. J. Am. Soc. Nephrol. 2000, 11, 632–639. [Google Scholar] [PubMed]
- Brambell, F.W.; Hemmings, W.A.; Morris, I.G. A theoretical model of gamma-globulin catabolism. Nature 1964, 203, 1352–1354. [Google Scholar] [CrossRef] [PubMed]
- Sarav, M.; Wang, Y.; Hack, B.K.; Chang, A.; Jensen, M.; Bao, L.; Quigg, R.J. Renal FcRn Reclaims Albumin but Facilitates Elimination of IgG. J. Am. Soc. Nephrol. 2009, 20, 1941–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuqayyas, L.; Balthasar, J.P. Application of knockout mouse models to investigate the influence of FcγR on the tissue distribution and elimination of 8C2, a murine IgG1 monoclonal antibody. Int. J. Pharm. 2012, 439, 8–16. [Google Scholar] [CrossRef]
- Lobo, E.D.; Hansen, R.J.; Balthasar, J.P. Antibody pharmacokinetics and pharmacodynamics. J. Pharm. Sci. 2004, 93, 2645–2668. [Google Scholar] [CrossRef]
- Schifferli, J.A.; Taylor, R.P. Physiological and pathological aspects of circulating immune complexes. Kidney Int. 1989, 35, 993–1003. [Google Scholar] [CrossRef] [Green Version]
- Emlen, W.; Carl, V.; Burdick, G. Mechanism of transfer of immune complexes from red blood cell CR1 to monocytes. Clin. Exp. Immunol. 1992, 89, 8–17. [Google Scholar] [CrossRef]
- Kosugi, I.; Muro, H.; Shirasawa, H.; Ito, I. Endocytosis of soluble IgG immune complex and its transport to lysosomes in hepatic sinusoidal endothelial cells. J. Hepatol. 1992, 16, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Gillis, C.; Gouel-Cheron, A.; Jonsson, F.; Bruhns, P. Contribution of Human FcgammaRs to Disease with Evidence from Human Polymorphisms and Transgenic Animal Studies. Front. Immunol. 2014, 5, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosales, C.; Uribe-Querol, E. Fc receptors: Cell activators of antibody functions. Adv. Biosci. Biotechnol. 2013, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.M.; Wormald, M.R.; Rudd, P.M.; Davey, G.P. Fc gamma receptors: Glycobiology and therapeutic prospects. J. Inflamm. Res. 2016, 9, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Strohl, W.R. Antibody interactions with the immune system A2. In Therapeutic Antibody Engineering; Strohl, L.M., Ed.; Woodhead Publishing: Amsterdam, The Netherlands, 2012; pp. 131–595. [Google Scholar]
- Israel, E.J.; Patel, V.K.; Taylor, S.F.; Marshak-Rothstein, A.; Simister, N.E. Requirement for a beta 2-microglobulin-associated Fc receptor for acquisition of maternal IgG by fetal and neonatal mice. J. Immunol. 1995, 154, 6246–6251. [Google Scholar]
- Christianson, G.J.; Brooks, W.; Vekasi, S.; Manolfi, E.A.; Niles, J.; Roopenian, S.L.; Roths, J.B.; Rothlein, R.; Roopenian, D.C. Beta 2-microglobulin-deficient mice are protected from hypergammaglobulinemia and have defective antibody responses because of increased IgG catabolism. J. Immunol. 1997, 159, 4781–4792. [Google Scholar]
- Wani, M.A.; Haynes, L.D.; Kim, J.; Bronson, C.L.; Chaudhury, C.; Mohanty, S.; Waldmann, T.A.; Robinson, J.M.; Anderson, C.L. Familial hypercatabolic hypoproteinemia caused by deficiency of the neonatal Fc receptor, FcRn, due to a mutant beta2-microglobulin gene. Proc. Natl. Acad. Sci. USA 2006, 103, 5084–5089. [Google Scholar] [CrossRef] [Green Version]
- Passot, C.; Azzopardi, N.; Renault, S.; Baroukh, N.; Arnoult, C.; Ohresser, M.; Boisdron-Celle, M.; Gamelin, E.; Watier, H.; Paintaud, G.; et al. Influence of FCGRT gene polymorphisms on pharmacokinetics of therapeutic antibodies. mAbs 2013, 5, 614–619. [Google Scholar] [CrossRef] [Green Version]
- Laegreid, W.W.; Heaton, M.P.; Keen, J.E.; Grosse, W.M.; Chitko-McKown, C.G.; Smith, T.P.; Keele, J.W.; Bennett, G.L.; Besser, T.E. Association of bovine neonatal Fc receptor alpha-chain gene (FCGRT) haplotypes with serum IgG concentration in newborn calves. Mamm. Genome 2002, 13, 704–710. [Google Scholar] [CrossRef]
- Zhang, R.; Zhao, Z.; Zhao, Y.; Kacskovics, I.; Eijk, M.; Groot, N.; Li, N.; Hammarstrom, L. Association of FcRn Heavy Chain Encoding Gene (FCGRT) Polymorphisms with IgG Content in Bovine Colostrum. Anim. Biotechnol. 2009, 20, 242–246. [Google Scholar] [CrossRef]
- Clawson, M.L.; Heaton, M.P.; Chitko-McKown, C.G.; Fox, J.M.; Smith, T.P.; Snelling, W.M.; Keele, J.W.; Laegreid, W.W. Beta-2-microglobulin haplotypes in U.S. beef cattle and association with failure of passive transfer in newborn calves. Mamm. Genome 2004, 15, 227–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Chonglong, W.; Zhengzhu, L.; Jingen, X.; Weixuan, F.; Wenwen, W.; Xiangdong, D.; Jianfeng, L.; Qin, Z. Tissues Expression, Polymorphisms Identification of FcRn Gene and Its Relationship with Serum Classical Swine Fever Virus Antibody Level in Pigs. Asian Australas. J. Anim. Sci. 2012, 25, 1089–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Z.H.; Shi, F.; Zhong, F.G.; Bai, D.P.; Zhang, X.Y. Analysis of Fcgrt gene polymorphism in indigenous Chinese sheep and its association with colostrum IgG concentration. Genet. Mol. Res. 2015, 14, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Sachs, U.J.; Socher, I.; Braeunlich, C.G.; Kroll, H.; Bein, G.; Santoso, S. A variable number of tandem repeats polymorphism influences the transcriptional activity of the neonatal Fc receptor alpha-chain promoter. Immunology 2006, 119, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Billiet, T.; Dreesen, E.; Cleynen, I.; Wollants, W.J.; Ferrante, M.; Van Assche, G.; Gils, A.; Vermeire, S. A Genetic Variation in the Neonatal Fc-Receptor Affects Anti-TNF Drug Concentrations in Inflammatory Bowel Disease. Am. J. Gastroenterol. 2016, 111, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Caulet, M.; Lecomte, T.; Bouche, O.; Rollin, J.; Gouilleux-Gruart, V.; Azzopardi, N.; Leger, J.; Borg, C.; Douillard, J.Y.; Manfredi, S.; et al. Bevacizumab Pharmacokinetics Influence Overall and Progression-Free Survival in Metastatic Colorectal Cancer Patients. Clin. Pharmacokinet. 2016, 55, 1381–1394. [Google Scholar] [CrossRef]
- Hogarth, P.M.; Anania, J.C.; Wines, B.D. The FcgammaR of humans and non-human primates and their interaction with IgG: Implications for induction of inflammation, resistance to infection and the use of therapeutic monoclonal antibodies. Curr. Top. Microbiol. Immunol. 2014, 382, 321–352. [Google Scholar] [CrossRef]
- Dijstelbloem, H.M.; van de Winkel, J.G.; Kallenberg, C.G. Inflammation in autoimmunity: Receptors for IgG revisited. Trends Immunol. 2001, 22, 510–516. [Google Scholar] [CrossRef]
- Luan, J.J.; Monteiro, R.C.; Sautes, C.; Fluteau, G.; Eloy, L.; Fridman, W.H.; Bach, J.F.; Garchon, H.J. Defective Fc gamma RII gene expression in macrophages of NOD mice: Genetic linkage with up-regulation of IgG1 and IgG2b in serum. J. Immunol. 1996, 157, 4707–4716. [Google Scholar]
- Radaev, S.; Sun, P. Recognition of immunoglobulins by Fcgamma receptors. Mol. Immunol. 2002, 38, 1073–1083. [Google Scholar] [CrossRef]
- Hadley, A.G.; Zupanska, B.; Kumpel, B.M.; Leader, K.A. The functional activity of Fc gamma RII and Fc gamma RIII on subsets of human lymphocytes. Immunology 1992, 76, 446–451. [Google Scholar] [PubMed]
- Karassa, F.B.; Trikalinos, T.A.; Ioannidis, J.P.A. The role of FcγRIIA and IIIA polymorphisms in autoimmune diseases. Biomed. Pharmacother. 2004, 58, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daeron, M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Kobayashi, T.; Sugita, N.; Tai, H.; Yoshie, H. The FcγRIIa polymorphism influences production of interleukin-1 by mononuclear cells. Int. J. Immunogenet. 2007, 34, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Siriboonrit, U.; Tsuchiya, N.; Sirikong, M.; Kyogoku, C.; Bejrachandra, S.; Suthipinittharm, P.; Luangtrakool, K.; Srinak, D.; Thongpradit, R.; Fujiwara, K.; et al. Association of Fcgamma receptor IIb and IIIb polymorphisms with susceptibility to systemic lupus erythematosus in Thais. Tissue Antigens 2003, 61, 374–383. [Google Scholar] [CrossRef]
- Pan, F.; Zhang, K.; Li, X.; Xu, J.; Hao, J.; Ye, D. Association of Fcgamma receptor IIB gene polymorphism with genetic susceptibility to systemic lupus erythematosus in Chinese populations—A family-based association study. J. Dermatol. Sci. 2006, 43, 35–41. [Google Scholar] [CrossRef]
- Pan, F.; Tang, X.; Zhang, K.; Li, X.; Xu, J.; Chen, H.; Ye, D.Q. Genetic susceptibility and haplotype analysis between Fcgamma receptor IIB and IIIA gene with systemic lupus erythematosus in Chinese population. Lupus 2008, 17, 733–738. [Google Scholar] [CrossRef]
- Jeon, J.Y.; Kim, K.Y.; Kim, B.S.; Jung, J.Y.; Kim, H.A.; Suh, C.H. FcgammaRIIB Gene Polymorphisms Are Associated with Disease Risk and Clinical Manifestations of Systemic Lupus Erythematosus in Koreans. Tohoku J. Exp. Med. 2015, 236, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Thabet, M.M.; Huizinga, T.W.; Marques, R.B.; Stoeken-Rijsbergen, G.; Bakker, A.M.; Kurreeman, F.A.; White, S.J.; Toes, R.E.; van der Helm-van Mil, A.H. Contribution of Fcgamma receptor IIIA gene 158V/F polymorphism and copy number variation to the risk of ACPA-positive rheumatoid arthritis. Ann. Rheum. Dis. 2009, 68, 1775–1780. [Google Scholar] [CrossRef]
- Kastbom, A.; Ahmadi, A.; Soderkvist, P.; Skogh, T. The 158V polymorphism of Fc gamma receptor type IIIA in early rheumatoid arthritis: Increased susceptibility and severity in male patients (the Swedish TIRA project). Rheumatology 2005, 44, 1294–1298. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; Miyazaki, K.; Shimohira, A.; Amano, N.; Okazaki, Y.; Nishimoto, T.; Akahoshi, T.; Munekata, S.; Kanoh, Y.; Ikeda, Y.; et al. Fcgamma receptor IIB gene polymorphism in adult Japanese patients with primary immune thrombocytopenia. Blood 2013, 122, 1991–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Sorge, N.M.; van der Pol, W.L.; Jansen, M.D.; Geleijns, K.P.; Kalmijn, S.; Hughes, R.A.; Rees, J.H.; Pritchard, J.; Vedeler, C.A.; Myhr, K.M.; et al. Severity of Guillain-Barre syndrome is associated with Fc gamma Receptor III polymorphisms. J. Neuroimmunol. 2005, 162, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, W.L.; van den Berg, L.H.; Scheepers, R.H.; van der Bom, J.G.; van Doorn, P.A.; van Koningsveld, R.; van den Broek, M.C.; Wokke, J.H.; van de Winkel, J.G. IgG receptor IIa alleles determine susceptibility and severity of Guillain-Barre syndrome. Neurology 2000, 54, 1661–1665. [Google Scholar] [CrossRef] [PubMed]
- Vedeler, C.A.; Raknes, G.; Myhr, K.M.; Nyland, H. IgG Fc-receptor polymorphisms in Guillain-Barre syndrome. Neurology 2000, 55, 705–707. [Google Scholar] [CrossRef]
- Van der Pol, W.L.; Jansen, M.D.; Kuks, J.B.; de Baets, M.; Leppers-van de Straat, F.G.; Wokke, J.H.; van de Winkel, J.G.; van den Berg, L.H. Association of the Fc gamma receptor IIA-R/R131 genotype with myasthenia gravis in Dutch patients. J. Neuroimmunol. 2003, 144, 143–147. [Google Scholar] [CrossRef]
