
Antibody-Based Approaches to Target Pancreatic Tumours


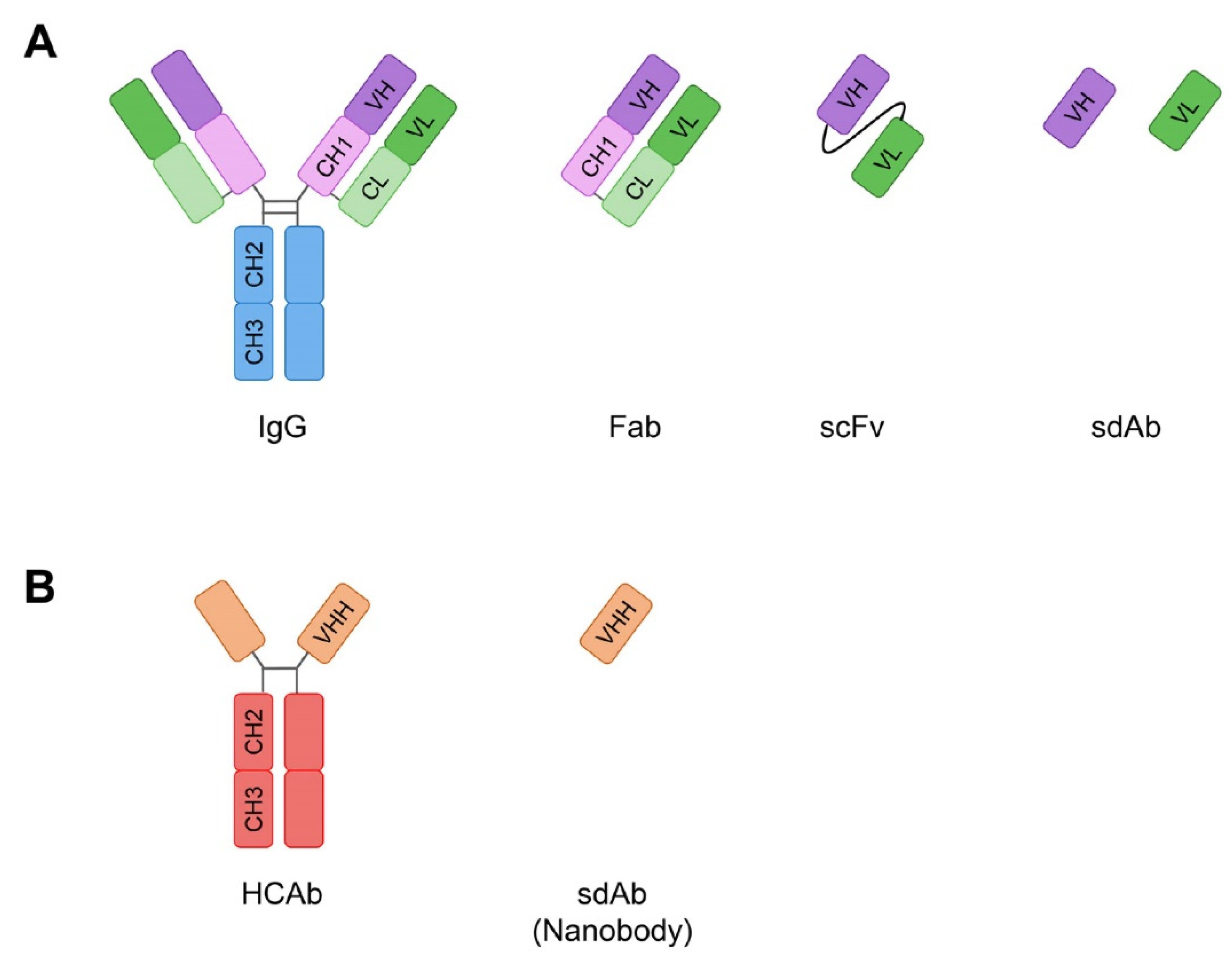{kind=link}
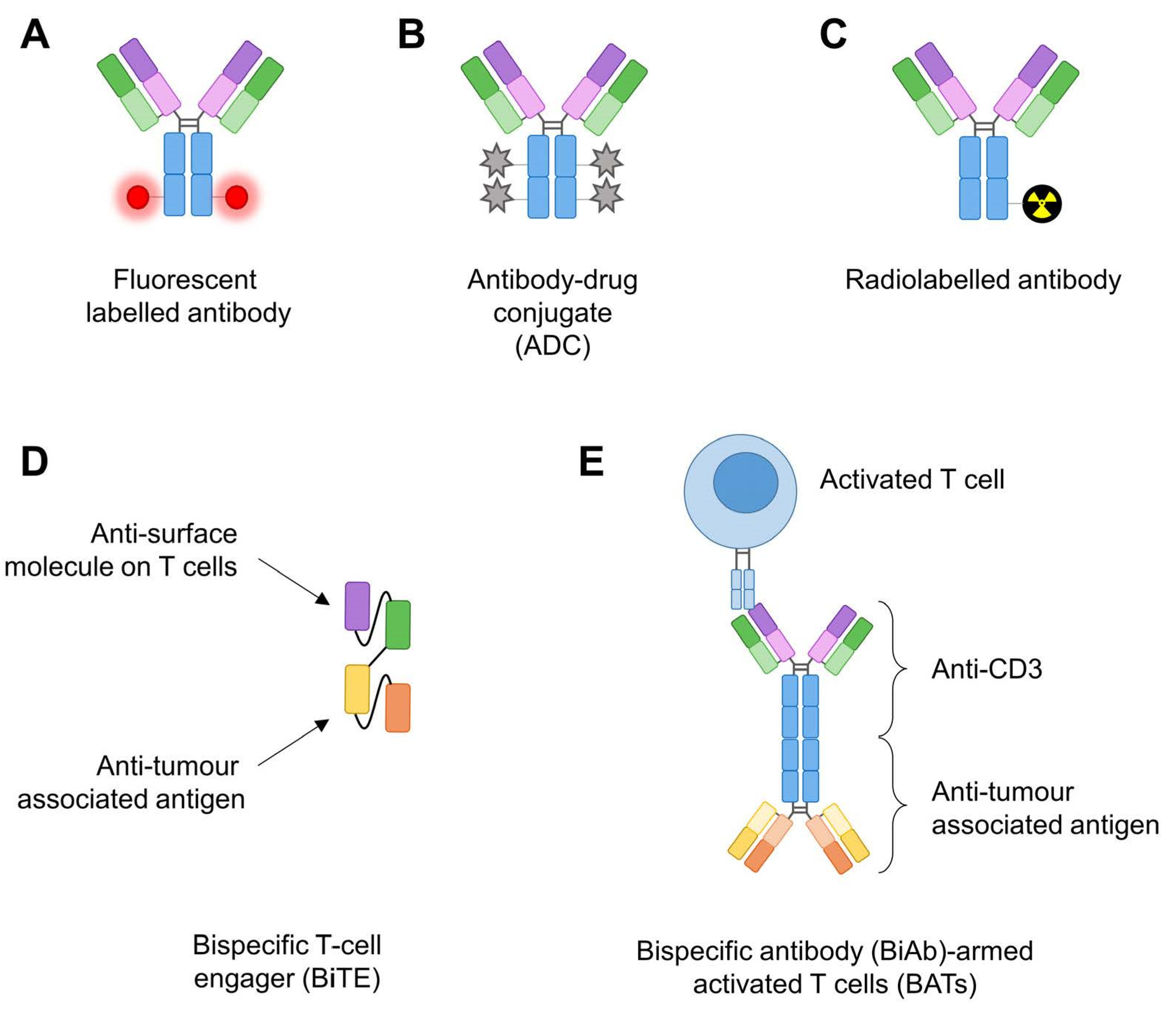{kind=link}
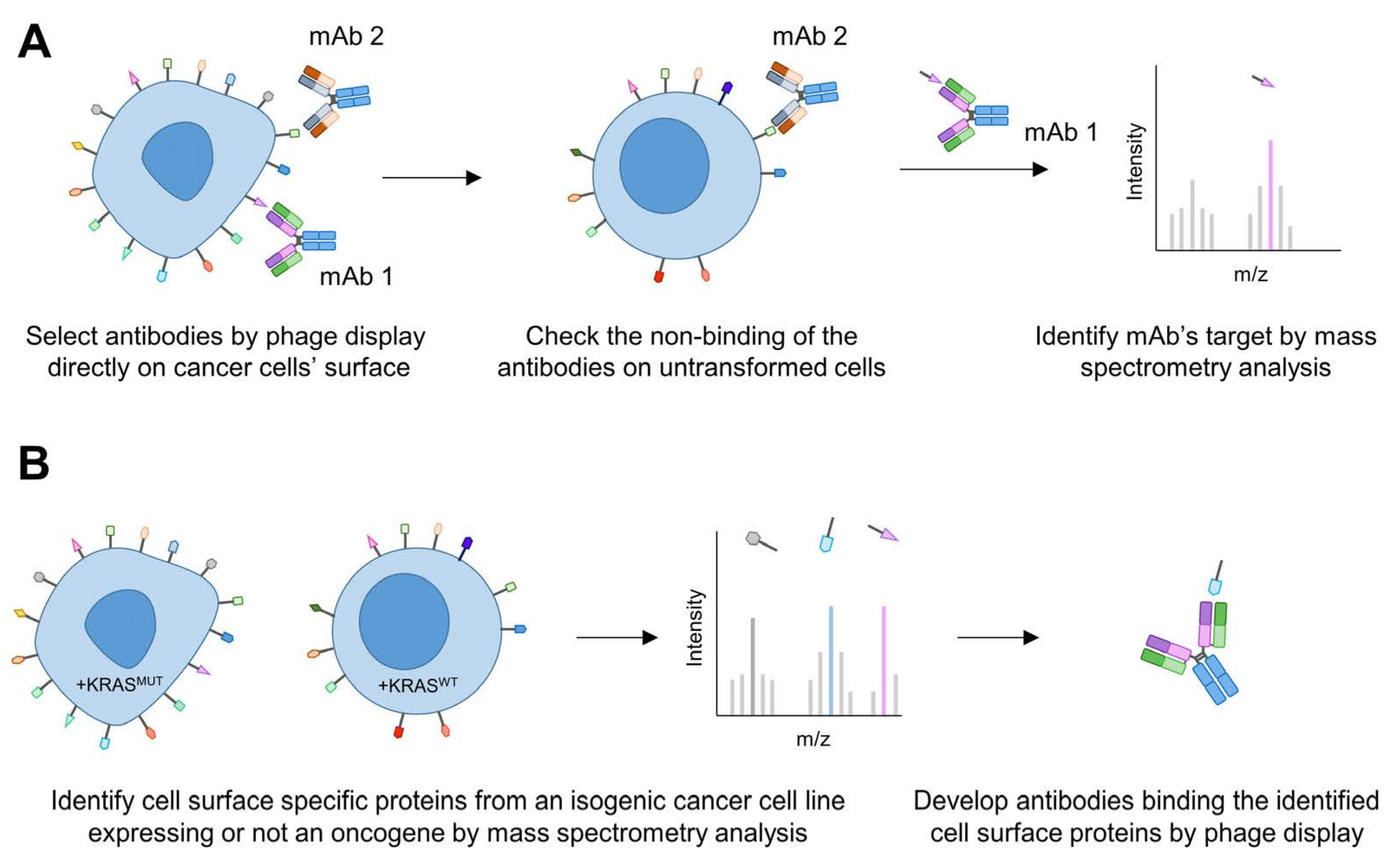{kind=link}
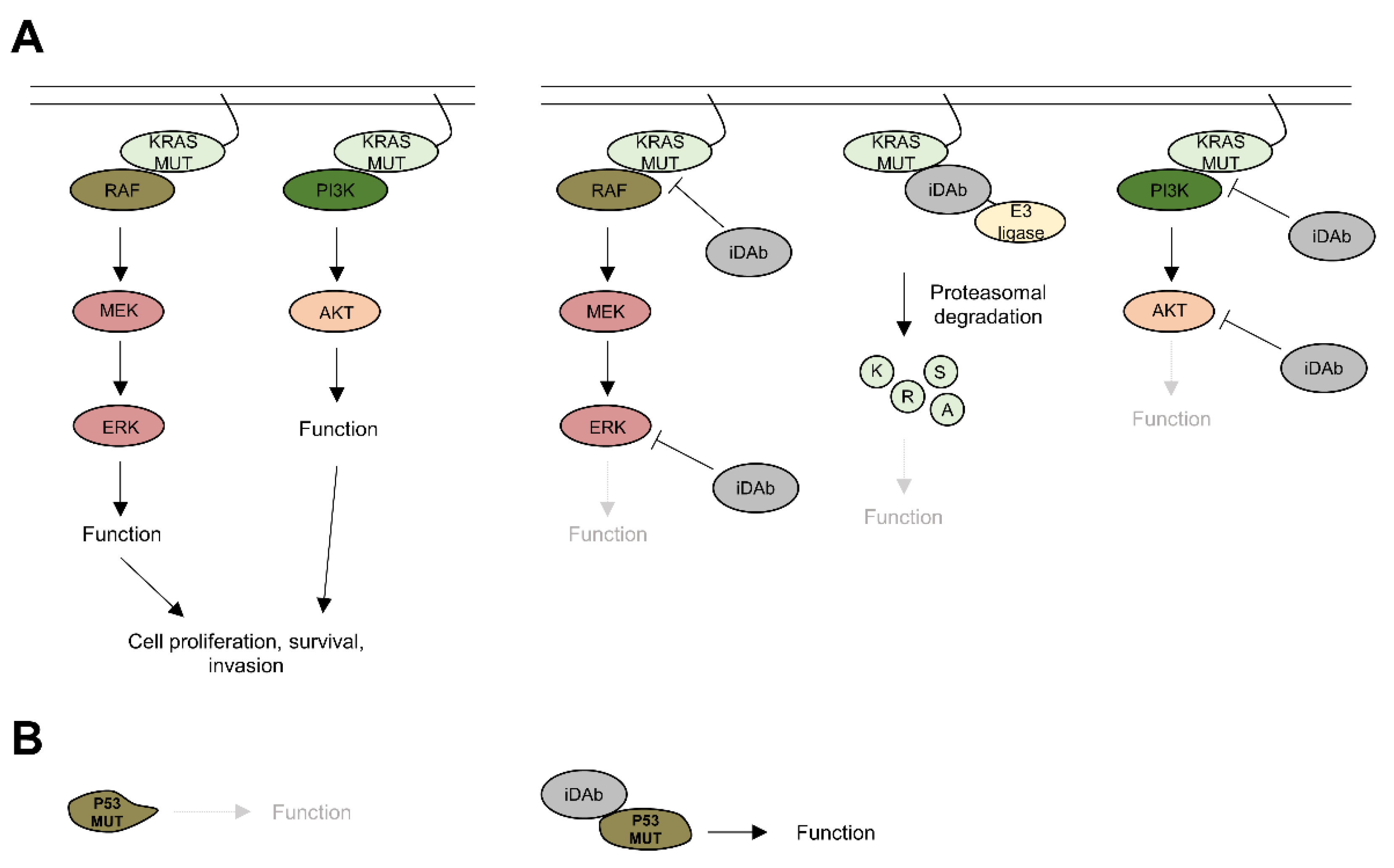{kind=link}
Abstract
:1. Introduction
2. Targeting the Surfaceome of Pancreatic Tumours
2.1. EGFR
2.2. Mesothelin
2.3. Mucins
2.4. CEA
2.5. Exploring the Surfaceome for the Discovery of Novel Targets and Biomarkers
3. Targeting Immune Checkpoints
4. Targeting Intracellular Proteins
4.1. KRAS
4.2. AKT
4.3. ERK
4.4. Alternative Strategies
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Christine Cripps, M.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Zhang, C.; Ötjengerdes, R.M.; Roewe, J.; Mejias-Estevez, R.; Marschall, A.L.J. Applying Antibodies Inside Cells: Principles and Recent Advances in Neurobiology, Virology and Oncology. BioDrugs 2020, 34, 435–462. [Google Scholar] [CrossRef] [Green Version]
- Weiner, L.M.; Surana, R.; Wang, S. Monoclonal antibodies: Versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010, 10, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Parakh, S.; Lee, S.T.; Gan, H.K.; Scott, A.M. Radiolabeled Antibodies for Cancer Imaging and Therapy. Cancers 2022, 14, 1454. [Google Scholar] [CrossRef] [PubMed]
- Dobson, L.; Reményi, I.; Tusnády, G.E. The human transmembrane proteome. Biol. Direct 2015, 10, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell. Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef]
- Overholser, J.P.; Prewett, M.C.; Hooper, A.T.; Waksal, H.W.; Hicklin, D.J. Epidermal growth factor receptor blockade by antibody IMC-C225 inhibits growth of a human pancreatic carcinoma xenograft in nude mice. Cancer 2000, 89, 74–82. [Google Scholar] [CrossRef]
- Graham, J.; Muhsin, M.; Kirkpatrick, P. Cetuximab . Nat. Rev. Drug. Discov. 2004, 3, 549–550. [Google Scholar]
- Bruns, C.J.; Harbison, M.T.; Davis, D.W.; A Portera, C.; Tsan, R.; McConkey, D.J.; Evans, D.B.; Abbruzzese, J.L.; Hicklin, D.J.; Radinsky, R. Epidermal growth factor receptor blockade with C225 plus gemcitabine results in regression of human pancreatic carcinoma growing orthotopically in nude mice by antiangiogenic mechanisms. Clin. Cancer Res. 2000, 6, 1936–1948. [Google Scholar]
- Philip, P.A.; Benedetti, J.; Corless, C.L.; Wong, R.; O’Reilly, E.M.; Flynn, P.J.; Rowland, K.M.; Atkins, J.N.; Mirtsching, B.C.; Rivkin, S.E.; et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J. Clin. Oncol. 2010, 28, 3605–3610. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.; van den Berg, N.S.; Martin, B.A.; Nishio, N.; Hart, Z.P.; van Keulen, S.; Fakurnejad, S.; Chirita, S.U.; Raymundo, R.C.; Yi, G.; et al. Tumour-specific fluorescence-guided surgery for pancreatic cancer using panitumumab-IRDye800CW: A phase 1 single-centre, open-label, single-arm, dose-escalation study. Lancet Gastroenterol. Hepatol. 2020, 5, 753–764. [Google Scholar] [CrossRef]
