IgA Nephropathy: Current Treatment and New Insights


Abstract
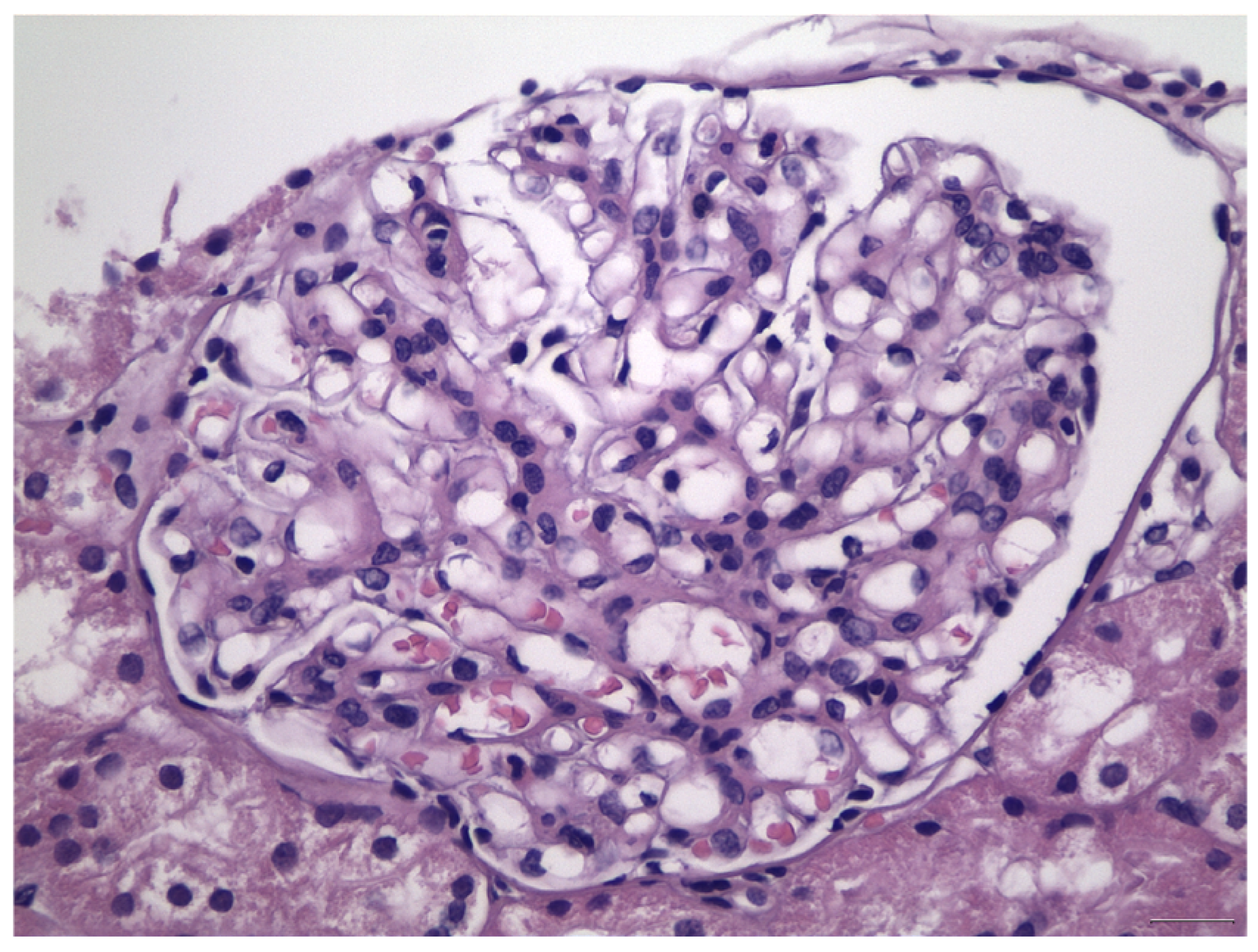
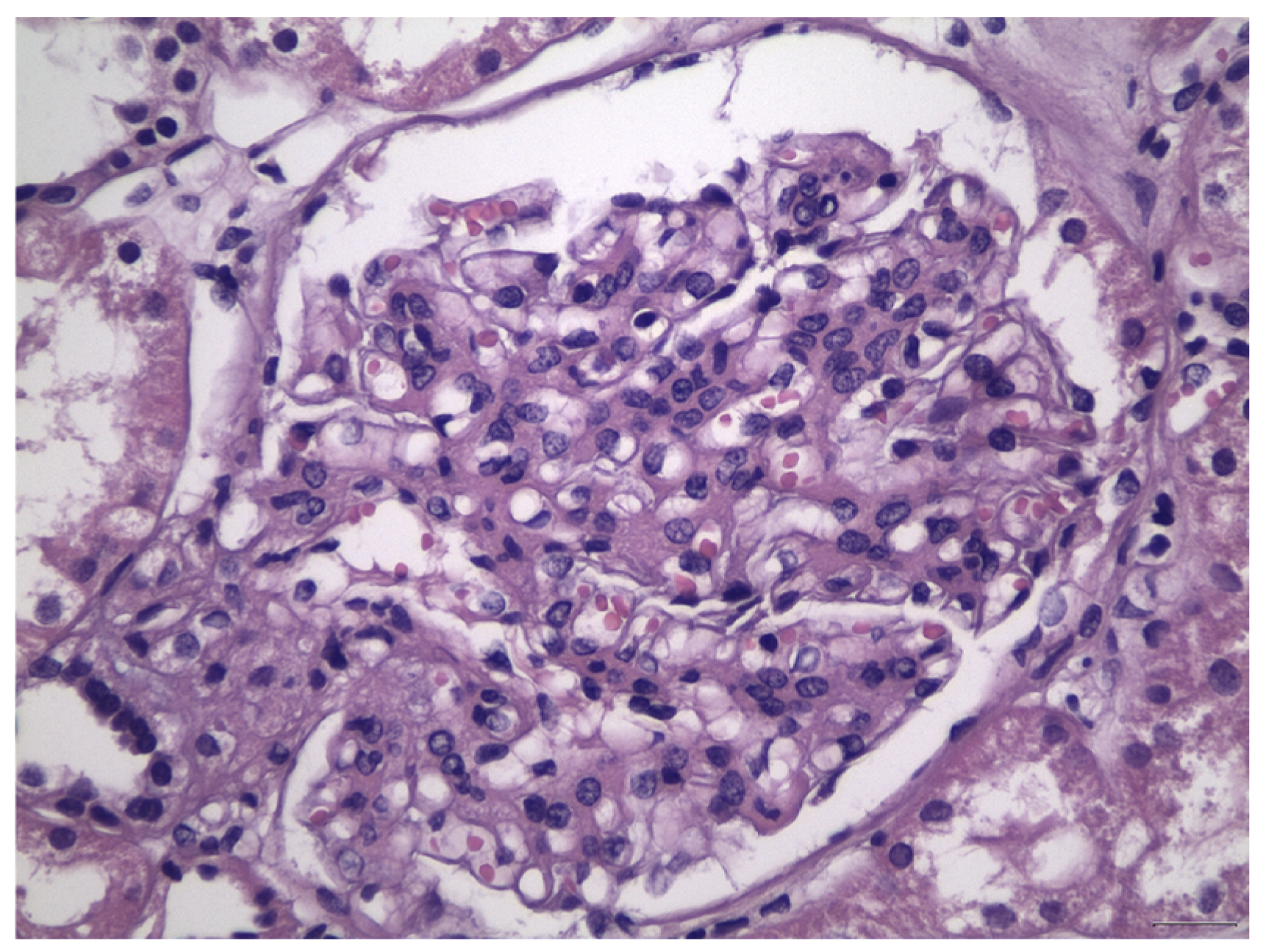
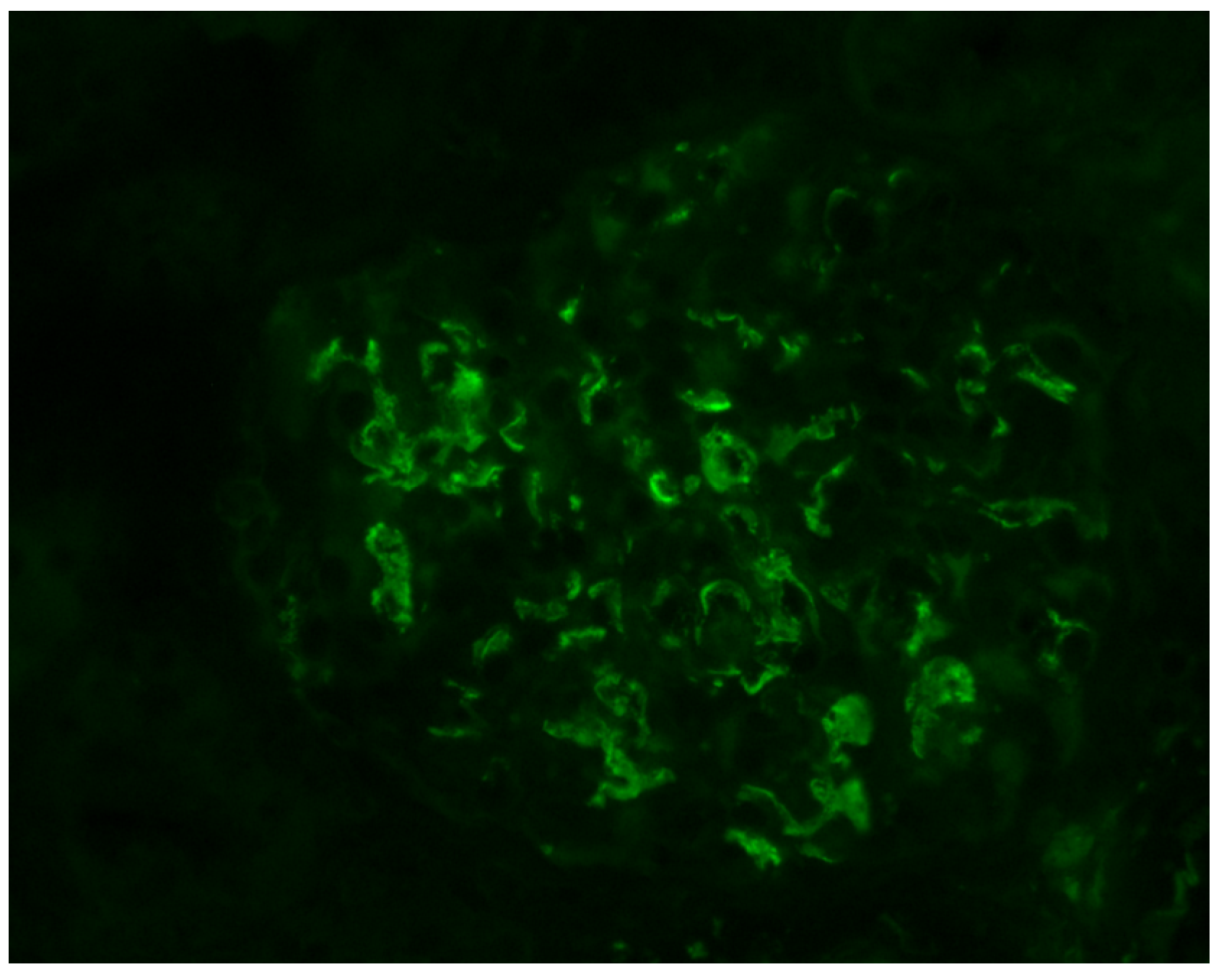
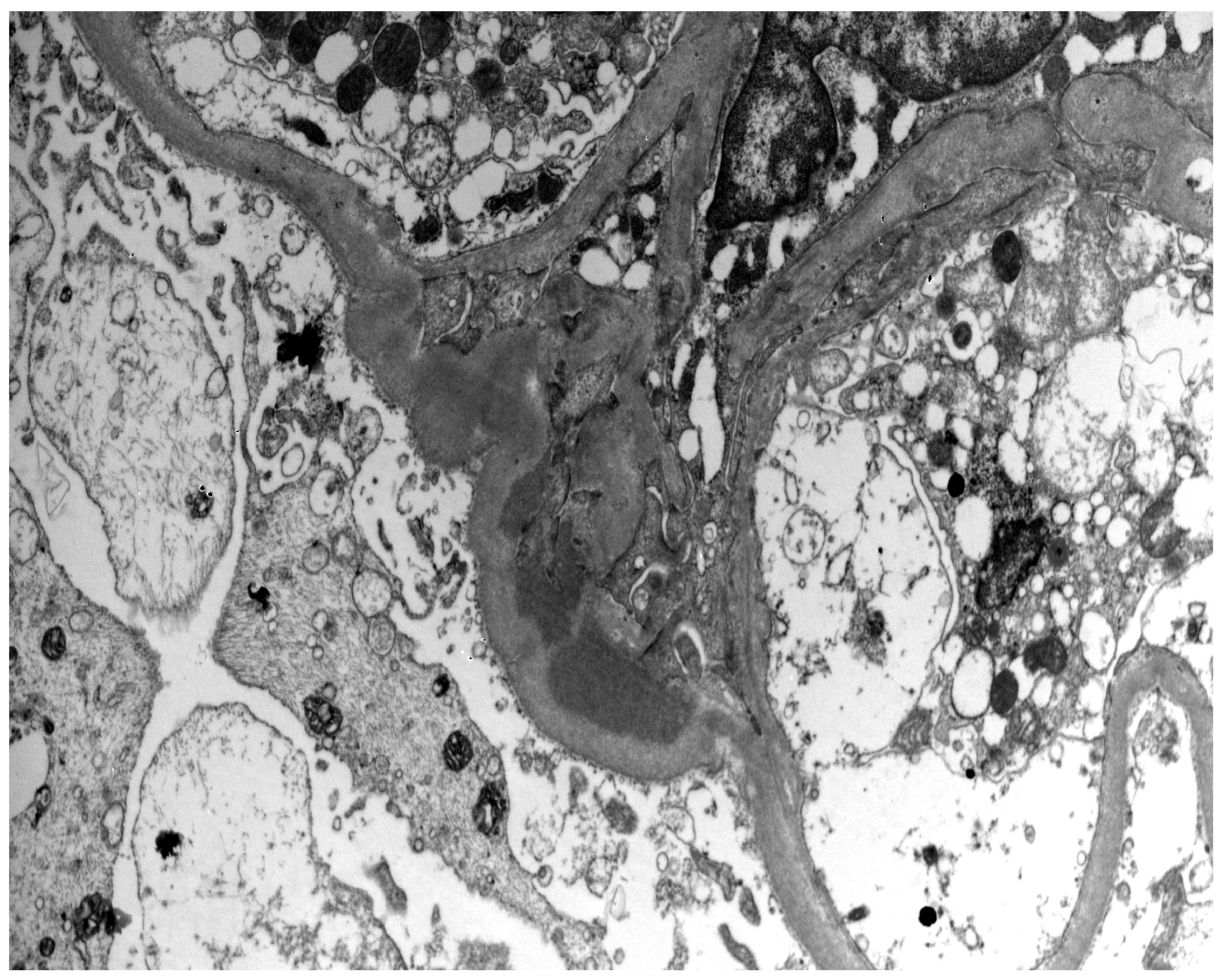
:1. Introduction
2. Risk Stratification for Disease Progression
3. Supportive Care in All Patients—The Role of SGLT2 Inhibitors
4. Immunosuppressive Therapy in High-Risk Patients
5. Other Investigational Agents
5.1. Inhibition of Immune Complex-Activated Complement Activity
5.2. Inhibition of BAFF/APRIL Signaling
5.3. Plasma Cell and B Cell Depletion
5.4. Inhibition of Endothelin A Receptor and Angiotensin II Subtype 1 Receptor
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wyatt, R.J.; Julian, B.A. IgA nephropathy. N. Engl. J. Med. 2013, 368, 2402–2414. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, R.J.; Julian, B.A.; Baehler, R.W.; Stafford, C.C.; McMorrow, R.G.; Ferguson, T.; Jackson, E.; Woodford, S.Y.; Miller, P.M.; Kritchevsky, S. Epidemiology of IgA nephropathy in central and eastern Kentucky for the period 1975 through 1994. Central Kentucky Region of the Southeastern United States IgA Nephropathy DATABANK Project. J. Am. Soc. Nephrol. 1998, 9, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Galla, J.H. IgA nephropathy. Kidney Int. 1995, 47, 377–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, S.C.; Goh, S.M.; Barratt, J. Is immunoglobulin A nephropathy different in different ethnic populations? Nephrology 2019, 24, 885–895. [Google Scholar] [CrossRef] [Green Version]
- Sanders, J.T.; Wyatt, R.J. IgA nephropathy and Henoch-Schönlein purpura nephritis. Curr. Opin. Pediatr. 2008, 20, 163–170. [Google Scholar] [CrossRef]
- Gutiérrez, E.; González, E.; Hernández, E.; Morales, E.; Martínez, M.Á.; Usera, G.; Jackson, E.; Woodford, S.Y.; Miller, P.M.; Kritchevsky, S. Factors that determine an incomplete recovery of renal function in macrohematuria-induced acute renal failure of IgA nephropathy. Clin. J. Am. Soc. Nephrol. 2007, 2, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Kiryluk, K.; Novak, J.; Moldoveanu, Z.; Herr, A.B.; Renfrow, M.B.; Wyatt, R.J.; Scolari, F.; Mestecky, J.; Gharavi, A.G.; et al. The pathophysiology of IgA nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1795–1803. [Google Scholar] [CrossRef] [Green Version]
- Sharmin, S.; Shimizu, Y.; Hagiwara, M.; Hirayama, K.; Koyama, A. Staphylococcus aureus antigens induce IgA-type glomerulonephritis in Balb/c mice. J. Nephrol. 2004, 17, 504–511. [Google Scholar]
- Waldo, F.B.; Britt, W.J.; Tomana, M.; Julian, B.A.; Mestecky, J. Non-specific mesangial staining with antibodies against cytomegalovirus in immunoglobulin-A nephropathy. Lancet 1989, 1, 129–131. [Google Scholar] [CrossRef]