- Taniuchi, S.; Masuda, M.; Yamamoto, A.; Hasui, M.; Tsuji, S.; Komiyama, Y.; Takahashi, H.; Kobayashi, Y. FcgammaRIII b and FcgammaRIIa polymorphism may affect the production of specific NA1 autoantibody and clinical course of autoimmune neutropenia of infancy. Hum. Immunol. 2001, 62, 408–413. [Google Scholar] [CrossRef]
- Tanaka, Y.; Suzuki, Y.; Tsuge, T.; Kanamaru, Y.; Horikoshi, S.; Monteiro, R.C.; Tomino, Y. FcgammaRIIa-131R allele and FcgammaRIIIa-176V/V genotype are risk factors for progression of IgA nephropathy. Nephrol. Dial. Transplant. 2005, 20, 2439–2445. [Google Scholar] [CrossRef] [Green Version]
- Bronner, I.M.; Hoogendijk, J.E.; de Visser, M.; van de Vlekkert, J.; Badrising, U.A.; Wintzen, A.R.; Uitdehaag, B.M.; Blokland-Fromme, M.; Leusen, J.H.; van der Pol, W.L. Association of the leukocyte immunoglobulin G (Fcgamma) receptor IIIa-158V/F polymorphism with inflammatory myopathies in Dutch patients. Tissue Antigens 2009, 73, 586–589. [Google Scholar] [CrossRef]
- Zhou, X.J.; Lv, J.C.; Bu, D.F.; Yu, L.; Yang, Y.R.; Zhao, J.; Cui, Z.; Yang, R.; Zhao, M.H.; Zhang, H. Copy number variation of FCGR3A rather than FCGR3B and FCGR2B is associated with susceptibility to anti-GBM disease. Int. Immunol. 2010, 22, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, S.; Wiener, H.; Shendre, A.; Kaslow, R.A.; Wu, J.; Olson, A.; Bowles, N.E.; Patel, H.; Edberg, J.C.; Portman, M.A. Role of activating FcgammaR gene polymorphisms in Kawasaki disease susceptibility and intravenous immunoglobulin response. Circulation. Cardiovasc. Genet. 2012, 5, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Hans, V.M.; Mehta, D.S. Genetic polymorphism of Fcgamma-receptors IIa, IIIa and IIIb in South Indian patients with generalized aggressive periodontitis. J. Oral Sci. 2011, 53, 467–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Sugita, N.; Kikuchi, A.; Iwanaga, R.; Hirano, E.; Shimada, Y.; Sasahara, J.; Tanaka, K.; Yoshie, H. FcgammaRIIB-nt645+25A/G gene polymorphism and periodontitis in Japanese women with preeclampsia. Int. J. Immunogenet. 2012, 39, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Omi, K.; Ohashi, J.; Patarapotikul, J.; Hananantachai, H.; Naka, I.; Looareesuwan, S.; Tokunaga, K. Fcgamma receptor IIA and IIIB polymorphisms are associated with susceptibility to cerebral malaria. Parasitol. Int. 2002, 51, 361–366. [Google Scholar] [CrossRef]
- Adu, B.; Dodoo, D.; Adukpo, S.; Hedley, P.L.; Arthur, F.K.; Gerds, T.A.; Larsen, S.O.; Christiansen, M.; Theisen, M. Fc gamma receptor IIIB (FcgammaRIIIB) polymorphisms are associated with clinical malaria in Ghanaian children. PLoS ONE 2012, 7, e46197. [Google Scholar] [CrossRef]
- Garcia, G.; Sierra, B.; Perez, A.B.; Aguirre, E.; Rosado, I.; Gonzalez, N.; Izquierdo, A.; Pupo, M.; Danay Diaz, D.R.; Sanchez, L.; et al. Asymptomatic dengue infection in a Cuban population confirms the protective role of the RR variant of the FcgammaRIIa polymorphism. Am. J. Trop. Med. Hyg. 2010, 82, 1153–1156. [Google Scholar] [CrossRef]
- Kangne, H.K.; Jijina, F.F.; Italia, Y.M.; Jain, D.L.; Nadkarni, A.H.; Gupta, M.; Pradhan, V.; Mukesh, R.D.; Ghosh, K.K.; Colah, R.B. The Fc receptor polymorphisms and expression of neutrophil activation markers in patients with sickle cell disease from Western India. BioMed Res. Int. 2013, 2013, 457656. [Google Scholar] [CrossRef]
- Abuqayyas, L.; Zhang, X.; Balthasar, J.P. Application of knockout mouse models to investigate the influence of FcgammaR on the pharmacokinetics and anti-platelet effects of MWReg30, a monoclonal anti-GPIIb antibody. Int. J. Pharm. 2013, 444, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Leabman, M.K.; Meng, Y.G.; Kelley, R.F.; DeForge, L.E.; Cowan, K.J.; Iyer, S. Effects of altered FcgammaR binding on antibody pharmacokinetics in cynomolgus monkeys. mAbs 2013, 5, 896–903. [Google Scholar] [CrossRef] [Green Version]
- Dall’Ozzo, S.; Tartas, S.; Paintaud, G.; Cartron, G.; Colombat, P.; Bardos, P.; Watier, H.; Thibault, G. Rituximab-dependent cytotoxicity by natural killer cells: Influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res. 2004, 64, 4664–4669. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.D.; Cho, S.-E.; Yang, C.-H.; Kim, J. Analysis of FcγRIII and IgG Fc Polymorphism Reveals Functional and Evolutionary Implications of Protein–Protein Interaction. J. Mol. Evol. 2001, 53, 1–9. [Google Scholar] [CrossRef]
- Wu, J.; Edberg, J.C.; Redecha, P.B.; Bansal, V.; Guyre, P.M.; Coleman, K.; Salmon, J.E.; Kimberly, R.P. A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J. Clin. Investig. 1997, 100, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Koene, H.R.; Kleijer, M.; Algra, J.; Roos, D.; von dem Borne, A.E.; de Haas, M. Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood 1997, 90, 1109–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartron, G.; Dacheux, L.; Salles, G.; Solal-Celigny, P.; Bardos, P.; Colombat, P.; Watier, H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002, 99, 754–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, W.K.; Levy, R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol. 2003, 21, 3940–3947. [Google Scholar] [CrossRef]
- Hatjiharissi, E.; Xu, L.; Santos, D.D.; Hunter, Z.R.; Ciccarelli, B.T.; Verselis, S.; Modica, M.; Cao, Y.; Manning, R.J.; Leleu, X.; et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the Fc{gamma}RIIIa-158 V/V and V/F polymorphism. Blood 2007, 110, 2561–2564. [Google Scholar] [CrossRef]
- Weng, W.K.; Weng, W.K.; Levy, R. Immunoglobulin G Fc receptor polymorphisms do not correlate with response to chemotherapy or clinical course in patients with follicular lymphoma. Leuk. Lymphoma 2009, 50, 1494–1500. [Google Scholar] [CrossRef] [Green Version]
- Ahlgrimm, M.; Pfreundschuh, M.; Kreuz, M.; Regitz, E.; Preuss, K.D.; Bittenbring, J. The impact of Fc-gamma receptor polymorphisms in elderly patients with diffuse large B-cell lymphoma treated with CHOP with or without rituximab. Blood 2011, 118, 4657–4662. [Google Scholar] [CrossRef]
- Paiva, M.; Marques, H.; Martins, A.; Ferreira, P.; Catarino, R.; Medeiros, R. FcgammaRIIa polymorphism and clinical response to rituximab in non-Hodgkin lymphoma patients. Cancer Genet. Cytogenet. 2008, 183, 35–40. [Google Scholar] [CrossRef]
- Congy-Jolivet, N.; Bolzec, A.; Ternant, D.; Ohresser, M.; Watier, H.; Thibault, G. Fc gamma RIIIa expression is not increased on natural killer cells expressing the Fc gamma RIIIa-158V allotype. Cancer Res. 2008, 68, 976–980. [Google Scholar] [CrossRef] [Green Version]
- Treon, S.P.; Hansen, M.; Branagan, A.R.; Verselis, S.; Emmanouilides, C.; Kimby, E.; Frankel, S.R.; Touroutoglou, N.; Turnbull, B.; Anderson, K.C.; et al. Polymorphisms in FcgammaRIIIA (CD16) receptor expression are associated with clinical response to rituximab in Waldenstrom’s macroglobulinemia. J. Clin. Oncol. 2005, 23, 474–481. [Google Scholar] [CrossRef] [Green Version]
- Robledo, G.; Marquez, A.; Davila-Fajardo, C.L.; Ortego-Centeno, N.; Rubio, J.L.; Garrido Ede, R.; Sanchez-Roman, J.; Garcia-Hernandez, F.J.; Rios-Fernandez, R.; Gonzalez-Escribano, M.F.; et al. Association of the FCGR3A-158F/V gene polymorphism with the response to rituximab treatment in Spanish systemic autoimmune disease patients. DNA Cell Biol. 2012, 31, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Farag, S.S.; Flinn, I.W.; Modali, R.; Lehman, T.A.; Young, D.; Byrd, J.C. Fc gamma RIIIa and Fc gamma RIIa polymorphisms do not predict response to rituximab in B-cell chronic lymphocytic leukemia. Blood 2004, 103, 1472–1474. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.; El Ghoul, Z.; Vermeire, S.; Dall’Ozzo, S.; Rutgeerts, P.; Paintaud, G.; Belaiche, J.; De Vos, M.; Van Gossum, A.; Colombel, J.F.; et al. Association between polymorphism in IgG Fc receptor IIIa coding gene and biological response to infliximab in Crohn’s disease. Aliment. Pharmacol. Ther. 2004, 19, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Chiba, T.; Sugai, T.; Habano, W. Association between tumor necrosis factor-alpha and Fc-gamma receptor polymorphisms with infliximab in Crohn’s disease. Hepatogastroenterology 2010, 57, 535–539. [Google Scholar]
- Ternant, D.; Berkane, Z.; Picon, L.; Gouilleux-Gruart, V.; Colombel, J.F.; Allez, M.; Louis, E.; Paintaud, G. Assessment of the Influence of Inflammation and FCGR3A Genotype on Infliximab Pharmacokinetics and Time to Relapse in Patients with Crohn’s Disease. Clin. Pharmacokinet. 2015, 54, 551–562. [Google Scholar] [CrossRef]
- Tout, M.; Gagez, A.L.; Lepretre, S.; Gouilleux-Gruart, V.; Azzopardi, N.; Delmer, A.; Mercier, M.; Ysebaert, L.; Laribi, K.; Gonzalez, H.; et al. Influence of FCGR3A-158V/F Genotype and Baseline CD20 Antigen Count on Target-Mediated Elimination of Rituximab in Patients with Chronic Lymphocytic Leukemia: A Study of FILO Group. Clin. Pharmacokinet. 2017, 56, 635–647. [Google Scholar] [CrossRef]
- Lopez-Albaitero, A.; Lee, S.C.; Morgan, S.; Grandis, J.R.; Gooding, W.E.; Ferrone, S.; Ferris, R.L. Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol. Immunother. 2009, 58, 1853–1864. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, J.; Zarate, R.; Bandres, E.; Boni, V.; Hernandez, A.; Sola, J.J.; Honorato, B.; Bitarte, N.; Garcia-Foncillas, J. Fc gamma receptor polymorphisms as predictive markers of Cetuximab efficacy in epidermal growth factor receptor downstream-mutated metastatic colorectal cancer. Eur. J. Cancer 2012, 48, 1774–1780. [Google Scholar] [CrossRef]
- Inoue, Y.; Hazama, S.; Iwamoto, S.; Miyake, Y.; Matsuda, C.; Tsunedomi, R.; Okayama, N.; Hinoda, Y.; Yamasaki, T.; Suehiro, Y.; et al. FcgammaR and EGFR polymorphisms as predictive markers of cetuximab efficacy in metastatic colorectal cancer. Mol. Diagn. Ther. 2014, 18, 541–548. [Google Scholar] [CrossRef]
- Negri, F.V.; Musolino, A.; Naldi, N.; Bortesi, B.; Missale, G.; Laccabue, D.; Zerbini, A.; Camisa, R.; Chernyschova, N.; Bisagni, G.; et al. Role of immunoglobulin G fragment C receptor polymorphism-mediated antibody-dependant cellular cytotoxicity in colorectal cancer treated with cetuximab therapy. Pharm. J. 2014, 14, 14–19. [Google Scholar] [CrossRef]
- Gashaw, I.; Ellinghaus, P.; Sommer, A.; Asadullah, K. What makes a good drug target? Drug Discov. Today 2012, 17, S24–S30. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.W.; Zeng, Q. Tapping the treasure of intracellular oncotargets with immunotherapy. FEBS Lett. 2014, 588, 350–355. [Google Scholar] [CrossRef] [Green Version]
- Mould, D.R.; Sweeney, K.R. The pharmacokinetics and pharmacodynamics of monoclonal antibodies—mechanistic modeling applied to drug development. Curr. Opin. Drug Discov. Dev. 2007, 10, 84–96. [Google Scholar]
- Mager, D.E. Target-mediated drug disposition and dynamics. Biochem. Pharmacol. 2006, 72, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mager, D.E.; Jusko, W.J. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J. Pharmacokinet. Pharmacodyn. 2001, 28, 507–532. [Google Scholar] [CrossRef] [PubMed]