- Li, Z.; Wang, M.; Yao, X.; Luo, W.; Qu, Y.; Yu, D.; Li, X.; Fang, J.; Huang, C. Development of a Novel EGFR-Targeting Antibody-Drug Conjugate for Pancreatic Cancer Therapy. Target. Oncol. 2019, 14, 93–105. [Google Scholar] [CrossRef] [PubMed]
- McDaid, W.J.; Greene, M.K.; Johnston, M.C.; Pollheimer, E.; Smyth, P.; McLaughlin, K.; Van Schaeybroeck, S.; Straubinger, R.M.; Longley, D.B.; Scott, C.J. Repurposing of Cetuximab in antibody-directed chemotherapy-loaded nanoparticles in EGFR therapy-resistant pancreatic tumours. Nanoscale 2019, 11, 20261–20273. [Google Scholar] [CrossRef] [PubMed]
- Reusch, U.; Sundaram, M.; Davol, P.A.; Olson, S.D.; Davis, J.B.; Demel, K.; Nissim, J.; Rathore, R.; Liu, P.Y.; Lum, L.G. Anti-CD3 x anti-epidermal growth factor receptor (EGFR) bispecific antibody redirects T-cell cytolytic activity to EGFR-positive cancers in vitro and in an animal model. Clin. Cancer Res. 2006, 12, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lum, L.G.; Thakur, A.; Choi, M.; Deol, A.; Kondadasula, V.; Schalk, D.; Fields, K.; Dufrense, M.; Philip, P.; Dyson, G.; et al. Clinical and immune responses to anti-CD3 x anti-EGFR bispecific antibody armed activated T cells (EGFR BATs) in pancreatic cancer patients. OncoImmunology 2020, 9, 1773201. [Google Scholar] [CrossRef]
- Blasco, M.T.; Navas, C.; Martín-Serrano, G.; Graña-Castro, O.; Lechuga, C.G.; Martín-Díaz, L.; Djurec, M.; Li, J.; Morales-Cacho, L.; Esteban-Burgos, L.; et al. Complete Regression of Advanced Pancreatic Ductal Adenocarcinomas upon Combined Inhibition of EGFR and C-RAF. Cancer Cell 2019, 35, 573–587. [Google Scholar] [CrossRef] [Green Version]
- Bera, T.K.; Pastan, I. Mesothelin Is Not Required for Normal Mouse Development or Reproduction. Mol. Cell. Biol. 2000, 20, 2902–2906. [Google Scholar] [CrossRef] [Green Version]
- Argani, P.; Iacobuzio-Donahue, C.; Ryu, B.; Rosty, C.; Goggins, M.; E Wilentz, R.; Murugesan, S.R.; Leach, S.D.; Jaffee, E.; Yeo, C.J.; et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: Identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin. Cancer Res. 2001, 7, 3862–3868. [Google Scholar]
- Chowdhury, P.S.; Vasmatzis, G.; Beers, R.; Lee, B.; Pastan, I. Improved stability and yield of a Fv-toxin fusion protein by computer design and protein engineering of the Fv. J. Mol. Biol. 1998, 281, 917–928. [Google Scholar] [CrossRef]
- Hassan, R.; Williams-Gould, J.; Steinberg, S.M.; Liewehr, D.J.; Yokokawa, J.; Tsang, K.Y.; Surawski, R.J.; Scott, T.; Camphausen, K.; Xin, X.; et al. Tumor-Directed Radiation and the Immunotoxin SS1P in the Treatment of Mesothelin-Expressing Tumor Xenografts. Clin. Cancer Res. 2006, 12, 4983–4988. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xiang, L.; Hassan, R.; Paik, C.H.; Carrasquillo, J.A.; Jang, B.-S.; Le, N.; Ho, M.; Pastan, I. Synergistic Antitumor Activity of Taxol and Immunotoxin SS1P in Tumor-Bearing Mice. Clin. Cancer Res. 2006, 12, 4695–4701. [Google Scholar] [CrossRef] [Green Version]