- Farooq, H.; Aemaz Ur Rehman, M.; Asmar, A.; Asif, S.; Mushtaq, A.; Qureshi, M.A. The pathogenesis of COVID-19-induced IgA nephropathy and IgA vasculitis: A systematic review. J. Taibah Univ. Med. Sci. 2022, 17, 1–13. [Google Scholar] [CrossRef]
- Zhang, Y.-M.; Zhou, X.-J.; Zhang, H. What Genetics Tells Us About the Pathogenesis of IgA Nephropathy: The Role of Immune Factors and Infection. Kidney Int. Rep. 2017, 2, 318–331. [Google Scholar] [CrossRef]
- Berger, J.; Hinglais, N. Intercapillary deposits of IgA-IgG. J. D’urologie Nephrol. 1968, 74, 694–695. [Google Scholar]
- Haas, M. Histologic subclassification of IgA nephropathy: A clinicopathologic study of 244 cases. Am. J. Kidney Dis. 1997, 29, 829–842. [Google Scholar] [CrossRef]
- Cattran, D.C.; Coppo, R.; Cook, H.T.; Feehally, J.; Roberts, I.S.D.; Troyanov, S.; Alpers, C.E.; Amore, A.; Barratt, J.; Berthoux, F.; et al. The Oxford classification of IgA nephropathy: Rationale, clinicopathological correlations, and classification. Kidney Int. 2009, 76, 534–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, I.S.D.; Cook, H.T.; Troyanov, S.; Alpers, C.E.; Amore, A.; Barratt, J.; Berthoux, F.; Bonsib, S.; Bruijn, J.A.; Cattran, D.C.; et al. The Oxford classification of IgA nephropathy: Pathology definitions, correlations, and reproducibility. Kidney Int. 2009, 76, 546–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennette, J.C.; Olson, J.L.; Silva, F.G.; D’Agati, V.D. Heptinstall’ s Pathology of the Kidney, 7th ed.; LWW: Philadelphia, PA, USA, 2015. [Google Scholar]
- Berthoux, F.; Mohey, H.; Laurent, B.; Mariat, C.; Afiani, A.; Thibaudin, L. Predicting the risk for dialysis or death in IgA nephropathy. J. Am. Soc. Nephrol. 2011, 22, 752–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donadio, J.V.; Bergstralh, E.J.; Grande, J.P.; Rademcher, D.M. Proteinuria patterns and their association with subsequent end-stage renal disease in IgA nephropathy. Nephrol. Dial. Transplant. 2002, 17, 1197–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbour, S.J.; Cattran, D.C.; Espino-Hernandez, G.; Hladunewich, M.A.; Reich, H.N. Identifying the ideal metric of proteinuria as a predictor of renal outcome in idiopathic glomerulonephritis. Kidney Int. 2015, 88, 1392–1401. [Google Scholar] [CrossRef] [Green Version]
- Pitcher, D.; Braddon, F.; Hendry, B.; Mercer, A.; Osmaston, K.; Saleem, M.A.; Steenkamp, R.; Wong, K.; Turner, A.N.; Wang, K.; et al. Long-Term Outcomes in IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 2023. ahead of print. [Google Scholar] [CrossRef]
- Reich, H.N.; Troyanov, S.; Scholey, J.W.; Cattran, D.C. Remission of proteinuria improves prognosis in IgA nephropathy. J. Am. Soc. Nephrol. 2007, 18, 3177–3183. [Google Scholar] [CrossRef] [Green Version]
- Rekola, S.; Bergstrand, A.; Bucht, H. Deterioration of GFR in IgA nephropathy as measured by 51Cr-EDTA clearance. Kidney Int. 1991, 40, 1050–1054. [Google Scholar] [CrossRef] [Green Version]