- Mager, D.E.; Krzyzanski, W. Quasi-equilibrium pharmacokinetic model for drugs exhibiting target-mediated drug disposition. Pharm. Res. 2005, 22, 1589–1596. [Google Scholar] [CrossRef]
- Gibiansky, L.; Gibiansky, E.; Kakkar, T.; Ma, P. Approximations of the target-mediated drug disposition model and identifiability of model parameters. J. Pharmacokinet. Pharmacodyn. 2008, 35, 573–591. [Google Scholar] [CrossRef]
- Lammerts van Bueren, J.J.; Bleeker, W.K.; Bogh, H.O.; Houtkamp, M.; Schuurman, J.; van de Winkel, J.G.; Parren, P.W. Effect of target dynamics on pharmacokinetics of a novel therapeutic antibody against the epidermal growth factor receptor: Implications for the mechanisms of action. Cancer Res. 2006, 66, 7630–7638. [Google Scholar] [CrossRef] [Green Version]
- Fujimori, K.; Covell, D.G.; Fletcher, J.E.; Weinstein, J.N. A modeling analysis of monoclonal antibody percolation through tumors: A binding-site barrier. J. Nucl. Med. 1990, 31, 1191–1198. [Google Scholar]
- Ackerman, M.E.; Pawlowski, D.; Wittrup, K.D. Effect of antigen turnover rate and expression level on antibody penetration into tumor spheroids. Mol. Cancer Ther. 2008, 7, 2233–2240. [Google Scholar] [CrossRef] [Green Version]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Antibody tumor penetration: Transport opposed by systemic and antigen-mediated clearance. Adv. Drug Deliv. Rev. 2008, 60, 1421–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, T.; Miyasaka, N.; Tatsuki, Y.; Yano, T.; Yoshinari, T.; Abe, T.; Koike, T. Baseline tumour necrosis factor alpha levels predict the necessity for dose escalation of infliximab therapy in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Mummadi, S.R.; Hatipoglu, U.S.; Gupta, M.; Bossard, M.K.; Xu, M.; Lang, D. Clinically significant variability of serum IgE concentrations in patients with severe asthma. J. Asthma 2012, 49, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Scheinberg, D.A.; Straus, D.J.; Yeh, S.D.; Divgi, C.; Garin-Chesa, P.; Graham, M.; Pentlow, K.; Coit, D.; Oettgen, H.F.; Old, L.J. A phase I toxicity, pharmacology, and dosimetry trial of monoclonal antibody OKB7 in patients with non-Hodgkin’s lymphoma: Effects of tumor burden and antigen expression. J. Clin. Oncol. 1990, 8, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Koon, H.B.; Severy, P.; Hagg, D.S.; Butler, K.; Hill, T.; Jones, A.G.; Waldmann, T.A.; Junghans, R.P. Antileukemic effect of daclizumab in CD25 high-expressing leukemias and impact of tumor burden on antibody dosing. Leuk. Res. 2006, 30, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Jagannath, S.; Velasquez, W.S.; Tucker, S.L.; Fuller, L.M.; McLaughlin, P.W.; Manning, J.T.; North, L.B.; Cabanillas, F.C. Tumor burden assessment and its implication for a prognostic model in advanced diffuse large-cell lymphoma. J. Clin. Oncol. 1986, 4, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Dayde, D.; Ternant, D.; Ohresser, M.; Lerondel, S.; Pesnel, S.; Watier, H.; Le Pape, A.; Bardos, P.; Paintaud, G.; Cartron, G. Tumor burden influences exposure and response to rituximab: Pharmacokinetic-pharmacodynamic modeling using a syngeneic bioluminescent murine model expressing human CD20. Blood 2009, 113, 3765–3772. [Google Scholar] [CrossRef] [Green Version]
- Boross, P.; Jansen, J.H.; de Haij, S.; Beurskens, F.J.; van der Poel, C.E.; Bevaart, L.; Nederend, M.; Golay, J.; van de Winkel, J.G.; Parren, P.W.; et al. The in vivo mechanism of action of CD20 monoclonal antibodies depends on local tumor burden. Haematologica 2011, 96, 1822–1830. [Google Scholar] [CrossRef]
- Coiffier, B.; Losic, N.; Ronn, B.B.; Lepretre, S.; Pedersen, L.M.; Gadeberg, O.; Frederiksen, H.; van Oers, M.H.; Wooldridge, J.; Kloczko, J.; et al. Pharmacokinetics and pharmacokinetic/pharmacodynamic associations of ofatumumab, a human monoclonal CD20 antibody, in patients with relapsed or refractory chronic lymphocytic leukaemia: A phase 1–2 study. Br. J. Haematol. 2010, 150, 58–71. [Google Scholar] [CrossRef]
- Gibiansky, E.; Gibiansky, L.; Carlile, D.J.; Jamois, C.; Buchheit, V.; Frey, N. Population Pharmacokinetics of Obinutuzumab (GA101) in Chronic Lymphocytic Leukemia (CLL) and Non-Hodgkin’s Lymphoma and Exposure-Response in CLL. CPT Pharmacomet. Syst. Pharmacol. 2014, 3, e144. [Google Scholar] [CrossRef] [Green Version]
- Bernadou, G.; Campone, M.; Merlin, J.L.; Gouilleux-Gruart, V.; Bachelot, T.; Lokiec, F.; Rezai, K.; Arnedos, M.; Dieras, V.; Jimenez, M.; et al. Influence of tumour burden on trastuzumab pharmacokinetics in HER2 positive non-metastatic breast cancer. Br. J. Clin. Pharmacol. 2016, 81, 941–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prevodnik, V.K.; Lavrencak, J.; Horvat, M.; Novakovic, B.J. The predictive significance of CD20 expression in B-cell lymphomas. Diagn. Pathol. 2011, 6, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossmann, E.D.; Lundin, J.; Lenkei, R.; Mellstedt, H.; Osterborg, A. Variability in B-cell antigen expression: Implications for the treatment of B-cell lymphomas and leukemias with monoclonal antibodies. Hematol. J. 2001, 2, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shi, B.Y.; Qian, Y.Y.; Cai, M.; Wang, Q. Short-term anti-CD25 monoclonal antibody administration down-regulated CD25 expression without eliminating the neogenetic functional regulatory T cells in kidney transplantation. Clin. Exp. Immunol. 2009, 155, 496–503. [Google Scholar] [CrossRef]
- Slavin, R.G.; Ferioli, C.; Tannenbaum, S.J.; Martin, C.; Blogg, M.; Lowe, P.J. Asthma symptom re-emergence after omalizumab withdrawal correlates well with increasing IgE and decreasing pharmacokinetic concentrations. J. Allergy Clin. Immunol. 2009, 123, 107–113. [Google Scholar] [CrossRef]
- Burstein, H.J.; Chen, Y.H.; Parker, L.M.; Savoie, J.; Younger, J.; Kuter, I.; Ryan, P.D.; Garber, J.E.; Chen, H.; Campos, S.M.; et al. VEGF as a marker for outcome among advanced breast cancer patients receiving anti-VEGF therapy with bevacizumab and vinorelbine chemotherapy. Clin. Cancer Res. 2008, 14, 7871–7877. [Google Scholar] [CrossRef] [Green Version]
- Spano, J.P.; Lagorce, C.; Atlan, D.; Milano, G.; Domont, J.; Benamouzig, R.; Attar, A.; Benichou, J.; Martin, A.; Morere, J.F.; et al. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann. Oncol. 2005, 16, 102–108. [Google Scholar] [CrossRef]
- Carlsson, J.; Nordgren, H.; Sjöström, J.; Wester, K.; Villman, K.; Bengtsson, N.O.; Ostenstad, B.; Lundqvist, H.; Blomqvist, C. HER2 expression in breast cancer primary tumours and corresponding metastases. Original data and literature review. Br. J. Cancer 2004, 90, 2344–2348. [Google Scholar] [CrossRef]
- Law, S.K. Antigen shedding and metastasis of tumour cells. Clin. Exp. Immunol. 1991, 85, 1–2. [Google Scholar] [CrossRef]
- Hagan, P.L.; Halpern, S.E.; Chen, A.; Krishnan, L.; Frincke, J.; Bartholomew, R.M.; David, G.S.; Carlo, D. In vivo kinetics of radiolabeled monoclonal anti-CEA antibodies in animal models. J. Nucl. Med. 1985, 26, 1418–1423. [Google Scholar]
- Watanabe, Y.; Endo, K.; Koizumi, M.; Kawamura, Y.; Saga, T.; Sakahara, H.; Kuroki, M.; Matsuoka, Y.; Konishi, J. Effect of tumor mass and antigenic nature on the biodistribution of labeled monoclonal antibodies in mice. Cancer Res. 1989, 49, 2884–2889. [Google Scholar] [PubMed]
- Pimm, M.V.; Durrant, L.G.; Baldwin, R.W. Influence of circulating antigen on the biodistribution and tumour localization of radiolabelled monoclonal antibody in a human tumour: Nude mouse xenograft model. Eur. J. Cancer Clin. Oncol. 1989, 25, 1325–1332. [Google Scholar] [CrossRef]
- Zhang, Y.; Pastan, I. High shed antigen levels within tumors: An additional barrier to immunoconjugate therapy. Clin. Cancer Res. 2008, 14, 7981–7986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, Q.; Perkins, A.C.; Frier, M.; Watson, S.; Lalani, E.N.; Symonds, E.M. The effect of circulating antigen on the biodistribution of the engineered human antibody hCTM01 in a nude mice model. Eur. J. Nucl. Med. 1997, 24, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Haisma, H.J.; Battaile, A.; Stradtman, E.W.; Knapp, R.C.; Zurawski, V.R., Jr. Antibody-antigen complex formation following injection of OC125 monoclonal antibody in patients with ovarian cancer. Int. J. Cancer. 1987, 40, 758–762. [Google Scholar] [CrossRef] [PubMed]
- McQuarrie, S.A.; Baum, R.P.; Niesen, A.; Madiyalakan, R.; Korz, W.; Sykes, T.R.; Sykes, C.J.; Hor, G.; McEwan, A.J.; Noujaim, A.A. Pharmacokinetics and radiation dosimetry of 99Tcm-labelled monoclonal antibody B43.13 in ovarian cancer patients. Nucl. Med. Commun. 1997, 18, 878–886. [Google Scholar] [CrossRef]
- Pastuskovas, C.V.; Mallet, W.; Clark, S.; Kenrick, M.; Majidy, M.; Schweiger, M.; Van Hoy, M.; Tsai, S.P.; Bennett, G.; Shen, B.Q.; et al. Effect of immune complex formation on the distribution of a novel antibody to the ovarian tumor antigen CA125. Drug Metab. Dispos. 2010, 38, 2309–2319. [Google Scholar] [CrossRef] [Green Version]
- Warren, R.; Doyle, D. Turnover of the surface proteins and the receptor for serum asialoglycoproteins in primary cultures of rat hepatocytes. J. Biol. Chem. 1981, 256, 1346–1355. [Google Scholar]
- Gerbin, C.S. Regulation of ERBB Receptors. Nat. Educ. 2010, 3, 36. [Google Scholar]
- Greig, M.J.; Niessen, S.; Weinrich, S.L.; Feng, J.L.; Shi, M.; Johnson, T.O. Effects of Activating Mutations on EGFR Cellular Protein Turnover and Amino Acid Recycling Determined Using SILAC Mass Spectrometry. Int. J. Cell Biol. 2015, 2015, 798936. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Glenewinkel, H.; Reinz, E.; Eils, R.; Brady, N.R. Systems biological analysis of epidermal growth factor receptor internalization dynamics for altered receptor levels. J. Biol. Chem. 2009, 284, 17243–17252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorkin, A.; Duex, J.E. Quantitative analysis of endocytosis and turnover of epidermal growth factor (EGF) and EGF receptor. Curr. Protoc. Cell Biol. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, R.P.; Eketjall, S.; Aineskog, H.; Ibanez, C.F. Distinct turnover of alternatively spliced isoforms of the RET kinase receptor mediated by differential recruitment of the Cbl ubiquitin ligase. J. Biol. Chem. 2005, 280, 13442–13449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gisbertz, I.A.; Schouten, H.C.; Bot, F.J.; Arends, J.W. Proliferation and apoptosis in primary gastric B-cell non-Hodgkin’s lymphoma. Histopathology 1997, 30, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Defoiche, J.; Debacq, C.; Asquith, B.; Zhang, Y.; Burny, A.; Bron, D.; Lagneaux, L.; Macallan, D.; Willems, L. Reduction of B cell turnover in chronic lymphocytic leukaemia. Br. J. Haematol. 2008, 143, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Rituxan®(rituximab) [package insert]. Genentech, Inc.: San Francisco, CA, USA. Available online: http://www.gene.com/download/pdf/rituxan_prescribing.pdf (accessed on 2 May 2016).