- Hassan, R.; Broaddus, V.C.; Wilson, S.; Liewehr, D.J.; Zhang, J. Anti–Mesothelin Immunotoxin SS1P in Combination with Gemcitabine Results in Increased Activity against Mesothelin-Expressing Tumor Xenografts. Clin. Cancer Res. 2007, 13, 7166–7171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, R.; Bullock, S.; Premkumar, A.; Kreitman, R.J.; Kindler, H.; Willingham, M.C.; Pastan, I. Phase I Study of SS1P, a Recombinant Anti-Mesothelin Immunotoxin Given as a Bolus I.V. Infusion to Patients with Mesothelin-Expressing Mesothelioma, Ovarian, and Pancreatic Cancers. Clin. Cancer Res. 2007, 13, 5144–5149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossoba, M.E.; Onda, M.; Taylor, J.; Massey, P.R.; Treadwell, S.; Sharon, E.; Hassan, R.; Pastan, I.; Fowler, D.H. Pentostatin Plus Cyclophosphamide Safely and Effectively Prevents Immunotoxin Immunogenicity in Murine Hosts. Clin. Cancer Res. 2011, 17, 3697–3705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollevoet, K.; Mason-Osann, E.; Liu, X.F.; Imhof-Jung, S.; Niederfellner, G.; Pastan, I. In vitro and in vivo activity of the low-immunogenic antimesothelin immunotoxin RG7787 in pancreatic cancer. Mol. Cancer Ther. 2014, 13, 2040–2049. [Google Scholar] [CrossRef] [Green Version]
- Kolyvas, E.; Rudloff, M.; Poruchynsky, M.; Landsman, R.; Hollevoet, K.; Venzon, D.; Alewine, C. Mesothelin-targeted immunotoxin RG7787 has synergistic anti-tumor activity when combined with taxanes. Oncotarget 2016, 8, 9189–9199. [Google Scholar] [CrossRef] [Green Version]
- Alewine, C.; Ahmad, M.; Peer, C.J.; Hu, Z.I.; Lee, M.-J.; Yuno, A.; Kindrick, J.D.; Thomas, A.; Steinberg, S.M.; Trepel, J.B.; et al. Phase I/II Study of the Mesothelin-targeted Immunotoxin LMB-100 with Nab-Paclitaxel for Patients with Advanced Pancreatic Adenocarcinoma. Clin. Cancer Res. 2020, 26, 828–836. [Google Scholar] [CrossRef]
- Hassan, R.; Blumenschein, G.R., Jr.; Moore, K.N.; Santin, A.D.; Kindler, H.L.; Nemunaitis, J.J.; Seward, S.M.; Thomas, A.; Kim, S.K.; Rajagopalan, P.; et al. First-in-Human, Multicenter, Phase I Dose-Escalation and Expansion Study of Anti-Mesothelin Antibody-Drug Conjugate Anetumab Ravtansine in Advanced or Metastatic Solid Tumors. J. Clin. Oncol. 2020, 38, 1824–1835. [Google Scholar] [CrossRef]
- Hassan, R.; Ebel, W.; Routhier, E.L.; Patel, R.; Kline, J.B.; Zhang, J.; Chao, Q.; Jacob, S.; Turchin, H.; Gibbs, L.; et al. Preclinical evaluation of MORAb-009, a chimeric antibody targeting tumor-associated mesothelin. Cancer Immun. 2007, 7, 20. [Google Scholar]
- Gubbels, J.A.; Belisle, J.; Onda, M.; Rancourt, C.; Migneault, M.; Ho, M.; Bera, T.K.; Connor, J.; Sathyanarayana, B.K.; Lee, B.; et al. Mesothelin-MUC16 binding is a high affinity, N-glycan dependent interaction that facilitates peritoneal metastasis of ovarian tumors. Mol. Cancer 2006, 5, 50. [Google Scholar] [CrossRef] [Green Version]
- Mizukami, T.; Kamachi, H.; Fujii, Y.; Matsuzawa, F.; Einama, T.; Kawamata, F.; Kobayashi, N.; Hatanaka, Y.; Taketomi, A. The anti-mesothelin monoclonal antibody amatuximab enhances the anti-tumor effect of gemcitabine against mesothelin-high expressing pancreatic cancer cells in a peritoneal metastasis mouse model. Oncotarget 2018, 9, 33844–33852. [Google Scholar] [CrossRef] [Green Version]