- Bobart, S.A.; Alexander, M.P.; Shawwa, K.; Vaughan, L.E.; Ghamrawi, R.; Sethi, S.; Cornell, L.; Glassock, R.J.; Fervenza, F.C.; Zand, L. The association of microhematuria with mesangial hypercellularity, endocapillary hypercellularity, crescent score and renal outcomes in immunoglobulin A nephropathy. Nephrol. Dial. Transplant. 2021, 36, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Le, W.B.; Liang, S.S.; Hu, Y.L.; Deng, K.P.; Bao, H.; Zeng, C.H.; ZhiHong Liu, Z.H. Long-term renal survival and related risk factors in patients with IgA nephropathy: Results from a cohort of 1155 cases in a Chinese adult population. Nephrol. Dial. Transplant. 2012, 27, 1479–1485. [Google Scholar] [CrossRef] [Green Version]
- Barbour, S.J.; Canney, M.; Coppo, R.; Zhang, H.; Liu, Z.H.; Suzuki, Y.; Matsuzaki, K.; Katafuchi, R.; Induruwage, D.; Er, L.; et al. Improving treatment decisions using personalized risk assessment from the International IgA Nephropathy Prediction Tool. Kidney Int. 2020, 98, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; Adler, S.G.; Barratt, J.; Bridoux, F.; Burdge, K.A.; Chan, T.M.; Cook, H.T.; Fervenza, F.C.; Gibson, K.L.; Glassock, R.J.; et al. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int. 2021, 100, S1–S276. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Liu, Y.; Perkovic, V.; Li, X.; Ninomiya, T.; Hou, W.; Zhao, N.; Liu, L.; Lv, J.; Zhang, H.; et al. Renin-Angiotensin System Inhibitors and Kidney and Cardiovascular Outcomes in Patients With CKD: A Bayesian Network Meta-analysis of Randomized Clinical Trials. Am. J. Kidney Dis. 2016, 67, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Praga, M.; Gutiérrez, E.; González, E.; Morales, E.; Hernandez, E. Treatment of IgA nephropathy with ACE inhibitors: A randomized and controlled trial. J. Am. Soc. Nephrol. 2003, 14, 1578–1583. [Google Scholar] [CrossRef] [Green Version]
- Li, P.K.T.; Leung, C.B.; Chow, K.M.; Cheng, Y.L.; Fung, S.K.S.; Mak, S.K.; Tang, A.W.-C.; Wong, T.Y.-H.; Yung, C.Y.; Yung, J.C.-U.; et al. Hong Kong study using valsartan in IgA nephropathy (HKVIN): A double-blind, randomized, placebo-controlled study. Am. J. Kidney Dis. 2006, 47, 751–760. [Google Scholar] [CrossRef]
- Ruggenenti, P.; Perna, A.; Gherardi, G.; Garini, G.; Zoccali, C.; Salvadori, M.; Scolari, F.; Schena, F.P.; Remuzzi, G. Renoprotective properties of ACE-inhibition in non-diabetic nephropathies with non-nephrotic proteinuria. Lancet 1999, 354, 359–364. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Perkins, B.A.; Fitchett, D.H.; Husain, M.; Cherney, D.Z.I. Sodium Glucose Cotransporter 2 Inhibitors in the Treatment of Diabetes Mellitus: Cardiovascular and Kidney Effects, Potential Mechanisms, and Clinical Applications. Circulation 2016, 134, 752–772. [Google Scholar] [CrossRef]