- Drebin, J.A.; Link, V.C.; Stern, D.F.; Weinberg, R.A.; Greene, M.I. Down-modulation of an oncogene protein product and reversion of the transformed phenotype by monoclonal antibodies. Cell 1985, 41, 697–706. [Google Scholar] [CrossRef]
- Krippendorff, B.F.; Kuester, K.; Kloft, C.; Huisinga, W. Nonlinear pharmacokinetics of therapeutic proteins resulting from receptor mediated endocytosis. J. Pharmacokinet. Pharmacodyn. 2009, 36, 239–260. [Google Scholar] [CrossRef] [Green Version]
- Fargion, S.; Carney, D.; Mulshine, J.; Rosen, S.; Bunn, P.; Jewett, P.; Cuttitta, F.; Gazdar, A.; Minna, J. Heterogeneity of cell surface antigen expression of human small cell lung cancer detected by monoclonal antibodies. Cancer Res. 1986, 46, 2633–2638. [Google Scholar]
- Edwards, P.A. Heterogeneous expression of cell-surface antigens in normal epithelia and their tumours, revealed by monoclonal antibodies. Br. J. Cancer 1985, 51, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Gerlinger, M.; Swanton, C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br. J. Cancer 2010, 103, 1139–1143. [Google Scholar] [CrossRef] [Green Version]
- Hand, P.H.; Nuti, M.; Colcher, D.; Schlom, J. Definition of antigenic heterogeneity and modulation among human mammary carcinoma cell populations using monoclonal antibodies to tumor-associated antigens. Cancer Res. 1983, 43, 728–735. [Google Scholar] [PubMed]
- Boyer, C.M.; Borowitz, M.J.; McCarty, K.S., Jr.; Kinney, R.B.; Everitt, L.; Dawson, D.V.; Ring, D.; Bast, R.C., Jr. Heterogeneity of antigen expression in benign and malignant breast and ovarian epithelial cells. Int. J. Cancer. 1989, 43, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Berchuck, A.; Olt, G.J.; Soisson, A.P.; Kamel, A.; Soper, J.T.; Boyer, C.M.; Clarke-Pearson, D.L.; Leslie, D.S.; Bast, R.C., Jr. Heterogeneity of antigen expression in advanced epithelial ovarian cancer. Am. J. Obstet. Gynecol. 1990, 162, 883–888. [Google Scholar] [CrossRef]
- Gui, T.; Bai, H.; Zeng, J.; Zhong, Z.; Cao, D.; Cui, Q.; Chen, J.; Yang, J.; Shen, K. Tumor heterogeneity in the recurrence of epithelial ovarian cancer demonstrated by polycomb group proteins. OncoTargets Ther. 2014, 7, 1705–1716. [Google Scholar] [CrossRef] [Green Version]
- Bitting, R.L.; Healy, P.; Halabi, S.; George, D.J.; Goodin, M.; Armstrong, A.J. Clinical phenotypes associated with circulating tumor cell enumeration in metastatic castration-resistant prostate cancer. Urol. Oncol. 2015, 33, 110.e1–110.e9. [Google Scholar] [CrossRef]
- Mroz, E.A.; Tward, A.D.; Pickering, C.R.; Myers, J.N.; Ferris, R.L.; Rocco, J.W. High intratumor genetic heterogeneity is related to worse outcome in patients with head and neck squamous cell carcinoma. Cancer 2013, 119, 3034–3042. [Google Scholar] [CrossRef]
- Sigalotti, L.; Fratta, E.; Coral, S.; Tanzarella, S.; Danielli, R.; Colizzi, F.; Fonsatti, E.; Traversari, C.; Altomonte, M.; Maio, M. Intratumor heterogeneity of cancer/testis antigens expression in human cutaneous melanoma is methylation-regulated and functionally reverted by 5-aza-2’-deoxycytidine. Cancer Res. 2004, 64, 9167–9171. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Fan, M.; Chen, X.; Wang, S.; Alsharif, M.J.; Wang, L.; Liu, L.; Deng, H. Intratumor genomic heterogeneity correlates with histological grade of advanced oral squamous cell carcinoma. Oral Oncol. 2006, 42, 740–744. [Google Scholar] [CrossRef]
- Diaz-Cano, S.J.; Blanes, A.; Rubio, J.; Matilla, A.; Wolfe, H.J. Molecular evolution and intratumor heterogeneity by topographic compartments in muscle-invasive transitional cell carcinoma of the urinary bladder. Lab. Investig. 2000, 80, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.C.; Xu, C.; Mitchell, R.M.; Zhang, B.; Zhao, D.; Li, Y.; Huang, X.; Fan, W.; Wang, H.; Lerma, L.A.; et al. Tumor evolution and intratumor heterogeneity of an oropharyngeal squamous cell carcinoma revealed by whole-genome sequencing. Neoplasia 2013, 15, 1371–1378. [Google Scholar] [CrossRef] [Green Version]
- Jakobsen, J.N.; Sorensen, J.B. Intratumor heterogeneity and chemotherapy-induced changes in EGFR status in non-small cell lung cancer. Cancer Chemother. Pharmacol. 2012, 69, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-C.; Li, X.-P. Clinical significance of intratumor heterogeneity for gynecological carcinoma. Chronic Dis. Transl. Med. 2015, 1, 14–17. [Google Scholar] [CrossRef] [Green Version]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Hammond, M.E.; Schwartz, J.N.; Hagerty, K.L.; Allred, D.C.; Cote, R.J.; Dowsett, M.; Fitzgibbons, P.L.; Hanna, W.M.; Langer, A.; et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J. Clin. Oncol. 2007, 25, 118–145. [Google Scholar] [CrossRef] [Green Version]
- Gajria, D.; Chandarlapaty, S. HER2-amplified breast cancer: Mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev. Anticancer Ther. 2011, 11, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Fessler, S.P.; Wotkowicz, M.T.; Mahanta, S.K.; Bamdad, C. MUC1* is a determinant of trastuzumab (Herceptin) resistance in breast cancer cells. Breast Cancer Res. Treat. 2009, 118, 113–124. [Google Scholar] [CrossRef]
- Chen, A.C.; Migliaccio, I.; Rimawi, M.; Lopez-Tarruella, S.; Creighton, C.J.; Massarweh, S.; Huang, C.; Wang, Y.C.; Batra, S.K.; Gutierrez, M.C.; et al. Upregulation of mucin4 in ER-positive/HER2-overexpressing breast cancer xenografts with acquired resistance to endocrine and HER2-targeted therapies. Breast Cancer Res. Treat. 2012, 134, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Ritter, C.A.; Perez-Torres, M.; Rinehart, C.; Guix, M.; Dugger, T.; Engelman, J.A.; Arteaga, C.L. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin. Cancer Res. 2007, 13, 4909–4919. [Google Scholar] [CrossRef] [Green Version]
- Ebbing, E.A.; Medema, J.P.; Damhofer, H.; Meijer, S.L.; Krishnadath, K.K.; van Berge Henegouwen, M.I.; Bijlsma, M.F.; van Laarhoven, H.W. ADAM10-mediated release of heregulin confers resistance to trastuzumab by activating HER3. Oncotarget 2016, 7, 10243–10254. [Google Scholar] [CrossRef] [PubMed]
- Sanabria-Figueroa, E.; Donnelly, S.M.; Foy, K.C.; Buss, M.C.; Castellino, R.C.; Paplomata, E.; Taliaferro-Smith, L.; Kaumaya, P.T.; Nahta, R. Insulin-like growth factor-1 receptor signaling increases the invasive potential of human epidermal growth factor receptor 2-overexpressing breast cancer cells via Src-focal adhesion kinase and forkhead box protein M1. Mol. Pharmacol. 2015, 87, 150–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shattuck, D.L.; Miller, J.K.; Carraway, K.L., 3rd; Sweeney, C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 2008, 68, 1471–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, E.; Vermeire, S.; Rutgeerts, P.; De Vos, M.; Van Gossum, A.; Pescatore, P.; Fiasse, R.; Pelckmans, P.; Reynaert, H.; D’Haens, G.; et al. A positive response to infliximab in Crohn disease: Association with a higher systemic inflammation before treatment but not with -308 TNF gene polymorphism. Scand. J. Gastroenterol. 2002, 37, 818–824. [Google Scholar] [CrossRef]
- Hlavaty, T.; Pierik, M.; Henckaerts, L.; Ferrante, M.; Joossens, S.; van Schuerbeek, N.; Noman, M.; Rutgeerts, P.; Vermeire, S. Polymorphisms in apoptosis genes predict response to infliximab therapy in luminal and fistulizing Crohn’s disease. Aliment. Pharmacol. Ther. 2005, 22, 613–626. [Google Scholar] [CrossRef]
- Cantor, M.J.; Nickerson, P.; Bernstein, C.N. The role of cytokine gene polymorphisms in determining disease susceptibility and phenotype in inflammatory bowel disease. Am. J. Gastroenterol. 2005, 100, 1134–1142. [Google Scholar] [CrossRef]
- Koss, K.; Satsangi, J.; Fanning, G.C.; Welsh, K.I.; Jewell, D.P. Cytokine (TNF alpha, LT alpha and IL-10) polymorphisms in inflammatory bowel diseases and normal controls: Differential effects on production and allele frequencies. Genes Immun. 2000, 1, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Medrano, L.M.; Taxonera, C.; Marquez, A.; Barreiro-de Acosta, M.; Gomez-Garcia, M.; Gonzalez-Artacho, C.; Perez-Calle, J.L.; Bermejo, F.; Lopez-Sanroman, A.; Martin Arranz, M.D.; et al. Role of TNFRSF1B polymorphisms in the response of Crohn’s disease patients to infliximab. Hum. Immunol. 2014, 75, 71–75. [Google Scholar] [CrossRef]
- Enevold, C.; Baslund, B.; Linde, L.; Josephsen, N.L.; Tarp, U.; Lindegaard, H.; Jacobsen, S.; Nielsen, C.H. Interleukin-6-receptor polymorphisms rs12083537, rs2228145, and rs4329505 as predictors of response to tocilizumab in rheumatoid arthritis. Pharm. Genom. 2014, 24, 401–405. [Google Scholar] [CrossRef]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. mAbs 2010, 2, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Schellekens, H. Factors influencing the immunogenicity of therapeutic proteins. Nephrol. Dial. Transplant. 2005, 20 (Suppl. 6), vi3–vi9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chirmule, N.; Jawa, V.; Meibohm, B. Immunogenicity to therapeutic proteins: Impact on PK/PD and efficacy. AAPS J. 2012, 14, 296–302. [Google Scholar] [CrossRef] [PubMed]
- U.S. Department of Health and Human Services Food and Drug Administration. Center for Drug Evaluation and Research. Immunogenicity Assessment for Therapeutic Protein Products. Rockville, MD. August 2014. Available online: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm338856.pdf (accessed on 2 December 2019).