- Hassan, R.; Cohen, S.J.; Phillips, M.; Pastan, I.; Sharon, E.; Kelly, R.J.; Schweizer, C.; Weil, S.; Laheru, D. Phase I Clinical Trial of the Chimeric Anti-Mesothelin Monoclonal Antibody MORAb-009 in Patients with Mesothelin-Expressing Cancers. Clin. Cancer Res. 2010, 16, 6132–6138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindenberg, L.; Thomas, A.; Adler, S.; Mena, E.; Kurdziel, K.; Maltzman, J.; Wallin, B.; Hoffman, K.; Pastan, I.; Paik, C.H.; et al. Safety and biodistribution of 111In-amatuximab in patients with mesothelin expressing cancers using Single Photon Emission Computed Tomography-Computed Tomography (SPECT-CT) imaging. Oncotarget 2015, 6, 4496–4504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akce, M.; Zaidi, M.Y.; Waller, E.K.; El-Rayes, B.F.; Lesinski, G.B. The Potential of CAR T Cell Therapy in Pancreatic Cancer. Front. Immunol. 2018, 9, 2166. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.H.; Jordan, A.C.; Tooker, E.; Lacey, S.F.; Chang, R.B.; Li, Y.; Venook, A.P.; Tempero, M.; Damon, L.; Fong, L.; et al. Dual Targeting of Mesothelin and CD19 with Chimeric Antigen Receptor-Modified T Cells in Patients with Metastatic Pancreatic. Cancer Mol. Ther. 2020, 28, 2367–2378. [Google Scholar] [CrossRef]
- Jonckheere, N.; Skrypek, N.; Van Seuningen, I. Mucins and Pancreatic Cancer. Cancers 2010, 2, 1794–1812. [Google Scholar] [CrossRef] [Green Version]
- Suh, H.; Pillai, K.; Morris, D.L. Mucins in pancreatic cancer: Biological role, implications in carcinogenesis and applications in diagnosis and therapy. Am. J. Cancer Res. 2017, 7, 1372–1383. [Google Scholar]
- Jonckheere, N.; Skrypek, N.; Van Seuningen, I. Mucins and tumor resistance to chemotherapeutic drugs. Biochim. et Biophys. Acta 2014, 1846, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; You, L.; Dai, M.; Zhao, Y. Mucins in pancreatic cancer: A well-established but promising family for diagnosis, prognosis and therapy. J. Cell. Mol. Med. 2020, 24, 10279–10289. [Google Scholar] [CrossRef]
- Danielczyk, A.; Stahn, R.; Faulstich, D.; Löffler, A.; Märten, A.; Karsten, U.; Goletz, S. PankoMab: A potent new generation anti-tumour MUC1 antibody. Cancer Immunol. Immunother. 2006, 55, 1337–1347. [Google Scholar] [CrossRef]
- Fiedler, W.; DeDosso, S.; Cresta, S.; Weidmann, J.; Tessari, A.; Salzberg, M.; Dietrich, B.; Baumeister, H.; Goletz, S.; Gianni, L.; et al. A phase I study of PankoMab-GEX, a humanised glyco-optimised monoclonal antibody to a novel tumour-specific MUC1 glycopeptide epitope in patients with advanced carcinomas. Eur. J. Cancer. 2016, 63, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daiichi Sankyo Enters Worldwide Licensing Agreement with Glycotope for Gatipotuzumab Antibody Drug Conjugate. Available online: https://www.daiichisankyo.com/media/press_release/detail/index_3243.html (accessed on 1 June 2022).