- Wheeler, D.C.; Toto, R.D.; Stefansson, B.V.; Jongs, N.; Chertow, G.M.; Greene, T.; Hou, F.F.; McMurray, J.J.; Pecoits-Filho, R.; Correa-Rotter, R.; et al. A pre-specified analysis of the DAPA-CKD trial demonstrates the effects of dapagliflozin on major adverse kidney events in patients with IgA nephropathy. Kidney Int. 2021, 100, 215–224. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.-F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- The EMPA-KIDNEY Collaborative Group; Herrington, W.G.; Staplin, N.; Wanner, C.; Green, J.B.; Hauske, S.J.; Preiss, D.; Judge, P.; Mayne, K.J.; Ng, S.Y.A.; et al. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2023, 388, 117–127. [Google Scholar] [CrossRef]
- Wong, M.G.; Lv, J.; Hladunewich, M.A.; Jha, V.; Hooi, L.S.; Monaghan, H.; Zhao, M.; Barbour, S.; Reich, H.N.; Cattran, D.; et al. The Therapeutic Evaluation of Steroids in IgA Nephropathy Global (TESTING) Study: Trial Design and Baseline Characteristics. Am. J. Nephrol. 2021, 52, 827–836. [Google Scholar] [CrossRef]
- Lv, J.; Wong, M.G.; Hladunewich, M.A.; Jha, V.; Hooi, L.S.; Monaghan, H.; Zhao, M.; Barbour, S.; Jardine, M.J.; Reich, H.N.; et al. Effect of Oral Methylprednisolone on Decline in Kidney Function or Kidney Failure in Patients with IgA Nephropathy: The TESTING Randomized Clinical Trial. JAMA 2022, 327, 1888–1898. [Google Scholar] [CrossRef]
- Lv, J.; Zhang, H.; Wong, M.G.; Jardine, M.J.; Hladunewich, M.; Jha, V.; Monaghan, H.; Zhao, M.; Barbour, S.; Reich, H.; et al. Effect of Oral Methylprednisolone on Clinical Outcomes in Patients with IgA Nephropathy: The TESTING Randomized Clinical Trial. JAMA 2017, 318, 432–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eitner, F.; Ackermann, D.; Hilgers, R.-D.; Floege, J. Supportive Versus Immunosuppressive Therapy of Progressive IgA nephropathy (STOP) IgAN trial: Rationale and study protocol. J. Nephrol. 2008, 21, 284–289. [Google Scholar] [PubMed]
- Rauen, T.; Wied, S.; Fitzner, C.; Eitner, F.; Sommerer, C.; Zeier, M.; Otte, B.; Panzer, U.; Budde, K.; Benck, U.; et al. After ten years of follow-up, no difference between supportive care plus immunosuppression and supportive care alone in IgA nephropathy. Kidney Int. 2020, 98, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Tesar, V.; Troyanov, S.; Bellur, S.; Verhave, J.C.; Cook, H.T.; Feehally, J.; Roberts, I.S.; Cattran, D.; Coppo, R. Corticosteroids in IgA Nephropathy: A Retrospective Analysis from the VALIGA Study. J. Am. Soc. Nephrol. 2015, 26, 2248–2258. [Google Scholar] [CrossRef] [Green Version]
- Manno, C.; Torres, D.D.; Rossini, M.; Pesce, F.; Schena, F.P. A randomized controlled clinical trial of corticosteroids plus ACE inhibitors with long-term follow-up in proteinuric IgA nephropathy. Nephrol. Dial. Transplant. 2009, 24, 3694–3701. [Google Scholar] [CrossRef]