- Kessler, M.; Goldsmith, D.; Schellekens, H. Immunogenicity of biopharmaceuticals. Nephrol. Dial. Transplant. 2006, 21 (Suppl. 5), v9–v12. [Google Scholar] [CrossRef] [PubMed]
- De Vries, M.K.; Wolbink, G.J.; Stapel, S.O.; de Groot, E.R.; Dijkmans, B.A.C.; Aarden, L.A.; van der Horst-Bruinsma, I.E. Inefficacy of infliximab in ankylosing spondylitis is correlated with antibody formation. Ann. Rheum. Dis. 2007, 66, 133–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Maas, A.; van den Bemt, B.J.; Wolbink, G.; van den Hoogen, F.H.; van Riel, P.L.; den Broeder, A.A. Low infliximab serum trough levels and anti-infliximab antibodies are prevalent in rheumatoid arthritis patients treated with infliximab in daily clinical practice: Results of an observational cohort study. BMC Musculoskelet. Disord. 2012, 13, 184. [Google Scholar] [CrossRef] [Green Version]
- Triguero, D.; Buciak, J.B.; Yang, J.; Pardridge, W.M. Blood-brain barrier transport of cationized immunoglobulin G: Enhanced delivery compared to native protein. Proc. Natl. Acad. Sci. USA 1989, 86, 4761–4765. [Google Scholar] [CrossRef] [Green Version]
- Ternant, D.; Aubourg, A.; Magdelaine-Beuzelin, C.; Degenne, D.; Watier, H.; Picon, L.; Paintaud, G. Infliximab pharmacokinetics in inflammatory bowel disease patients. Ther. Drug Monit. 2008, 30, 523–529. [Google Scholar] [CrossRef]
- Xu, Z.H.; Lee, H.; Vu, T.; Hu, C.; Yan, H.; Baker, D.; Hsu, B.; Pendley, C.; Wagner, C.; Davis, H.M.; et al. Population pharmacokinetics of golimumab in patients with ankylosing spondylitis: Impact of body weight and immunogenicity. Int. J. Clin. Pharmacol. Ther. 2010, 48, 596–607. [Google Scholar] [CrossRef]
- Xu, Z.; Vu, T.; Lee, H.; Hu, C.; Ling, J.; Yan, H.; Baker, D.; Beutler, A.; Pendley, C.; Wagner, C.; et al. Population pharmacokinetics of golimumab, an anti-tumor necrosis factor-alpha human monoclonal antibody, in patients with psoriatic arthritis. J. Clin. Pharmacol. 2009, 49, 1056–1070. [Google Scholar] [CrossRef]
- Zhu, Y.W.; Mendelsohn, A.; Pendley, C.; Davis, H.M.; Zhou, H. Population pharmacokinetics of ustekinumab in patients with active psoriatic arthritis. Int. J. Clin. Pharmacol. Ther. 2010, 48, 830–846. [Google Scholar] [CrossRef]
- Zhu, Y.; Hu, C.; Lu, M.; Liao, S.; Marini, J.C.; Yohrling, J.; Yeilding, N.; Davis, H.M.; Zhou, H. Population pharmacokinetic modeling of ustekinumab, a human monoclonal antibody targeting IL-12/23p40, in patients with moderate to severe plaque psoriasis. J. Clin. Pharmacol. 2009, 49, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Bihorel, S.; Fiedler-Kelly, J.; Ludwig, E.; Sloan-Lancaster, J.; Raddad, E. Population Pharmacokinetic Modeling of LY2189102 after Multiple Intravenous and Subcutaneous Administrations. AAPS J. 2014, 16, 1009–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, L.; Hang, Y.; Othman, A.A.; Nestorov, I.; Tran, J.Q. Population Pharmacokinetics of Daclizumab High-Yield Process in Healthy Volunteers and Subjects with Multiple Sclerosis: Analysis of Phase I-III Clinical Trials. Clin. Pharmacokinet. 2016, 55, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Hussein, Z.; Hassan, R.; Wustner, J.; Maltzman, J.D.; Wallin, B.A. Population pharmacokinetics and exposure-response relationship of amatuximab, an anti-mesothelin monoclonal antibody, in patients with malignant pleural mesothelioma and its application in dose selection. Cancer Chemother. Pharmacol. 2016, 77, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Stroh, M.; Winter, H.; Marchand, M.; Claret, L.; Eppler, S.; Ruppel, J.; Abidoye, O.; Teng, S.L.; Lin, W.T.; Dayog, S.; et al. Clinical Pharmacokinetics and Pharmacodynamics of Atezolizumab in Metastatic Urothelial Carcinoma. Clin. Pharmacol. Ther. 2017, 102, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yan, L.; Yao, Z.; Roskos, L.K. Population Pharmacokinetics and Pharmacodynamics of Benralizumab in Healthy Volunteers and Patients with Asthma. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Brinks, V.; Jiskoot, W.; Schellekens, H. Immunogenicity of therapeutic proteins: The use of animal models. Pharm. Res. 2011, 28, 2379–2385. [Google Scholar] [CrossRef] [Green Version]
- Van Meer, P.J.; Kooijman, M.; Brinks, V.; Gispen-de Wied, C.C.; Silva-Lima, B.; Moors, E.H.; Schellekens, H. Immunogenicity of mAbs in non-human primates during nonclinical safety assessment. mAbs 2013, 5, 810–816. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Hickling, T.; Kraynov, E.; Kuang, B.; Parng, C.; Vicini, P. A mathematical model of the effect of immunogenicity on therapeutic protein pharmacokinetics. AAPS J. 2013, 15, 1141–1154. [Google Scholar] [CrossRef] [Green Version]
- Kathman, S., Jr.; Thway, T.M.; Zhou, L.; Lee, S.; Yu, S.; Ma, M.; Chirmule, N.; Jawa, V. Utility of a Bayesian Mathematical Model to Predict the Impact of Immunogenicity on Pharmacokinetics of Therapeutic Proteins. AAPS J. 2016, 18, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Maini, R.N.; Breedveld, F.C.; Kalden, J.R.; Smolen, J.S.; Davis, D.; Macfarlane, J.D.; Antoni, C.; Leeb, B.; Elliott, M.J.; Woody, J.N.; et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheum. 1998, 41, 1552–1563. [Google Scholar] [CrossRef]
- Kovarik, J.M.; Pescovitz, M.D.; Sollinger, H.W.; Kaplan, B.; Legendre, C.; Salmela, K.; Book, B.K.; Gerbeau, C.; Girault, D.; Somberg, K. Differential influence of azathioprine and mycophenolate mofetil on the disposition of basiliximab in renal transplant patients. Clin. Transplant. 2001, 15, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Pescovitz, M.D.; Bumgardner, G.; Gaston, R.S.; Kirkman, R.L.; Light, S.; Patel, I.H.; Nieforth, K.; Vincenti, F. Pharmacokinetics of daclizumab and mycophenolate mofetil with cyclosporine and steroids in renal transplantation. Clin. Transplant. 2003, 17, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, A.; Punzi, L.; Marchesoni, A.; Lubrano, E.; Mathieu, A.; Cantini, F.; Olivieri, I.; Salvarani, C.; Scarpa, R.; Scrivo, R.; et al. Switching from infliximab or etanercept to adalimumab in resistant or intolerant patients with spondyloarthritis: A 4-year study. Rheumatology 2010, 49, 1107–1111. [Google Scholar] [CrossRef] [Green Version]
- Reich, K.; Nestle, F.O.; Papp, K.; Ortonne, J.-P.; Evans, R.; Guzzo, C.; Li, S.; Dooley, L.T.; Griffiths, C.E.M. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: A phase III, multicentre, double-blind trial. Lancet 2005, 366, 1367–1374. [Google Scholar] [CrossRef]
- Baert, F.; Noman, M.; Vermeire, S.; Van Assche, G.; D’Haens, G.; Carbonez, A.; Rutgeerts, P. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn’s disease. N. Engl. J. Med. 2003, 348, 601–608. [Google Scholar] [CrossRef]
- Bartelds, G.M.; Krieckaert, C.L.; Nurmohamed, M.T.; van Schouwenburg, P.A.; Lems, W.F.; Twisk, J.W.; Dijkmans, B.A.; Aarden, L.; Wolbink, G.J. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA 2011, 305, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Menting, S.P.; van Lumig, P.P.; de Vries, A.C.; van den Reek, J.M.; van der Kleij, D.; de Jong, E.M.; Spuls, P.I.; Lecluse, L.L. Extent and consequences of antibody formation against adalimumab in patients with psoriasis: One-year follow-up. JAMA Dermatol. 2014, 150, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Ternant, D.; Karmiris, K.; Vermeire, S.; Desvignes, C.; Azzopardi, N.; Bejan-Angoulvant, T.; van Assche, G.; Paintaud, G. Pharmacokinetics of adalimumab in Crohn’s disease. Eur. J. Clin. Pharmacol. 2015, 71, 1155–1157. [Google Scholar] [CrossRef]
- Berends, S.E.; Strik, A.S.; Van Selm, J.C.; Lowenberg, M.; Ponsioen, C.Y.; D’Haens, G.R.; Mathot, R.A. Explaining Interpatient Variability in Adalimumab Pharmacokinetics in Patients with Crohn’s Disease. Ther. Drug Monit. 2018, 40, 202–211. [Google Scholar] [CrossRef]
- Tysabri: Package insert. Food and Drug Administration: Silver springs, MD, USA. 2012. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/125104s0576lbl.pdf (accessed on 2 December 2019).
- Wang, D.D.; Zhang, S.; Zhao, H.; Men, A.Y.; Parivar, K. Fixed dosing versus body size-based dosing of monoclonal antibodies in adult clinical trials. J. Clin. Pharmacol. 2009, 49, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Jorga, K.; Xin, Y.; Jin, D.; Zheng, Y.; Damico-Beyer, L.A.; Gupta, M.; Tang, M.; Allison, D.E.; Lu, D.; et al. A guide to rational dosing of monoclonal antibodies. Clin. Pharmacokinet. 2012, 51, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Yang, B.B.; Wang, Y.M.; Peterson, M.; Narayanan, A.; Sutjandra, L.; Rodriguez, R.; Chow, A. Population pharmacokinetic analysis of panitumumab in patients with advanced solid tumors. J. Clin. Pharmacol. 2009, 49, 1142–1156. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Shi, R.; Li, C.; Parivar, K.; Wang, D.D. Fixed dosing versus body size-based dosing of therapeutic peptides and proteins in adults. J. Clin. Pharmacol. 2012, 52, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Weitman, E.S.; Aschen, S.Z.; Farias-Eisner, G.; Albano, N.; Cuzzone, D.A.; Ghanta, S.; Zampell, J.C.; Thorek, D.; Mehrara, B.J. Obesity Impairs Lymphatic Fluid Transport and Dendritic Cell Migration to Lymph Nodes. PLoS ONE 2013, 8, e70703. [Google Scholar] [CrossRef]
- Jeevanandam, M.; Horowitz, G.D.; Lowry, S.F.; Brennan, M.F. Cancer cachexia and protein metabolism. Lancet 1984, 1, 1423–1426. [Google Scholar] [CrossRef]
- Lebwohl, M.; Yeilding, N.; Szapary, P.; Wang, Y.; Li, S.; Zhu, Y.; Reich, K.; Langley, R.G.; Papp, K.A. Impact of weight on the efficacy and safety of ustekinumab in patients with moderate to severe psoriasis: Rationale for dosing recommendations. J. Am. Acad. Dermatol. 2010, 63, 571–579. [Google Scholar] [CrossRef]
- Stelara®(ustekinumab) [package insert]. Janssen Biotech, Inc.: Horsham, PA, USA. Available online: http://www.stelarainfo.com/pdf/PrescribingInformation.pdf#zoom=100 (accessed on 2 December 2019).
- Narwal, R.; Roskos, L.K.; Robbie, G.J. Population pharmacokinetics of sifalimumab, an investigational anti-interferon-alpha monoclonal antibody, in systemic lupus erythematosus. Clin. Pharmacokinet. 2013, 52, 1017–1027. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.; Quartino, A.; Li, J.; Jin, J.; Wada, D.R.; Li, H.; Cortes, J.; McNally, V.; Ross, G.; Visich, J.; et al. Population pharmacokinetic and covariate analysis of pertuzumab, a HER2-targeted monoclonal antibody, and evaluation of a fixed, non-weight-based dose in patients with a variety of solid tumors. Cancer Chemother. Pharmacol. 2014, 74, 819–829. [Google Scholar] [CrossRef]
- Lowe, P.J.; Georgiou, P.; Canvin, J. Revision of omalizumab dosing table for dosing every 4 instead of 2 weeks for specific ranges of bodyweight and baseline IgE. Regul. Toxicol. Pharmacol. 2015, 71, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Kornmann, O.; Watz, H.; Fuhr, R.; Krug, N.; Erpenbeck, V.J.; Kaiser, G. Omalizumab in patients with allergic (IgE-mediated) asthma and IgE/bodyweight combinations above those in the initially approved dosing table. Pulm. Pharmacol. Ther. 2014, 28, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Fadiran, E.O.; Zhang, L. Effects of Sex Differences in the Pharmacokinetics of Drugs and Their Impact on the Safety of Medicines in Women. In Medicines for Women; Harrison-Woolrych, M., Ed.; Springer: Cham, Switzerland, 2015; pp. 41–68. [Google Scholar] [CrossRef]