- Gold, D.V.; Lew, K.; Maliniak, R.; Hernandez, M.; Cardillo, T. Characterization of monoclonal antibody PAM4 reactive with a pancreatic cancer mucin. Int. J. Cancer 1994, 57, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Gold, D.V.; Cardillo, T.; Goldenberg, D.M.; Sharkey, R.M. Localization of pancreatic cancer with radiolabeled monoclonal antibody PAM4. Crit. Rev. Oncol. Hematol. 2001, 39, 147–154. [Google Scholar] [CrossRef]
- Cardillo, T.M.; Ying, Z.; Gold, D.V. Therapeutic advantage of (90)yttrium- versus (131)iodine-labeled PAM4 antibody in experimental pancreatic cancer. Clin. Cancer Res. 2001, 7, 3186–3192. [Google Scholar] [PubMed]
- Gold, D.V.; Karanjawala, Z.; Modrak, D.E.; Goldenberg, D.M.; Hruban, R.H. PAM4-reactive MUC1 is a biomarker for early pancreatic adenocarcinoma. Clin. Cancer Res. 2007, 13, 7380–7387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørensen, A.L.; Reis, C.A.; Tarp, M.A.; Mandel, U.; Ramachandran, K.; Sankaranarayanan, V.; Schwientek, T.; Graham, R.; Taylor-Papadimitriou, J.; Hollingsworth, M.A.; et al. Chemoenzymatically synthesized multimeric Tn/STn MUC1 glycopeptides elicit cancer-specific anti-MUC1 antibody responses and override tolerance. Glycobiology 2006, 16, 96–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posey, A.D., Jr.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef] [Green Version]
- Gautam, S.K.; Kumar, S.; Cannon, A.; Hall, B.; Bhatia, R.; Nasser, M.W.; Mahapatra, S.; Batra, S.K.; Jain, M. MUC4 mucin- a therapeutic target for pancreatic ductal adenocarcinoma. Expert Opin. Ther. Targets 2017, 21, 657–669. [Google Scholar] [CrossRef]
- Bafna, S.; Kaur, S.; Momi, N.; Batra, S.K. Pancreatic cancer cells resistance to gemcitabine: The role of MUC4 mucin. Br. J. Cancer 2009, 101, 1155–1161. [Google Scholar] [CrossRef]
- Manne, A.; Esnakula, A.; Abushahin, L.; Tsung, A. Understanding the Clinical Impact of MUC5AC Expression on Pancreatic Ductal Adenocarcinoma. Cancers 2021, 13, 3059. [Google Scholar] [CrossRef]
- Sawada, T.; Nishihara, T.; Yamamoto, A.; Teraoka, H.; Yamashita, Y.; Okamura, T.; Ochi, H.; Ho, J.J.L.; Kim, Y.-S.; Hirakawa, K. Preoperative Clinical Radioimmunodetection of Pancreatic Cancer by111In-labeled Chimeric Monoclonal Antibody Nd2. Jpn. J. Cancer Res. 1999, 90, 1179–1186. [Google Scholar] [CrossRef]
- Nakata, N.; Kobashi, N.; Okumura, Y.; Sato, M.; Matono, M.; Otsuki, K.; Tanaka, A.; Hayashi, A. Radiation dosimetry and efficacy of an 89Zr/225Ac-labeled humanized anti-MUC5AC antibody. Nucl. Med. Biol. 2022, 108, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.A.; Hollandsworth, H.M.; Nishino, H.; Amirfakhri, S.; Lwin, T.M.; Lowy, A.M.; Kaur, S.; Natarajan, G.; Mallya, K.; Hoffman, R.M.; et al. Fluorescent Anti-MUC5AC Brightly Targets Pancreatic Cancer in a Patient-derived Orthotopic Xenograft. Vivo 2021, 36, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.G.; Bai, X.F.; Mao, Y.L.; Shao, Y.F.; Wu, J.X.; Shan, Y.; Wang, C.F.; Wang, J.; Tian, Y.T.; Liu, Q.; et al. The clinical value of serum CEA, CA19-9, and CA242 in the diagnosis and prognosis of pancreatic cancer. Eur. J. Surg. Oncol. 2005, 31, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Rizeq, B.; Zakaria, Z.; Ouhtit, A. Towards understanding the mechanisms of actions of carcinoembryonic antigen-related cell adhesion molecule 6 in cancer progression. Cancer Sci. 2018, 109, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dam, M.A.; Vuijk, F.A.; Stibbe, J.A.; Houvast, R.D.; Luelmo, S.A.; Crobach, S.; Shahbazi Feshtali, S.; de Geus-Oei, L.F.; Bonsing, B.A.; Sier, C.F.; et al. Overview and Future Perspectives on Tumor-Targeted Positron Emission Tomography and Fluorescence Imaging of Pancreatic Cancer in the Era of Neoadjuvant Therapy. Cancers 2021, 13, 6088. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Kishiwada, M.; Hayasaki, A.; Chipaila, J.; Maeda, K.; Noguchi, D.; Gyoten, K.; Fujii, T.; Iizawa, Y.; Tanemura, A.; et al. Role of Serum Carcinoma Embryonic Antigen (CEA) Level in Localized Pancreatic Adenocarcinoma: CEA Level Before Operation is a Significant Prognostic Indicator in Patients with Locally Advanced Pancreatic Cancer Treated With Neoadjuvant Therapy Followed by Surgical Resection: A Retrospective Analysis. Ann. Surg. 2022, 275, e698–e707. [Google Scholar]
- Knutson, S.; Raja, E.; Bomgarden, R.; Nlend, M.; Chen, A.; Kalyanasundaram, R.; Desai, S. Development and Evaluation of a Fluorescent Antibody-Drug Conjugate for Molecular Imaging and Targeted Therapy of Pancreatic Cancer. PLoS ONE 2016, 11, e0157762. [Google Scholar] [CrossRef] [Green Version]
- Lwin, T.M.; Murakami, T.; Miyake, K.; Yazaki, P.J.; Shivley, J.E.; Hoffman, R.M.; Bouvet, M. Tumor-Specific Labeling of Pancreatic Cancer Using a Humanized Anti-CEA Antibody Conjugated to a Near-Infrared Fluorophore. Ann. Surg. Oncol. 2018, 25, 1079–1085. [Google Scholar] [CrossRef]
- Gutowski, M.; Framery, B.; Boonstra, M.C.; Garambois, V.; Quenet, F.; Dumas, K.; Scherninski, F.; Cailler, F.; Vahrmeijer, A.L.; Pèlegrin, A. SGM-101: An innovative near-infrared dye-antibody conjugate that targets CEA for fluorescence-guided surgery. Surg. Oncol. 2017, 26, 153–162. [Google Scholar] [CrossRef]
- Hoogstins, C.E.S.; Boogerd, L.S.F.; Mulder, B.G.S.; Mieog, J.S.D.; Swijnenburg, R.J.; Van De Velde, C.J.H.; Farina Sarasqueta, A.; Bonsing, B.A.; Framery, B.; Pèlegrin, A.; et al. Image-Guided Surgery in Patients with Pancreatic Cancer: First Results of a Clinical Trial Using SGM-101, a Novel Carcinoembryonic Antigen-Targeting, Near-Infrared Fluorescent Agent. Ann. Surg. Oncol. 2018, 25, 3350–3357. [Google Scholar] [CrossRef] [PubMed]