- Lv, J.; Zhang, H.; Chen, Y.; Li, G.; Jiang, L.; Singh, A.K.; Wanget, H. Combination therapy of prednisone and ACE inhibitor versus ACE-inhibitor therapy alone in patients with IgA nephropathy: A randomized controlled trial. Am. J. Kidney Dis. 2009, 53, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, C.; Bolasco, P.G.; Fogazzi, G.B.; Andrulli, S.; Altieri, P.; Ponticelli, C.; Locatelli, F. Corticosteroids in IgA nephropathy: A randomized controlled trial. Lancet 1999, 353, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Rauen, T.; Eitner, F.; Fitzner, C.; Sommerer, C.; Zeier, M.; Otte, B.; Panzer, U.; Peters, H.; Benck, U.; Mertens, P.R.; et al. Intensive supportive care plus immunosuppression in IgA nephropathy. N. Engl. J. Med. 2015, 373, 2225–2236. [Google Scholar] [CrossRef] [PubMed]
- Fellström, B.C.; Barratt, J.; Cook, H.; Coppo, R.; Feehally, J.; de Fijter, J.W.; Floege, J.; Hetzel, G.; Jardine, A.G.; Locatelli, F.; et al. Targeted-release budesonide versus placebo in patients with IgA nephropathy (NEFIGAN): A double-blind, randomized, placebo-controlled phase 2b trial. Lancet 2017, 389, 2117–2127. [Google Scholar] [CrossRef] [Green Version]
- Natale, P.; Palmer, S.C.; Ruospo, M.; Saglimbene, V.M.; Craig, J.C.; Vecchio, M.; Samuels, J.A.; Molony, D.A.; Schena, F.P.; Strippoli, G.F.; et al. Immunosuppressive agents for treating IgA nephropathy. Cochrane Database Syst. Rev. 2020, 3, CD003965. [Google Scholar] [CrossRef]
- Maes, B.D.; Oyen, R.; Claes, K.; Evenepoel, P.; Kuypers, D.; Vanwalleghem, J.; Van Damme, B.; Vanrenterghem, Y.F. Mycophenolate mofetil in IgA nephropathy: Results of a 3-year prospective placebo-controlled randomized study. Kidney Int. 2004, 65, 1842–1849. [Google Scholar] [CrossRef]
- Frisch, G.; Lin, J.; Rosenstock, J.; Markowitz, G.; D’Agati, V.; Radhakrishnan, J.; Preddie, D.; Crew, J.; Valeri, A.; Appel, G. Mycophenolate mofetil (MMF) vs placebo in patients with moderately advanced IgA nephropathy: A double-blind randomized controlled trial. Nephrol. Dial. Transplant. 2005, 20, 2139–2145. [Google Scholar] [CrossRef] [Green Version]
- Hou, F.F.; Xie, D.; Wang, J.; Xu, X.; Yang, X.; Ai, J.; Nie, S.; Liang, M.; Wang, G.; Jia, N. Effectiveness of Mycophenolate Mofetil Among Patients with Progressive IgA Nephropathy: A Randomized Clinical Trial. JAMA Netw. Open 2023, 6, e2254054. [Google Scholar] [CrossRef]
- Barratt, J.; Lafayette, R.; Kristensen, J.; Stone, A.; Cattran, D.; Floege, J.; Tesar, V.; Trimarchi, H.; Zhang, H.; Eren, N.; et al. Results from part A of the multi-center, double-blind, randomized, placebo-controlled NefIgArd trial, which evaluated targeted-release formulation of budesonide for the treatment of primary immunoglobulin A nephropathy. Kidney Int. 2023, 103, 391–402. [Google Scholar] [CrossRef]