- Friedman, D.; Netti, F.; Schreiber, A.D. Effect of estradiol and steroid analogues on the clearance of immunoglobulin G-coated erythrocytes. J. Clin. Investig. 1985, 75, 162–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, A.D.; Nettl, F.M.; Sanders, M.C.; King, M.; Szabolcs, P.; Friedman, D.; Gomez, F. Effect of endogenous and synthetic sex steroids on the clearance of antibody-coated cells. J. Immunol. 1988, 141, 2959–2966. [Google Scholar] [PubMed]
- Gomez, F.; Ruiz, P.; Briceno, F.; Lopez, R.; Michan, A. Treatment with progesterone analogues decreases macrophage Fcgamma receptors expression. Clin. Immunol. Immunopathol. 1998, 89, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Adamski, F.M.; King, A.T.; Demmer, J. Expression of the Fc receptor in the mammary gland during lactation in the marsupial Trichosurus vulpecula (brushtail possum). Mol. Immunol. 2000, 37, 435–444. [Google Scholar] [CrossRef]
- Martin, M.G.; Wu, S.V.; Walsh, J.H. Hormonal control of intestinal Fc receptor gene expression and immunoglobulin transport in suckling rats. J. Clin. Investig. 1993, 91, 2844–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderpump, M.P.J. The epidemiology of thyroid disease. Br. Med Bull. 2011, 99, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Seitz, K.; Fasanmade, A.; Ford, J.; Williamson, P.; Xu, W.; Davis, H.M.; Zhou, H. Population pharmacokinetics of infliximab in patients with ankylosing spondylitis. J. Clin. Pharmacol. 2008, 48, 681–695. [Google Scholar] [CrossRef]
- Fasanmade, A.A.; Adedokun, O.J.; Ford, J.; Hernandez, D.; Johanns, J.; Hu, C.; Davis, H.M.; Zhou, H. Population pharmacokinetic analysis of infliximab in patients with ulcerative colitis. Eur. J. Clin. Pharmacol. 2009, 65, 1211–1228. [Google Scholar] [CrossRef] [Green Version]
- Buurman, D.J.; Maurer, J.M.; Keizer, R.J.; Kosterink, J.G.; Dijkstra, G. Population pharmacokinetics of infliximab in patients with inflammatory bowel disease: Potential implications for dosing in clinical practice. Aliment. Pharmacol. Ther. 2015, 42, 529–539. [Google Scholar] [CrossRef]
- Frey, N.; Grange, S.; Woodworth, T. Population pharmacokinetic analysis of tocilizumab in patients with rheumatoid arthritis. J. Clin. Pharmacol. 2010, 50, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Murawski, N.; Wiesen, M.H.; Held, G.; Poeschel, V.; Zeynalova, S.; Wenger, M.; Nickenig, C.; Peter, N.; Lengfelder, E.; et al. The role of sex and weight on rituximab clearance and serum elimination half-life in elderly patients with DLBCL. Blood 2012, 119, 3276–3284. [Google Scholar] [CrossRef] [PubMed]
- Gisselbrecht, C.; Schmitz, N.; Mounier, N.; Singh Gill, D.; Linch, D.C.; Trneny, M.; Bosly, A.; Milpied, N.J.; Radford, J.; Ketterer, N.; et al. Rituximab maintenance therapy after autologous stem-cell transplantation in patients with relapsed CD20(+) diffuse large B-cell lymphoma: Final analysis of the collaborative trial in relapsed aggressive lymphoma. J. Clin. Oncol. 2012, 30, 4462–4469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carella, A.M.; de Souza, C.A.; Luminari, S.; Marcheselli, L.; Chiappella, A.; di Rocco, A.; Cesaretti, M.; Rossi, A.; Rigacci, L.; Gaidano, G.; et al. Prognostic role of gender in diffuse large B-cell lymphoma treated with rituximab containing regimens: A Fondazione Italiana Linfomi/Grupo de Estudos em Molestias Onco-Hematologicas retrospective study. Leuk. Lymphoma 2013, 54, 53–57. [Google Scholar] [CrossRef]
- Sun, Y.N.; Lu, J.F.; Joshi, A.; Compton, P.; Kwon, P.; Bruno, R.A. Population pharmacokinetics of efalizumab (humanized monoclonal anti-CD11a antibody) following long-term subcutaneous weekly dosing in psoriasis subjects. J. Clin. Pharmacol. 2005, 45, 468–476. [Google Scholar] [CrossRef]
- Othman, A.A.; Tran, J.Q.; Tang, M.T.; Dutta, S. Population pharmacokinetics of daclizumab high-yield process in healthy volunteers: Integrated analysis of intravenous and subcutaneous, single-and multiple-dose administration. Clin. Pharmacokinet. 2014, 53, 907–918. [Google Scholar] [CrossRef]
- Hayashi, N.; Tsukamoto, Y.; Sallas, W.M.; Lowe, P.J. A mechanism-based binding model for the population pharmacokinetics and pharmacodynamics of omalizumab. Br. J. Clin. Pharmacol. 2007, 63, 548–561. [Google Scholar] [CrossRef] [Green Version]
- Honma, W.; Gautier, A.; Paule, I.; Yamaguchi, M.; Lowe, P.J. Ethnic sensitivity assessment of pharmacokinetics and pharmacodynamics of omalizumab with dosing table expansion. Drug Metab. Pharmacokinet. 2016, 31, 173–184. [Google Scholar] [CrossRef]
- Hicks, C.; Miele, L.; Koganti, T.; Young-Gaylor, L.; Rogers, D.; Vijayakumar, V.; Megason, G. Analysis of Patterns of Gene Expression Variation within and between Ethnic Populations in Pediatric B-ALL. Cancer Inform. 2013, 12, 155–173. [Google Scholar] [CrossRef] [Green Version]
- Moul, J.W.; Connelly, R.R.; Mooneyhan, R.M.; Zhang, W.E.I.; Sesterhenn, I.A.; Mostofi, F.K.; McLeod, D.G. Racial differences in tumor volume and prostrate specific antigen among radical prostatectomy patients. J. Urol. 1999, 162, 394–397. [Google Scholar] [CrossRef]
- Zhou, H.; Tsukamoto, Y.; Davis, H.M. Should Clinical Pharmacokinetic Bridging Studies Between Caucasian and Asian Populations Be Required for Approval of Monoclonal Antibodies? J. Clin. Pharmacol. 2012, 52, 1273–1276. [Google Scholar] [CrossRef]
- Ince, I.; de Wildt, S.N.; Tibboel, D.; Danhof, M.; Knibbe, C.A. Tailor-made drug treatment for children: Creation of an infrastructure for data-sharing and population PK-PD modeling. Drug Discov. Today 2009, 14, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, A.L.; Yu, A.L.; Reaman, G.; Ingle, A.M.; Secola, R.; Adamson, P.C. A phase II study of Campath-1H in children with relapsed or refractory acute lymphoblastic leukemia: A Children’s Oncology Group report. Pediatr. Blood Cancer 2009, 53, 978–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovarik, J.M.; Offner, G.; Broyer, M.; Niaudet, P.; Loirat, C.; Mentser, M.; Lemire, J.; Crocker, J.F.; Cochat, P.; Clark, G.; et al. A rational dosing algorithm for basiliximab (Simulect) in pediatric renal transplantation based on pharmacokinetic-dynamic evaluations. Transplantation 2002, 74, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Buckwalter, M.; Dowell, J.A.; Korth-Bradley, J.; Gorovits, B.; Mayer, P.R. Pharmacokinetics of gemtuzumab ozogamicin as a single-agent treatment of pediatric patients with refractory or relapsed acute myeloid leukemia. J. Clin. Pharmacol. 2004, 44, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Bender, J.L.G.; Adamson, P.C.; Reid, J.M.; Xu, L.; Baruchel, S.; Shaked, Y.; Kerbel, R.S.; Cooney-Qualter, E.M.; Stempak, D.; Chen, H.X.; et al. Phase I Trial and Pharmacokinetic Study of Bevacizumab in Pediatric Patients With Refractory Solid Tumors: A Children’s Oncology Group Study. J. Clin. Oncol. 2008, 26, 399–405. [Google Scholar] [CrossRef]
- Burns, J.C.; Best, B.M.; Mejias, A.; Mahony, L.; Fixler, D.E.; Jafri, H.S.; Melish, M.E.; Jackson, M.A.; Asmar, B.I.; Lang, D.J.; et al. Infliximab treatment of intravenous immunoglobulin-resistant Kawasaki disease. J. Pediatr. 2008, 153, 833–838. [Google Scholar] [CrossRef] [Green Version]
- Trippett, T.M.; Herzog, C.; Whitlock, J.A.; Wolff, J.; Kuttesch, J.; Bagatell, R.; Hunger, S.P.; Boklan, J.; Smith, A.A.; Arceci, R.J.; et al. Phase I and pharmacokinetic study of cetuximab and irinotecan in children with refractory solid tumors: A study of the pediatric oncology experimental therapeutic investigators’ consortium. J. Clin. Oncol. 2009, 27, 5102–5108. [Google Scholar] [CrossRef] [Green Version]
- Lowe, P.J.; Tannenbaum, S.; Gautier, A.; Jimenez, P. Relationship between omalizumab pharmacokinetics, IgE pharmacodynamics and symptoms in patients with severe persistent allergic (IgE-mediated) asthma. Br. J. Clin. Pharmacol. 2009, 68, 61–76. [Google Scholar] [CrossRef] [Green Version]
- McMillan, D.C.; Watson, W.S.; O’Gorman, P.; Preston, T.; Scott, H.R.; McArdle, C.S. Albumin concentrations are primarily determined by the body cell mass and the systemic inflammatory response in cancer patients with weight loss. Nutr. Cancer 2001, 39, 210–213. [Google Scholar] [CrossRef] [Green Version]
- Chaudhury, C.; Brooks, C.L.; Carter, D.C.; Robinson, J.M.; Anderson, C.L. Albumin binding to FcRn: Distinct from the FcRn-IgG interaction. Biochemistry 2006, 45, 4983–4990. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.L.; Chaudhury, C.; Kim, J.; Bronson, C.L.; Wani, M.A.; Mohanty, S. Perspective—FcRn transports albumin: Relevance to immunology and medicine. Trends Immunol. 2006, 27, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Fasanmade, A.A.; Adedokun, O.J.; Blank, M.; Zhou, H.; Davis, H.M. Pharmacokinetic properties of infliximab in children and adults with Crohn’s disease: A retrospective analysis of data from 2 phase III clinical trials. Clin. Ther. 2011, 33, 946–964. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.M.; Lum, B.L.; Gimenez, V.; Kelsey, S.; Allison, D. Rationale for fixed dosing of pertuzumab in cancer patients based on population pharmacokinetic analysis. Pharm. Res. 2006, 23, 1275–1284. [Google Scholar] [CrossRef]
- Bruno, R.; Washington, C.B.; Lu, J.F.; Lieberman, G.; Banken, L.; Klein, P. Population pharmacokinetics of trastuzumab in patients with HER2+ metastatic breast cancer. Cancer Chemother. Pharmacol. 2005, 56, 361–369. [Google Scholar] [CrossRef]
- Fasanmade, A.A.; Adedokun, O.J.; Olson, A.; Strauss, R.; Davis, H.M. Serum albumin concentration: A predictive factor of infliximab pharmacokinetics and clinical response in patients with ulcerative colitis. Int. J. Clin. Pharmacol. Ther. 2010, 48, 297–308. [Google Scholar] [CrossRef]
- Engler, F.A.; Zheng, B.; Balthasar, J.P. Investigation of the influence of nephropathy on monoclonal antibody disposition: A pharmacokinetic study in a mouse model of diabetic nephropathy. Pharm. Res. 2014, 31, 1185–1193. [Google Scholar] [CrossRef]
- Rustom, R.; Grime, J.S.; Costigan, M.; Maltby, P.; Hughes, A.; Shenkin, A.; Critchley, M.; Bone, J.M. Proximal renal tubular peptide catabolism, ammonia excretion and tubular injury in patients with proteinuria: Before and after lisinopril. Clin. Sci. 1998, 94, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Chen, J. Diabetic Nephropathy: Scope of the Problem. In Diabetes and Kidney Disease; Lerma, V.E., Batuman, V., Eds.; Springer: New York, NY, USA, 2014; pp. 9–14. [Google Scholar] [CrossRef]
- Lemley, K.V.; Blouch, K.; Abdullah, I.; Boothroyd, D.B.; Bennett, P.H.; Myers, B.D.; Nelson, R.G. Glomerular permselectivity at the onset of nephropathy in type 2 diabetes mellitus. J. Am. Soc. Nephrol. 2000, 11, 2095–2105. [Google Scholar]
- Mohan, S.; Kalia, K.; Mannari, J. Association between urinary IgG and relative risk for factors affecting proteinuria in type 2 diabetic patients. Indian J. Clin. Biochem. IJCB 2012, 27, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Chadha, G.S.; Morris, M.E. Effect of Type 2 Diabetes Mellitus and Diabetic Nephropathy on IgG Pharmacokinetics and Subcutaneous Bioavailability in the Rat. AAPS J. 2015, 17, 965–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, J.; Barnett, E.V.; MacDonald, N.S.; Klinenberg, J.R. Altered immunoglobulin metabolism in systemic lupus erythematosus and heumatoid arthritis. J. Clin. Investig. 1970, 49, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Wochner, R.D. Hypercatabolism of normal IgG; an unexplained immunoglobulin abnormality in the connective tissue diseases. J. Clin. Investig. 1970, 49, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Pop, L.M.; Ghetie, V. Hypercatabolism of IgG in mice with lupus-like syndrome. Lupus 2005, 14, 458–466. [Google Scholar] [CrossRef]