- Lwin, T.M.; Turner, M.A.; Nishino, H.; Amirfakhri, S.; Hernot, S.; Hoffman, R.M.; Bouvet, M. Fluorescent Anti-CEA Nanobody for Rapid Tumor-Targeting and Imaging in Mouse Models of Pancreatic Cancer. Biomolecules 2022, 12, 711. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, G.; Akahori, Y.; Morita, M.; Sumitomo, M.; Sato, N.; Muramatsu, C.; Eguchi, K.; Matsuda, K.; Takasaki, A.; Tanaka, M.; et al. Comprehensive screening for antigens overexpressed on carcinomas via isolation of human mAbs that may be therapeutic. Proc. Natl. Acad. Sci. USA 2008, 105, 7287–7292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurosawa, G.; Sumitomo, M.; Ukai, Y.; Subere, J.; Muramatsu, C.; Eguchi, K.; Tanaka-Hashiba, M.; Sugiura, M.; Ando, M.; Sato, N.; et al. Selection and analysis of anti-cancer antibodies for cancer therapy obtained from antibody phage library. Cancer Sci. 2010, 102, 175–181. [Google Scholar] [CrossRef]
- Sugyo, A.; Tsuji, A.B.; Sudo, H.; Nagatsu, K.; Koizumi, M.; Ukai, Y.; Kurosawa, G.; Zhang, M.-R.; Kurosawa, Y.; Saga, T. Evaluation of 89Zr-Labeled Human Anti-CD147 Monoclonal Antibody as a Positron Emission Tomography Probe in a Mouse Model of Pancreatic Cancer. PLoS ONE 2013, 8, e61230. [Google Scholar] [CrossRef] [Green Version]
- Town, J.; Pais, H.; Harrison, S.; Stead, L.F.; Bataille, C.; Bunjobpol, W.; Zhang, J.; Rabbitts, T.H. Exploring the surfaceome of Ewing sarcoma identifies a new and unique therapeutic target. Proc. Natl. Acad. Sci. USA 2016, 113, 3603–3608. [Google Scholar] [CrossRef] [Green Version]
- Pais, H.; Ruggero, K.; Zhang, J.; Al-Assar, O.; Bery, N.; Bhuller, R.; Weston, V.; Kearns, P.; Mecucci, C.; Miller, A.; et al. Surfaceome interrogation using an RNA-seq approach highlights leukemia initiating cell biomarkers in an LMO2 T cell transgenic model. Sci. Rep. 2019, 9, 5760. [Google Scholar] [CrossRef]
- Martinko, A.J.; Truillet, C.; Julien, O.; Diaz, J.E.; Horlbeck, M.A.; Whiteley, G.; Blonder, J.; Weissman, J.S.; Bandyopadhyay, S.; Evans, M.J.; et al. Targeting RAS-driven human cancer cells with antibodies to upregulated and essential cell-surface proteins. eLife 2018, 7, e31098. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, A.S.; Erdjument-Bromage, H.; Tempst, P.; Craik, C.S.; Moasser, M.M. Adhesion signaling by a novel mitotic substrate of src kinases. Oncogene 2005, 24, 5333–5343. [Google Scholar] [CrossRef] [Green Version]
- Casar, B.; Rimann, I.; Kato, H.; Shattil, S.J.; Quigley, J.P.; Deryugina, E.I. In vivo cleaved CDCP1 promotes early tumor dissemination via complexing with activated beta1 integrin and induction of FAK/PI3K/Akt motility signaling. Oncogene 2014, 33, 255–268. [Google Scholar] [CrossRef] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H., Jr. A Review of Cancer Immunotherapy: From the Past to the Present, to the Future. Curr. Oncol. 2020, 27, S87–S97. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, D.J.; Finetti, P.; Lopresti, A.; Gilabert, M.; Poizat, F.; Turrini, O.; Raoul, J.L.; Delpero, J.R.; Moutardier, V.; Birnbaum, D.; et al. Prognostic value of PDL1 expression in pancreatic cancer. Oncotarget 2016, 7, 71198–71210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contardi, E.; Palmisano, G.L.; Tazzari, P.L.; Martelli, A.M.; Falà, F.; Fabbi, M.; Kato, T.; Lucarelli, E.; Donati, D.; Polito, L.; et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int. J. Cancer 2005, 117, 538–550. [Google Scholar] [CrossRef]
- Sobhani, N.; Tardiel-Cyril, D.R.; Davtyan, A.; Generali, D.; Roudi, R.; Li, Y. CTLA-4 in Regulatory T Cells for Cancer Immunotherapy. Cancers 2021, 13, 1440. [Google Scholar] [CrossRef]
- Bengsch, F.; Knoblock, D.M.; Liu, A.; McAllister, F.; Beatty, G.L. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol. Immunother. 2017, 66, 1609–1617. [Google Scholar] [CrossRef]
- Patnaik, A.; Kang, S.P.; Rasco, D.; Papadopoulos, K.P.; Elassaiss-Schaap, J.; Beeram, M.; Drengler, R.; Chen, C.; Smith, L.; Espino, G.; et al. Phase I Study of Pembrolizumab (MK-3475; Anti–PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 4286–4293. [Google Scholar] [CrossRef] [Green Version]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Schmiechen, Z.C.; Stromnes, I.M. Mechanisms Governing Immunotherapy Resistance in Pancreatic Ductal Adenocarcinoma. Front Immunol. 2020, 11, 613815. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Łuksza, M.; Sethna, Z.M.; Rojas, L.A.; Lihm, J.; Bravi, B.; Elhanati, Y.; Soares, K.; Amisaki, M.; Dobrin, A.; Hoyos, D.; et al. Neoantigen quality predicts immunoediting in survivors of pancreatic cancer. Nature 2022, 606, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Hendifar, A.; Tuli, R.; Chuang, J.; Cho, M.; Chung, V.; Li, D.; Salgia, R. Combination systemic therapies with immune checkpoint inhibitors in pancreatic cancer: Overcoming resistance to single-agent checkpoint blockade. Clin. Transl. Med. 2018, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2017, 118, 312–324. [Google Scholar] [CrossRef] [Green Version]
- Gabitass, R.F.; Annels, N.E.; Stocken, D.D.; Pandha, H.A.; Middleton, G.W. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol. Immunother. 2011, 60, 1419–1430. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Li, Y.; Niu, Z.; Zong, Y.; Wang, M.; Yao, L.; Lu, Z.; Liao, Q.; Zhao, Y. Atorvastatin (Lipitor) attenuates the effects of aspirin on pancreatic cancerogenesis and the chemotherapeutic efficacy of gemcitabine on pancreatic cancer by promoting M2 polarized tumor associated macrophages. J. Exp. Clin. Cancer Res. 2016, 35, 33. [Google Scholar] [CrossRef] [Green Version]