- Lopez-Martinez, M.; Torres, I.; Bermejo, S.; Moreso, F.; Garcia-Carro, C.; Vergara, A.; Ramos, N.; Perello, M.; Gabaldon, A.; Azancot, M.A.; et al. Enteric Budesonide in Transplant and Native IgA Nephropathy: Real-World Clinical Practice. Transpl. Int. 2022, 35, 10693. [Google Scholar] [CrossRef]
- Ismail, G.; Obrişcă, B.; Jurubiţă, R.; Andronesi, A.; Sorohan, B.; Vornicu, A.; Sinescu, I.; Hârza, M. Budesonide versus systemic corticosteroids in IgA Nephropathy: A retrospective, propensity-matched comparison. Medicine 2020, 99, e21000. [Google Scholar] [CrossRef]
- Maillard, N.; Wyatt, R.J.; Julian, B.A.; Kiryluk, K.; Gharavi, A.; Fremeaux-Bacchi, V.; Novak, J. Current Understanding of the Role of Complement in IgA Nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1503–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, A.; Rastaldi, M.P.; Calvaresi, N.; Oortwijn, B.D.; Schlagwein, N.; van Gijlswijk-Janssen, D.J.; Stahl, G.L.; Matsushita, M.; Fujita, T.; van Kooten, C.; et al. Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. J. Am. Soc. Nephrol. 2006, 17, 1724–1734. [Google Scholar] [CrossRef] [Green Version]
- Stangou, M.; Alexopoulos, E.; Pantzaki, A.; Leonstini, M.; Memmos, D. C5b-9 glomerular deposition and tubular alpha3beta1-integrin expression are implicated in the development of chronic lesions and predict renal function outcome in immunoglobulin A nephropathy. Scand. J. Urol. Nephrol. 2008, 42, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Hu, Z.; Wang, Y.; Hu, D.; Yang, Q.; Li, Y.; Dai, W.; Zhu, F.; Yang, J.; Wang, M.; et al. Severe glomerular C3 deposition indicates severe renal lesions and a poor prognosis in patients with immunoglobulin A nephropathy. Histopathology 2021, 78, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Zhu, Y.; Wang, X.; Ren, J.; Guo, H.; Huang, B.; Wang, S.; Wang, P.; Liu, Y.; Liu, Y.; et al. Predictive prognostic value of glomerular C3 deposition in IgA nephropathy. J. Nephrol. 2023, 36, 495–505. [Google Scholar] [CrossRef]
- Bruchfeld, A.; Magin, H.; Nachman, P.; Parikh, S.; Lafayette, R.; Potarca, A.; Miao, S.; Bekker, P. C5a receptor inhibitor avacopan in immunoglobulin A nephropathy-an open-label pilot study. Clin. Kidney J. 2022, 15, 922–928. [Google Scholar] [CrossRef]
- Barratt, J.; Rovin, B.; Zhang, H.; Kashihara, N.; Maes, B.; Rizk, D.; Trimarchi, H.; Sprangers, B.; Meier, M.; Kollins, D.; et al. POS-546 efficacy and safety of iptacopan in iga nephropathy: Results of a randomized double-blind placebo-controlled phase 2 study at 6 months. Kidney Int. Rep. 2022, 7, S236. [Google Scholar] [CrossRef]
- Lafayette, R.A.; Rovin, B.H.; Reich, H.N.; Tumlin, J.A.; Floege, J.; Barratt, J. Safety, Tolerability and Efficacy of Narsoplimab, a Novel MASP-2 Inhibitor for the Treatment of IgA Nephropathy. Kidney Int. Rep. 2020, 5, 2032–2041. [Google Scholar] [CrossRef]