- Frieri, M.; Heuser, W.; Bliss, J. Efficacy of novel monoclonal antibody belimumab in the treatment of lupus nephritis. J. Pharmacol. Pharmacother. 2015, 6, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Struemper, H.; Chen, C.; Cai, W. Population pharmacokinetics of belimumab following intravenous administration in patients with systemic lupus erythematosus. J. Clin. Pharmacol. 2013, 53, 711–720. [Google Scholar] [CrossRef]
- Karstila, K.; Korpela, M.; Sihvonen, S.; Mustonen, J. Prognosis of clinical renal disease and incidence of new renal findings in patients with rheumatoid arthritis: Follow-up of a population-based study. Clin. Rheumatol. 2007, 26, 2089–2095. [Google Scholar] [CrossRef]
- Fervenza, F.C.; Cosio, F.G.; Erickson, S.B.; Specks, U.; Herzenberg, A.M.; Dillon, J.J.; Leung, N.; Cohen, I.M.; Wochos, D.N.; Bergstralh, E.; et al. Rituximab treatment of idiopathic membranous nephropathy. Kidney Int. 2008, 73, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Counsilman, C.E.; Jol-van der Zijde, C.M.; Stevens, J.; Cransberg, K.; Bredius, R.G.; Sukhai, R.N. Pharmacokinetics of rituximab in a pediatric patient with therapy-resistant nephrotic syndrome. Pediatr. Nephrol. 2015, 30, 1367–1370. [Google Scholar] [CrossRef] [Green Version]
- Kaaroud, H.; Fatma, L.B.; Beji, S.; Jeribi, A.; Maiz, H.B.; Moussa, F.B.; Goucha, R.; Turki, S.; Kheder, A. Interstitial and glomerular renal involvement in sarcoidosis. Saudi J. Kidney Dis. Transplant. 2008, 19, 67–71. [Google Scholar]
- Pezzoli, A.; Pascali, E. Monoclonal Bence Jones proteinuria in chronic lymphocytic leukaemia. Scand. J. Haematol. 1986, 36, 18–24. [Google Scholar] [CrossRef]
- Naderi, A.S.; Reilly, R.F. Primary care approach to proteinuria. J. Am. Board Fam. Med. 2008, 21, 569–574. [Google Scholar] [CrossRef]
- Yang, Y.; Li, T.R.; Balthasar, J.P. Investigation of the Influence of Protein-Losing Enteropathy on Monoclonal Antibody Pharmacokinetics in Mice. AAPS J. 2017, 19, 1791–1803. [Google Scholar] [CrossRef] [PubMed]
- Karbach, U.; Ewe, K.; Bodenstein, H. Alpha 1-antitrypsin, a reliable endogenous marker for intestinal protein loss and its application in patients with Crohn’s disease. Gut 1983, 24, 718–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosson, V.F.; Ng, V.W.; Lehle, M.; Lum, B.L. Population pharmacokinetics and exposure-response analyses of trastuzumab in patients with advanced gastric or gastroesophageal junction cancer. Cancer Chemother. Pharmacol. 2014, 73, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Jin, J.; Maia, M.; Lowe, J.; Sersch, M.A.; Allison, D.E. Lower exposure and faster clearance of bevacizumab in gastric cancer and the impact of patient variables: Analysis of individual data from AVAGAST phase III trial. AAPS J. 2014, 16, 1056–1063. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.K.; Rha, S.Y.; Tassone, P.; Barriuso, J.; Yu, R.; Szado, T.; Garg, A.; Bang, Y.J. A phase IIa dose-finding and safety study of first-line pertuzumab in combination with trastuzumab, capecitabine and cisplatin in patients with HER2-positive advanced gastric cancer. Br. J. Cancer 2014, 111, 660–666. [Google Scholar] [CrossRef] [Green Version]
- Brandse, J.F.; van den Brink, G.R.; Wildenberg, M.E.; van der Kleij, D.; Rispens, T.; Jansen, J.M.; Mathot, R.A.; Ponsioen, C.Y.; Lowenberg, M.; D’Haens, G.R. Loss of Infliximab Into Feces Is Associated With Lack of Response to Therapy in Patients With Severe Ulcerative Colitis. Gastroenterology 2015, 149, 350–355. [Google Scholar] [CrossRef] [Green Version]
- Neurodegeneration: Antibodies fight Parkinson’s. Nature 2014, 511, 267. [CrossRef] [Green Version]
- Zhang, Y.; Pardridge, W.M. Mediated efflux of IgG molecules from brain to blood across the blood-brain barrier. J. Neuroimmunol. 2001, 114, 168–172. [Google Scholar] [CrossRef]
- Schlachetzki, F.; Zhu, C.; Pardridge, W.M. Expression of the neonatal Fc receptor (FcRn) at the blood-brain barrier. J. Neurochem. 2002, 81, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Balthasar, J.P. Investigation of the influence of FcRn on the distribution of IgG to the brain. AAPS J. 2009, 11, 553–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuqayyas, L.; Balthasar, J.P. Investigation of the role of FcgammaR and FcRn in mAb distribution to the brain. Mol. Pharm. 2013, 10, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Cooper, P.R.; Ciambrone, G.J.; Kliwinski, C.M.; Maze, E.; Johnson, L.; Li, Q.; Feng, Y.; Hornby, P.J. Efflux of monoclonal antibodies from rat brain by neonatal Fc receptor, FcRn. Brain Res. 2013, 1534, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of amyloid-β peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30. [Google Scholar] [CrossRef]
- Bowman, G.L.; Kaye, J.A.; Moore, M.; Waichunas, D.; Carlson, N.E.; Quinn, J.F. Blood-brain barrier impairment in Alzheimer disease: Stability and functional significance. Neurology 2007, 68, 1809–1814. [Google Scholar] [CrossRef] [Green Version]
- Erickson, M.A.; Banks, W.A. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2013, 33, 1500–1513. [Google Scholar] [CrossRef] [Green Version]
- Imbach, P.; Barandun, S.; d’Apuzzo, V.; Baumgartner, C.; Hirt, A.; Morell, A.; Rossi, E.; Schoni, M.; Vest, M.; Wagner, H.P. High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet 1981, 1, 1228–1231. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. The antiinflammatory activity of IgG: The intravenous IgG paradox. J. Exp. Med. 2007, 204, 11–15. [Google Scholar] [CrossRef]
- Hansen, R.J.; Balthasar, J.P. Intravenous immunoglobulin mediates an increase in anti-platelet antibody clearance via the FcRn receptor. Thromb. Haemost. 2002, 88, 898–899. [Google Scholar] [CrossRef]
- Jin, F.; Tayab, Z.R.; Balthasar, J.P. Pharmacokinetic and pharmacodynamic effects of high-dose monoclonal antibody therapy in a rat model of immune thrombocytopenia. AAPS J. 2005, 7, E895–E902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuqayyas, L.; Balthasar, J.P. Pharmacokinetic mAb-mAb interaction: Anti-VEGF mAb decreases the distribution of anti-CEA mAb into colorectal tumor xenografts. AAPS J. 2012, 14, 445–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastuskovas, C.V.; Mundo, E.E.; Williams, S.P.; Nayak, T.K.; Ho, J.; Ulufatu, S.; Clark, S.; Ross, S.; Cheng, E.; Parsons-Reponte, K.; et al. Effects of anti-VEGF on pharmacokinetics, biodistribution, and tumor penetration of trastuzumab in a preclinical breast cancer model. Mol. Cancer Ther. 2012, 11, 752–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, V.A.; Balthasar, J.P. Sorafenib Decreases Tumor Exposure to an Anti-carcinoembryonic Antigen Monoclonal Antibody in a Mouse Model of Colorectal Cancer. AAPS J. 2016. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.M.; Ferguson, T.R.; Pickering, L.M.; Edmonds, K.; James, M.G.; Thomas, K.; Banerji, U.; Berns, B.; de Boer, C.; Gore, M.E. A phase I/II trial of sorafenib and infliximab in advanced renal cell carcinoma. Br. J. Cancer 2010, 103, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Anderson, S.K.; Lafky, J.M.; Uhm, J.H.; Giannini, C.; Kumar, S.K.; Kimlinger, T.K.; Northfelt, D.W.; Flynn, P.J.; Jaeckle, K.A.; et al. Phase II study of bevacizumab in combination with sorafenib in recurrent glioblastoma (N0776): A north central cancer treatment group trial. Clin. Cancer Res. 2013, 19, 4816–4823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.L.; Kang, Y.K.; He, A.R.; Lim, H.Y.; Ryoo, B.Y.; Hung, C.H.; Sheen, I.S.; Izumi, N.; Austin, T.; Wang, Q.; et al. Safety and efficacy of tigatuzumab plus sorafenib as first-line therapy in subjects with advanced hepatocellular carcinoma: A phase 2 randomized study. J. Hepatol. 2015, 63, 896–904. [Google Scholar] [CrossRef]
- Zhu, A.X.; Park, J.O.; Ryoo, B.Y.; Yen, C.J.; Poon, R.; Pastorelli, D.; Blanc, J.F.; Chung, H.C.; Baron, A.D.; Pfiffer, T.E.; et al. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 859–870. [Google Scholar] [CrossRef]
- Marsh, A.M.; Lo, L.; Cohen, R.A.; Feusner, J.H. Sorafenib and bevacizumab for recurrent metastatic hepatoblastoma: Stable radiographic disease with decreased AFP. Pediatr. Blood Cancer 2012, 59, 939–940. [Google Scholar] [CrossRef]
- Castellano, D.; Capdevila, J.; Sastre, J.; Alonso, V.; Llanos, M.; Garcia-Carbonero, R.; Manzano Mozo, J.L.; Sevilla, I.; Duran, I.; Salazar, R. Sorafenib and bevacizumab combination targeted therapy in advanced neuroendocrine tumour: A phase II study of Spanish Neuroendocrine Tumour Group (GETNE0801). Eur. J. Cancer 2013, 49, 3780–3787. [Google Scholar] [CrossRef]
- Galal, K.M.; Khaled, Z.; Mourad, A.M. Role of cetuximab and sorafenib in treatment of metastatic colorectal cancer. Indian J. Cancer 2011, 48, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Pan, X.; Layman, R.; Lustberg, M.B.; Mrozek, E.; Macrae, E.R.; Wesolowski, R.; Carothers, S.; Puhalla, S.; Shapiro, C.L.; et al. A Phase II study of bevacizumab in combination with trastuzumab and docetaxel in HER2 positive metastatic breast cancer. Investig. New Drugs 2014, 32, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Muselaers, C.H.; Stillebroer, A.B.; Desar, I.M.; Boers-Sonderen, M.J.; van Herpen, C.M.; de Weijert, M.C.; Langenhuijsen, J.F.; Oosterwijk, E.; Leenders, W.P.; Boerman, O.C.; et al. Tyrosine kinase inhibitor sorafenib decreases 111In-girentuximab uptake in patients with clear cell renal cell carcinoma. J. Nucl. Med. 2014, 55, 242–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong, M.N.; Matera, E.L.; Mathe, D.; Evesque, A.; Valsesia-Wittmann, S.; Clemenceau, B.; Dumontet, C. Effect of kinase inhibitors on the therapeutic properties of monoclonal antibodies. mAbs 2015, 7, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Roit, F.; Engelberts, P.J.; Taylor, R.P.; Breij, E.C.; Gritti, G.; Rambaldi, A.; Introna, M.; Parren, P.W.; Beurskens, F.J.; Golay, J. Ibrutinib interferes with the cell-mediated anti-tumor activities of therapeutic CD20 antibodies: Implications for combination therapy. Haematologica 2015, 100, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Skarzynski, M.; Niemann, C.U.; Lee, Y.S.; Martyr, S.; Maric, I.; Salem, D.; Stetler-Stevenson, M.; Marti, G.E.; Calvo, K.R.; Yuan, C.; et al. Interactions between Ibrutinib and Anti-CD20 Antibodies: Competing Effects on the Outcome of Combination Therapy. Clin. Cancer Res. 2016, 22, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winiarska, M.; Bil, J.; Wilczek, E.; Wilczynski, G.M.; Lekka, M.; Engelberts, P.J.; Mackus, W.J.M.; Gorska, E.; Bojarski, L.; Stoklosa, T.; et al. Statins Impair Antitumor Effects of Rituximab by Inducing Conformational Changes of CD20. PLoS Med. 2008, 5, e64. [Google Scholar] [CrossRef] [Green Version]
- Tumlin, J.A.; Galphin, C.M.; Rovin, B.H. Advanced Diabetic Nephropathy with Nephrotic Range Proteinuria: A Pilot Study of the Long-Term Efficacy of Subcutaneous ACTH Gel on Proteinuria, Progression of CKD, and Urinary Levels of VEGF and MCP-1. J. Diabetes Res. 2013, 2013, 489869. [Google Scholar] [CrossRef] [Green Version]
- Praluent (Alirocumab) [package insert]. Regeneron Pharmaceuticals Inc.: Tarrytown, NY, USA. Available online: http://www.regeneron.com/Praluent/Praluent-fpi.pdf (accessed on 26 July 2016).