- Amit, M.; Gil, Z. Macrophages increase the resistance of pancreatic adenocarcinoma cells to gemcitabine by upregulating cytidine deaminase. OncoImmunology 2013, 2, e27231. [Google Scholar] [CrossRef] [Green Version]
- Kamath, S.D.; Kalyan, A.; Kircher, S.; Nimeiri, H.; Fought, A.J.; Benson, A.; Mulcahy, M. Ipilimumab and Gemcitabine for Advanced Pancreatic Cancer: A Phase Ib Study. Oncologist 2019, 25, e808–e815. [Google Scholar] [CrossRef] [Green Version]
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schütz, E.; Khemka, V. Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Investig. New Drugs 2018, 36, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Procureur, A.; Simonaggio, A.; Bibault, J.-E.; Oudard, S.; Vano, Y.-A. Enhance the Immune Checkpoint Inhibitors Efficacy with Radiotherapy Induced Immunogenic Cell Death: A Comprehensive Review and Latest Developments. Cancers 2021, 13, 678. [Google Scholar] [CrossRef] [PubMed]
- Daguenet, E.; Louati, S.; Wozny, A.-S.; Vial, N.; Gras, M.; Guy, J.-B.; Vallard, A.; Rodriguez-Lafrasse, C.; Magné, N. Radiation-induced bystander and abscopal effects: Important lessons from preclinical models. Br. J. Cancer 2020, 123, 339–348. [Google Scholar] [CrossRef]
- Sharabi, A.B.; Nirschl, C.J.; Kochel, C.M.; Nirschl, T.R.; Francica, B.J.; Velarde, E.; Deweese, T.L.; Drake, C.G. Stereotactic Radiation Therapy Augments Antigen-Specific PD-1-Mediated Antitumor Immune Responses via Cross-Presentation of Tumor Antigen. Cancer Immunol. Res. 2015, 3, 345–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Probst, H.C.; Vuong, V.; Landshammer, A.; Muth, S.; Yagita, H.; Schwendener, R.; Pruschy, M.; Knuth, A.; van den Broek, M. Radiotherapy promotes tumor-specific effector CD8+ T cells via dendritic cell activation. J. Immunol. 2012, 189, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.-X. Irradiation and anti–PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef]
- Park, S.S.; Dong, H.; Liu, X.; Harrington, S.M.; Krco, C.J.; Grams, M.P.; Mansfield, A.S.; Furutani, K.M.; Olivier, K.R.; Kwon, E.D. PD-1 Restrains Radiotherapy-Induced Abscopal Effect. Cancer Immunol. Res. 2015, 3, 610–619. [Google Scholar] [CrossRef] [Green Version]
- Azad, A.; Yin Lim, S.; D’Costa, Z.; Jones, K.; Diana, A.; Sansom, O.J.; Kruger, P.; Liu, S.; McKenna, W.G.; Dushek, O.; et al. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol. Med. 2017, 9, 167–180. [Google Scholar] [CrossRef]
- Parikh, A.R.; Szabolcs, A.; Allen, J.N.; Clark, J.W.; Wo, J.Y.; Raabe, M.; Thel, H.; Hoyos, D.; Mehta, A.; Arshad, S.; et al. Radiation therapy enhances immunotherapy response in microsatellite stable colorectal and pancreatic adenocarcinoma in a phase II trial. Nat. Cancer 2021, 2, 1124–1135. [Google Scholar] [CrossRef]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Jackson, V.; Hamada, H.; Pardoll, D.; Mulligan, R.C. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar] [CrossRef] [Green Version]
- Soares, K.C.; Rucki, A.A.; Wu, A.A.; Olino, K.; Xiao, Q.; Chai, Y.; Wamwea, A.; Bigelow, E.; Lutz, E.; Liu, L.; et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J. Immunother. 2015, 38, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.A., Jr.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.A.; Bever, K.M.; Ho, W.J.; Fertig, E.J.; Niu, N.; Zheng, L.; Parkinson, R.M.; Durham, J.N.; Onners, B.L.; Ferguson, A.K.; et al. A Phase II Study of Allogeneic GM-CSF–Transfected Pancreatic Tumor Vaccine (GVAX) with Ipilimumab as Maintenance Treatment for Metastatic Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 5129–5139. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, V.P.; Rojas, L.A.; Sethna, Z.; Soares, K.; Derhovanessian, E.; Mueller, F.; Yadav, M.; Basturk, O.; Gonen, M.; Wei, A.C.-C.; et al. Phase I trial of adjuvant autogene cevumeran, an individualized mRNA neoantigen vaccine, for pancreatic ductal adenocarcinoma. J. Clin. Oncol. 2022, 40, 2516. [Google Scholar] [CrossRef]
- Kraehenbuehl, L.; Weng, C.-H.; Eghbali, S.; Wolchok, J.D.; Merghoub, T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat. Rev. Clin. Oncol. 2021, 19, 37–50. [Google Scholar] [CrossRef]
- Ino, Y.; Oguro, S.; Yamazaki-Itoh, R.; Hori, S.; Shimada, K.; Hiraoka, N. Reliable evaluation of tumor-infiltrating lymphocytes in pancreatic cancer tissue biopsies. Oncotarget 2019, 10, 1149–1159. [Google Scholar] [CrossRef]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Jullien, D.; Vignard, J.; Fedor, Y.; Bery, N.; Olichon, A.; Crozatier, M.; Erard, M.; Cassard, H.; Ducommun, B.; Salles, B.; et al. Chromatibody, a novel non-invasive molecular tool to explore and manipulate chromatin in living cells. J. Cell Sci. 2016, 129, 2673–2683. [Google Scholar] [CrossRef] [Green Version]
- Panza, P.; Maier, J.; Schmees, C.; Rothbauer, U.; Söllner, C. Live imaging of endogenous protein dynamics in zebrafish using chromobodies. Development 2015, 142, 1879–1884. [Google Scholar] [CrossRef] [Green Version]
- Bery, N.; Legg, S.; Debreczeni, J.; Breed, J.; Embrey, K.; Stubbs, C.; Kolasinska-Zwierz, P.; Barrett, N.; Marwood, R.; Watson, J.; et al. KRAS-specific inhibition using a DARPin binding to a site in the allosteric lobe. Nat. Commun. 2019, 10, 2607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Williams, R.L.; Rabbitts, T.H. Tumour prevention by a single antibody domain targeting the interaction of signal transduction proteins with RAS. EMBO J. 2007, 26, 3250–3259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bery, N.; Keller, L.; Soulié, M.; Gence, R.; Iscache, A.-L.; Cherier, J.; Cabantous, S.; Sordet, O.; Lajoie-Mazenc, I.; Pedelacq, J.-D.; et al. A Targeted Protein Degradation Cell-Based Screening for Nanobodies Selective toward the Cellular RHOB GTP-Bound Conformation. Cell Chem. Biol. 2019, 26, 1544–1558. [Google Scholar] [CrossRef] [PubMed]