- Samy, E.; Wax, S.; Huard, B.; Hess, H.; Schneider, P. Targeting BAFF and APRIL in systemic lupus erythematosus and other antibody-associated diseases. Int. Rev. Immunol. 2017, 36, 3–19. [Google Scholar] [CrossRef]
- Zhai, Y.-L.; Zhu, L.; Shi, S.-F.; Liu, L.-J.; Lv, J.-C.; Zhang, H. Increased APRIL Expression Induces IgA1 Aberrant Glycosylation in IgA Nephropathy. Medicine 2016, 95, e3099. [Google Scholar] [CrossRef] [PubMed]
- Mathur, M.; Barratt, J.; Suzuki, Y.; Engler, F.; Pasetti, M.F.; Yarbrough, J.; Sloan, S.; Oldach, D. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of VIS649 (Sibeprenlimab), an APRIL-Neutralizing IgG2 Monoclonal Antibody, in Healthy Volunteers. Kidney Int. Rep. 2022, 7, 993–1003. [Google Scholar] [CrossRef]
- Barratt, J.; Tumlin, J.; Suzuki, Y.; Kao, A.; Aydemir, A.; Pudota, K.; Jin, H.; Gühring, H.; Appel, G. Randomized Phase II JANUS Study of Atacicept in Patients with IgA Nephropathy and Persistent Proteinuria. Kidney Int. Rep. 2022, 7, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Liu, L.; Hao, C.; Li, G.; Fu, P.; Xing, G.; Zheng, H.; Chen, N.; Wang, C.; Luo, P.; et al. Randomized Phase 2 Trial of Telitacicept in Patients with IgA Nephropathy with Persistent Proteinuria. Kidney Int. Rep. 2023, 8, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Lafayette, R.A.; Canetta, P.A.; Rovin, B.H.; Appel, G.B.; Novak, J.; Nath, K.A.; Sethi, S.; Tumlin, J.A.; Mehta, K.; Hogan, M.; et al. A Randomized, Controlled Trial of Rituximab in IgA Nephropathy with Proteinuria and Renal Dysfunction. J. Am. Soc. Nephrol. 2017, 28, 1306–1313. [Google Scholar] [CrossRef] [Green Version]
- Hartono, C.; Chung, M.; Perlman, A.S.; Chevalier, J.M.; Serur, D.; Seshan, S.V.; Muthukumar, T. Bortezomib for Reduction of Proteinuria in IgA Nephropathy. Kidney Int. Rep. 2018, 3, 861–866. [Google Scholar] [CrossRef] [Green Version]
- Raina, R.; Chauvin, A.; Chakraborty, R.; Nair, N.; Shah, H.; Krishnappa, V.; Kusumi, K. The Role of Endothelin and Endothelin Antagonists in Chronic Kidney Disease. Kidney Dis. 2020, 6, 22–34. [Google Scholar] [CrossRef]
- Kohan, D.E.; Barton, M. Endothelin and endothelin antagonists in chronic kidney disease. Kidney Int. 2014, 86, 896–904. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Medication | Initial Dose | Taper | Total Exposure |
|---|---|---|---|---|
| TESTING 2022 (Lv) [36] | Methylprednisolone | 0.4 mg/kg orally once daily (maximum dose: 32 mg/day) for 2 months | Reduce daily dose by 4 mg every month for 4 months | 6 months |
| TESTING 2017 (Lv) [37] | Methylprednisolone | 0.6 to 0.8 mg/kg orally once daily (maximum dose: 48 mg/day) for 2 months | Reduce daily dose by 8 mg every month for 4 months | 6 months |
| Manno et al. [41] | Prednisone | 1 mg/kg orally per day (maximum dose: 75 mg/day) for 2 months | Reduce daily dose by 0.2 mg/kg every month for 4 months | 6 months |