- Cilliers, C.; Guo, H.; Liao, J.; Christodolu, N.; Thurber, G.M. Multiscale Modeling of Antibody-Drug Conjugates: Connecting Tissue and Cellular Distribution to Whole Animal Pharmacokinetics and Potential Implications for Efficacy. AAPS J. 2016. [Google Scholar] [CrossRef] [Green Version]
- Garrett, M.; Ruiz-Garcia, A.; Parivar, K.; Boni, J. Characterization of inotuzumab ozogamicin time dependent clearance in relapsed/refractory acute lymphoblastic leukemia patients by nonlinear mixed-effects analysis. Clin. Pharm. 2016, 99 (Suppl. 1), S5–S107. [Google Scholar]


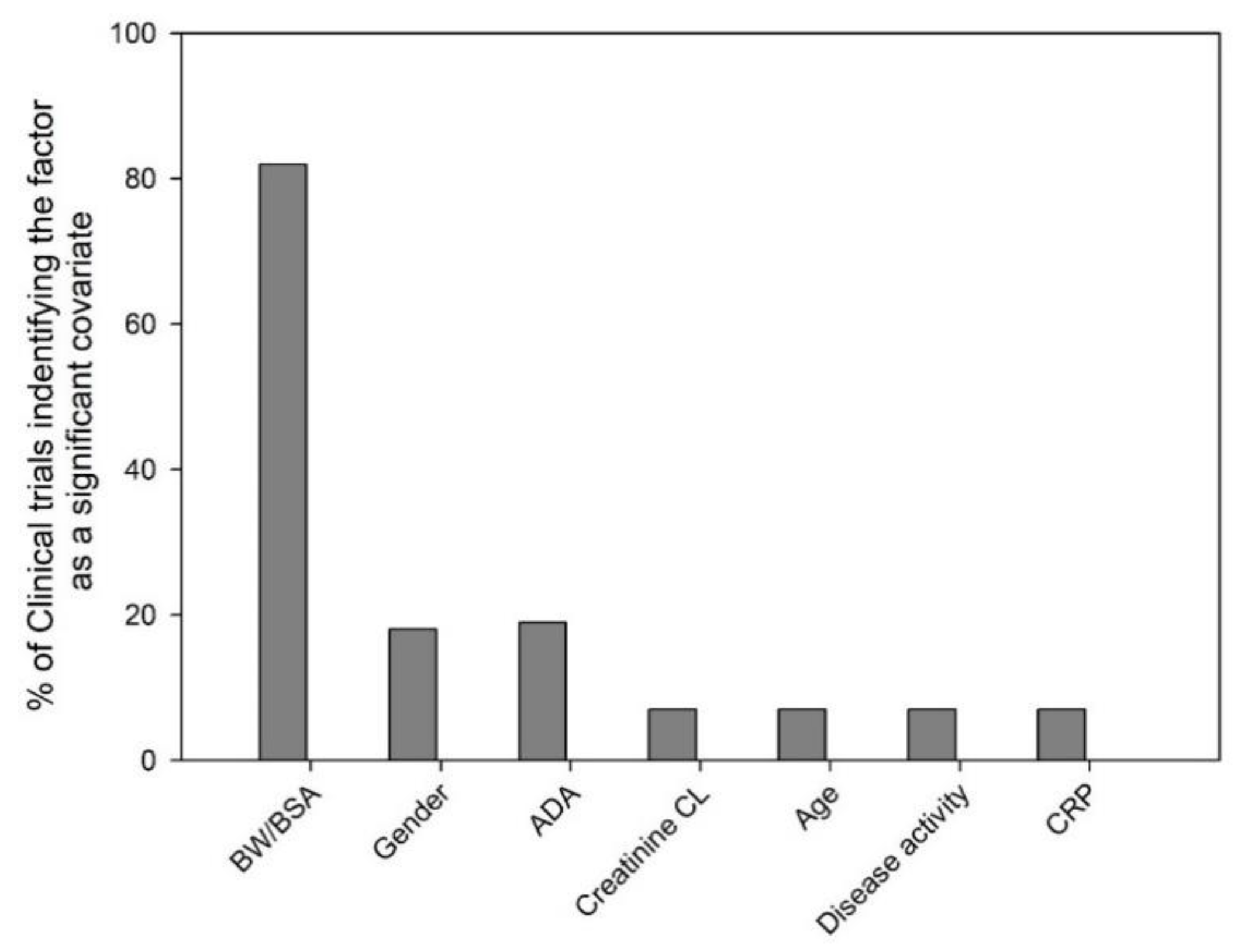{kind=link}
| Species | Model Protein | Molecular Weight (kDa) | Site of Injection | % Dose Recovery in Lymph | Selected Lymph Duct/Node | Does the Lymphatic System Contribute to SC Absorption? | Ref. |
|---|---|---|---|---|---|---|---|
| Sheep | 5-fluoro-2′deoxyuridine | 0.246 | Lower part of right hind leg | 4.0 ± 1.5 | Efferent duct of the popliteal lymph node | Yes | [17] |
| Sheep | Insulin | 5.80 | Interdigital space of hind leg | 17.3 ± 1 | Efferent duct of the popliteal lymph node | Yes | [18] |
| Sheep | Inulin | 5.20 | Lower part of right hind leg | 21.0 ± 7.1 | Efferent duct of the popliteal lymph node | Yes | [17] |
| Sheep | Cytochrome c | 12.3 | Lower part of right hind leg | 38.6 ± 6.7 | Efferent duct of the popliteal lymph node | Yes | [17] |
| Sheep | Recombinant methionyl human leptin | 16.2 | Interdigital space of hind leg | 34.4 ± 9.7 | Efferent duct of the popliteal lymph node | Yes | [19] |
| Sheep | Human recombinant interferon-2α | 19.0 | Lower part of right hind leg | 59.5 ± 7.1 | Efferent duct of the popliteal lymph node | Yes | [17] |
| Sheep | Human growth Hormone | 22.0 | Interdigital space of hind leg | 61.7 ± 8.5 | Efferent duct of the popliteal lymph node | Yes | [20] |
| Sheep | Recombinant human epoetin-α | 30.4 | Interdigital space of hind leg | 83.9 ± 6.6 | Efferent duct of the popliteal lymph node | Yes | [21] |
| Sheep | Darbepoetin- α | 37.0 | Interdigital space of hind leg | 90.2 ± 4.4 | Efferent duct of the popliteal lymph node | Yes | [22] |
| Dog | PEGylated recombinant human erythropoietin α | 60.4 | Lower left region of hind leg | 20 | Thoracic lymph duct | Yes | [23] |
| Dog | PEGylated neuromedin-U receptor agonist MRL-1 | 46.0 | Popliteal region of hind limb | 72.9 | Thoracic lymph duct | Yes | [24] |
| Rabbit | Interferon- α2 | 19.2 | Hind leg | 0.10 ± 0.06 | Thoracic lymph duct | No | [26] |
| Rat | Recombinant human tumor necrosis factor-α | 45.0 | Back region | 4.7 ± 3.4 | Thoracic lymph duct | No | [27] |
| Rat | Bovine insulin | 5.60 | Lateral side of thigh | 0.072 ± 0.0016 | Thoracic lymph duct | No | [28] |
| Rat | Recombinant human erythropoietin α | 30.4 | Lateral side of thigh | 1.44 ± 0.26 | Thoracic lymph duct | No | [28] |
| Rat | Bovine serum albumin | 66.0 | Lateral side of thigh | 2.15 ± 1.08 | Thoracic lymph duct | No | [28] |
| Rat | PEGylated poly-L-lysine Lys16 (PEG2000)32 | 68.0 | Lower right hind leg | 29 | Thoracic lymph duct | Yes | [29] |
| Rat | PEGylated recombinant human erythropoietin α | 60.4 | Lower left hind leg | 23.8 ± 1.08 | Thoracic lymph duct | Yes | [23] |
| Rat | PEGylated neuromedin-U receptor agonist MRL-1 | 46.0 | Lower left hind leg | 26.7 ± 9.0 | Thoracic lymph duct | Yes | [24] |
| Rat | Trastuzumab | 149 | Inner left hind leg | 26.7 ± 10.4 | Thoracic lymph duct | Yes | [30] |
| Human Fc Gamma Receptors | |||||||||||
| Name | FcγRI (CD 64) | FcγRIIA (CD 32A) | FcγRIIB (CD 32B) | FcγRIIC (CD 32C) | FcγRIIIA (CD 16A) | FcγRIIIB (CD 16B) | |||||
| Gene [117] | FCGRT1A | FCGRT2A | FCGRT2B | FCGRT2C | FCGRT3A | FCGRT3B | |||||
| Alleles [117] | - | H131 | R131 | I232 | T232 | Q13 | Stop13 | V158 | F158 | NA1 | |
| NA2 | |||||||||||
| SH | |||||||||||
| Affinity for Ligand [117] (M−1) | IgG1 | 6 × 107 | 5 × 106 | 3 × 106 | 3 × 108 | ND | 1 × 105 | NB | 2 × 105 | 1 × 105 | 2 × 105 |
| IgG2 | NB | 4 × 105 | 1 × 105 | 1 × 105 | ND | 2 × 104 | NB | 7 × 104 | 3 × 104 | NB | |
| IgG3 | 6 × 107 | 9 × 105 | 9 × 105 | 9 × 105 | ND | 2 × 105 | NB | 1 × 107 | 8 × 106 | 1 × 106 | |
| IgG4 | 3 × 107 | 2 × 105 | 2 × 105 | 2 × 105 | ND | 2 × 105 | NB | 2 × 105 | 2 × 105 | NB | |
| Cell Distribution [117,118] | Macrophages | Macrophages | Macrophages | Macrophages | Macrophages | Neutrophils | |||||
| Eosinophils | Eosinophils | Eosinophils | Neutrophils | Dendritic cells | |||||||
| Dendritic cells | Dendritic cells | Dendritic cells | NK cells | Basophils | |||||||
| Neutrophils | Neutrophils | Neutrophils | Mast cells | ||||||||
| Mast cells | Mast cells | NK cells | |||||||||
| Platelets | B cells | ||||||||||
| Class | Activation | Activation | Inhibition | Activation | Activation | Decoy | |||||
| Activation (not clear) | |||||||||||
| Function [119,120] | Effector cell activation | Effector cell activation | Inhibition of effector activity | Co-activation receptor for FcγRIIIA, ADCC | Effector cell activation | Unknown | |||||
| Phagocytosis | Phagocytosis | Phagocytosis | |||||||||
| Degranulation | ADCC | ||||||||||
| ADCC | |||||||||||
| Mouse Fc Gamma Receptors | |||||||||||
| Name | FcγRI (CD 64) | FcγRIIB (CD 32B) | FcγRIII (CD 16) | FcγRIV (CD 16-2) | |||||||
| Affinity for Ligand [118] (M−1) | IgG1 | NB | 3.3 × 106 | 0.3 × 106 | NB | ||||||
| IgG2a | 1.6 × 108 | 0.4 × 106 | 0.7 × 106 | 2.9 × 107 | |||||||
| IgG2b | NB | 2.2 × 106 | 0.6 × 106 | 1.7 × 107 | |||||||
| Cell Distribution [118] | Monocytes | B cells | Monocytes | Monocytes | |||||||
| Macrophages | Dendritic cells | Macrophages | Macrophages | ||||||||
| Dendritic cells | Neutrophils | Neutrophils | |||||||||
| Dendritic cells | Dendritic cells | ||||||||||
| NK cells | |||||||||||
| Class | Activation | Inhibition | Activation | Activation | |||||||
| Target | Disease | Number of Patients | Target Expression | Unit | Fold Range in Expression | Approved mAb | Ref. |
|---|---|---|---|---|---|---|---|
| CD-20 | Chronic lymphocytic leukemia | 31 | 2737–115623 | MESF | 42 | Rituximab | [208] |
| Ofatumumab | |||||||
| CD-20 | Diffuse large B-cell lymphomas | 64 | 3549–679577 | MESF | 191 | Rituximab | [208] |
| Ibritumomab | |||||||
| CD-20 | Follicular Lymphoma | 56 | 8460–445755 | MESF | 52 | tiuxetan | [208] |
| CD-20 | Mantle Cell Lymphoma | 34 | 8826–423799 | MESF | 48 | Tositumomab | [208] |
| CD-20 | Marginal Zone Lymphoma | 18 | 3615–207034 | MESF | 57 | (I-131) | [208] |
| CD-52 | Chronic lymphocytic leukemia | 5 | 371303 ± 117212 | Receptor Number | - | Alemtuzumab | [209] |
| TNF-α | Rheumatoid Arthritis | 327 | 0.92–9.68 | pg/Ml | 10.5 | Adalimumab | [198] |
| Certolizumab | |||||||
| Golimumab | |||||||
| Infliximab | |||||||
| CD25 | Kidney Transplantation | 14 | 57.1 ± 12.7 | Mean % of CD25 + within CD4 + T cells | - | Basiliximab | [210] |
| IgE | Asthma | 245 | 51–1692 | ng/mL | 33 | Omalizumab | [211] |
| VEGF | Advanced Breast Cancer | 56 | 12.5–445 (plasma) | pg/mL | 35 | Bevacizumab | [212] |
| EGFR | Colorectal Adenocarcinoma | 143 | 1 (10%) | IHC score (% of patient) | - | Cetuximab | [213] |
| 2 (32%) | Panitumumab | ||||||
| 3 (55%) | |||||||
| HER-2 | Breast Cancer | 47 | 1 (49%) | IHC score (% of patient) | - | Trastuzumab | [214] |
| 2 (6%) | Pertuzumab | ||||||
| 3 (55%) |
| mAb | Type | Antigen | Route | Disease | % Immunogenicity (n = Total Number of Patients) | Impact on PK | Ref. |
|---|---|---|---|---|---|---|---|
| Infliximab | Chimeric IgG1k | TNF-α | IV infusion | AS | 25% (n = 8) | NQ | [272] |
| RA | 33% (n = 143) | NQ | [273] | ||||
| Psoriasis | 19–22% (n = 264) | NQ | [293] | ||||
| CD | 61% (n = 125) | NQ | [294] | ||||
| IBD | 15% (n = 33) | 2.7 fold ↑ in CL; 34% ↓ in t1/2 | [275] | ||||
| Adalimumab | Human IgG1k | TNF-α | IV | RA | 28% (n = 272) | 1.4 fold ↓ in mAb, median concentration | [295] |
| Psoriasis | 49% (n = 80) | Significant ↓ in mAb, Cmin | [296] | ||||
| CD | 13–18% (n = 65 to 96) | 4–5.5 fold ↑ in CL | [297,298] | ||||
| Natalizumab | Humanized IgG4k | α4-Integrin | IV infusion | MS | 9–82% (n = 2195) | 3 fold ↑ in CL | [299] |
| CD | 5–10% (n = 1414) | NQ | [299] | ||||
| Golimumab | Human IgG1k | TNF-α | SC | PA | 2.90% (n = 337) | Antibody to mAb significant covariate on CL/F | [277] |
| AS | 3.10% (n = 312) | 36% ↑ in median CL/F | [276] | ||||
| Ustekinumab | Human IgG1k | IL-12/IL-23 | SC | Psoriasis | 3.20% (n = 1937) | 35.5% ↑ in median CL/F | [279] |
| PA | 9.20% (n = 130) | 42% ↑ in median CL/F | [278] | ||||
| Anti-IL-1β | Humanized IgG4 | IL-1β | SC | T2DM | 36.7% (n = 79) | ||
| IV | RA | 2.1% (n = 96) | 37.6% ↑ in CL | [280] | |||
| Daclizumab | Humanized IgG1 | IL-2 Receptor α | SC | Remitting relapsing MS | 0.80% (n = 17139) | 19% ↑ in median CL | [281] |
| Amatuximab | Chimeric IgG1k | Mesothelin | IV infusion | Unresectable malignant pleural mesothelioma | 24.60% (n = 199) | 1.5 fold ↑ in CL | [282] |
| Atezolizumab | Humanized IgG1 | PD-L1 | IV infusion | Metastatic Urothelial Carcinoma | 31.70% (n = 139) | 16% ↑ in median CL | [283] |
| Benralizumab | Humanized IgG1 | IL-5 receptor α | SC | Asthma | 9.50% (n = 200) | 4.6 fold ↑ in median CL | [284] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, V.A.; Balthasar, J.P. Understanding Inter-Individual Variability in Monoclonal Antibody Disposition. Antibodies 2019, 8, 56. https://doi.org/10.3390/antib8040056
Thomas VA, Balthasar JP. Understanding Inter-Individual Variability in Monoclonal Antibody Disposition. Antibodies. 2019; 8(4):56. https://doi.org/10.3390/antib8040056
Chicago/Turabian StyleThomas, Veena A., and Joseph P. Balthasar. 2019. "Understanding Inter-Individual Variability in Monoclonal Antibody Disposition" Antibodies 8, no. 4: 56. https://doi.org/10.3390/antib8040056