| Lv et al. [42] | Prednisone | 0.8 to 1 mg/kg orally per day for 2 months | Reduce daily dose by 5 to 10 mg every 2 weeks for 4 months | 6 months |
| Pozzi et al. [43] STOP-IgA (Rauen) [44] | Methylprednisolone (IV) and Prednisolone/prednisone (oral) | Methylprednisolone 1 g IV for 3 days at the start of months 1, 3, and 5 and Prednisolone or prednisone 0.5 mg/kg orally every other day on remaining days for 6 months | None | 6 months |
| NEFIGAN (Fellström) [45] | TRF-budesonide | 16 mg orally daily for 9 months | Reduce dose to 8 mg once daily for 2 weeks, then discontinue | 9 months |
| Agent | Phase | Registration No | Mechanism of Action |
|---|---|---|---|
| Inhibition of Immune Complex-Activated Complement Activity | |||
| Avacopan | II | NCT02384317 | anti-C5a receptor antagonist |
| Ravulizumab | II | NCT04564339 | long-acting C5-blocking antibody |
| Cemdisiran | II | NCT03841448 | small interfering RNA-targeting C5 |
| APL-2 | II | NCT03453619 | cyclic peptide inhibitor of C3 and C3b |
| Iptacopan | III | NCT04578834 | small-molecule inhibitor of complement factor B |
| IONIS-FB-LRx | II | NCT04014335 | antisense inhibitor of complement factor B messenger ribonucleic acid |
| Narsoplimab | III | NCT03608033 | human monoclonal antibody against (MASP-2) |
| Inhibition of BAFF/APRIL Signaling | |||
| Blisibimod | II/III | NCT02062684 | monoclonal antibody against both soluble and membrane BAFF |
| Sibeprenlimab | NCT05248646 | monoclonal IgG2κ antibody targeting APRIL | |
| BION-1301 | I/II | NCT03945318 | monoclonal IgG4 antibody targeting APRIL |
| Atacicept | IIb | NCT04716231 | BAFF/APRIL dual inhibitor |
| Telitacicept | II | NCT04905212 | BAFF/APRIL dual inhibitor |
| Plasma Cell Depletion | |||
| Felzartamab | II | NCT05065970 | monoclonal IgG1 antibody targeting CD38 |
| Bortezomib | NA | NCT05383547 | proteasome inhibitor that depletes plasma cells |
| Inhibition of Endothelin A Receptor and Angiotensin II Subtype 1 Receptor | |||
| Sparsentan | III | NCT03762850 | endothelin A receptor and angiotensin II subtype 1 receptor inhibitor |
| Atrasentan | III | NCT04573478 | endothelin A receptor antagonist |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrou, D.; Kalogeropoulos, P.; Liapis, G.; Lionaki, S. IgA Nephropathy: Current Treatment and New Insights. Antibodies 2023, 12, 40. https://doi.org/10.3390/antib12020040
Petrou D, Kalogeropoulos P, Liapis G, Lionaki S. IgA Nephropathy: Current Treatment and New Insights. Antibodies. 2023; 12(2):40. https://doi.org/10.3390/antib12020040
Chicago/Turabian StylePetrou, Dimitra, Petros Kalogeropoulos, George Liapis, and Sophia Lionaki. 2023. "IgA Nephropathy: Current Treatment and New Insights" Antibodies 12, no. 2: 40. https://doi.org/10.3390/antib12020040