- Bery, N.; Miller, A.; Rabbitts, T. A potent KRAS macromolecule degrader specifically targeting tumours with mutant KRAS. Nat. Commun. 2020, 11, 3233. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug. Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Rabbitts, T.H. Intrabodies based on intracellular capture frameworks that bind the RAS protein with high affinity and impair oncogenic transformation. EMBO J. 2003, 22, 1025–1035. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Rabbitts, T.H. Interfering with RAS–effector protein interactions prevent RAS-dependent tumour initiation and causes stop–start control of cancer growth. Oncogene 2010, 29, 6064–6070. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.M.; Choi, D.K.; Jung, K.; Bae, J.; Kim, J.S.; Park, S.W.; Song, K.H.; Kim, Y.S. Antibody targeting intracellular oncogenic Ras mutants exerts anti-tumour effects after systemic administration. Nat. Commun. 2017, 8, 15090. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.-M.; Kim, J.-S.; Park, S.-W.; Jun, S.-Y.; Kweon, H.-J.; Choi, D.-K.; Lee, D.; Cho, Y.B.; Kim, Y.-S. Direct targeting of oncogenic RAS mutants with a tumor-specific cytosol-penetrating antibody inhibits RAS mutant–driven tumor growth. Sci. Adv. 2020, 6, eaay2174. [Google Scholar] [CrossRef] [Green Version]
- Bellacosa, A.; Kumar, C.C.; Di Cristofano, A.; Testa, J.R. Activation of AKT Kinases in Cancer: Implications for Therapeutic Targeting. Adv. Cancer Res. 2005, 94, 29–86. [Google Scholar] [CrossRef]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.-H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, I.; Edl, J.; Biswas, S.; Lin, P.C.; Mernaugh, R.; Arteaga, C.L. Proapoptotic Activity of Cell-Permeable Anti-Akt Single-Chain Antibodies. Cancer Res. 2005, 65, 2815–2824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merckaert, T.; Zwaenepoel, O.; Gevaert, K.; Gettemans, J. Development and characterization of protein kinase B/AKT isoform-specific nanobodies. PLoS ONE 2020, 15, e0240554. [Google Scholar] [CrossRef] [PubMed]
- Merckaert, T.; Zwaenepoel, O.; Gevaert, K.; Gettemans, J. An AKT2-specific nanobody that targets the hydrophobic motif induces cell cycle arrest, autophagy and loss of focal adhesions in MDA-MB-231 cells. Biomed. Pharmacother. 2020, 133, 111055. [Google Scholar] [CrossRef]
- Klomp, J.E.; Klomp, J.A.; Der, C.J. The ERK mitogen-activated protein kinase signaling network: The final frontier in RAS signal transduction. Biochem. Soc. Trans. 2021, 49, 253–267. [Google Scholar] [CrossRef]
- Kummer, L.; Parizek, P.; Rube, P.; Millgramm, B.; Prinz, A.; Mittl, P.R.; Kaufholz, M.; Zimmermann, B.; Herberg, F.W.; Plückthun, A. Structural and functional analysis of phosphorylation-specific binders of the kinase ERK from designed ankyrin repeat protein libraries. Proc. Natl. Acad. Sci. USA 2012, 109, E2248–E2257. [Google Scholar] [CrossRef] [Green Version]
- Sabapathy, K.; Lane, D.P. Understanding p53 functions through p53 antibodies. J. Mol. Cell. Biol. 2019, 11, 317–329. [Google Scholar] [CrossRef] [Green Version]
- Caron de Fromentel, C.; Gruel, N.; Venot, C.; Debussche, L.; Conseiller, E.; Dureuil, C.; Teillaud, J.L.; Tocque, B.; Bracco, L. Restoration of transcriptional activity of p53 mutants in human tumour cells by intracellular expression of anti-p53 single chain Fv fragments. Oncogene 1999, 18, 551–557. [Google Scholar] [CrossRef] [Green Version]
- Weisbart, R.H.; Hansen, J.E.; Chan, G.; Wakelin, R.; Chang, S.S.; Heinze, E.; Miller, C.W.; Koeffler, P.H.; Yang, F.; Cole, G.M.; et al. Antibody-mediated transduction of p53 selectively kills cancer cells. Int. J. Oncol. 2004, 25, 1867–1873. [Google Scholar] [CrossRef]
- Weisbart, R.H.; Wakelin, R.; Chan, G.; Miller, C.W.; Koeffler, P.H. Construction and expression of a bispecific single-chain antibody that penetrates mutant p53 colon cancer cells and binds p53. Int. J. Oncol. 2004, 25, 1113–1118. [Google Scholar]
- Tal, P.; Eizenberger, S.; Cohen, E.; Goldfinger, N.; Pietrokovski, S.; Oren, M.; Rotter, V. Cancer therapeutic approach based on conformational stabilization of mutant p53 protein by small peptides. Oncotarget 2016, 7, 11817–11837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosein, A.N.; Dougan, S.K.; Aguirre, A.J.; Maitra, A. Translational advances in pancreatic ductal adenocarcinoma therapy. Nat. Cancer 2022, 3, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Sunami, Y.; Boker, V.; Kleeff, J. Targeting and Reprograming Cancer-Associated Fibroblasts and the Tumor Microenvironment in Pancreatic Cancer. Cancers 2021, 13, 697. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Rouanet, M.; Lebrin, M.; Gross, F.; Bournet, B.; Cordelier, P.; Buscail, L. Gene Therapy for Pancreatic Cancer: Specificity, Issues and Hopes. Int. J. Mol. Sci. 2017, 18, 1231. [Google Scholar] [CrossRef] [Green Version]
- Mulligan, M.J.; Lyke, K.E.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Raabe, V.; Bailey, R.; Swanson, K.A.; et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 2020, 586, 589–593. [Google Scholar] [CrossRef]
- Bery, N.; Bataille, C.J.; Russell, A.; Hayes, A.; Raynaud, F.; Milhas, S.; Anand, S.; Tulmin, H.; Miller, A.; Rabbitts, T.H. A cell-based screening method using an intracellular antibody for discovering small molecules targeting the translocation protein LMO2. Sci. Adv. 2021, 7, eabg1950. [Google Scholar] [CrossRef]
- Bery, N.; Cruz-Migoni, A.; Bataille, C.J.; Quevedo, C.E.; Tulmin, H.; Miller, A.; Russell, A.; Phillips, S.E.; Carr, S.B.; Rabbitts, T.H. BRET-based RAS biosensors that show a novel small molecule is an inhibitor of RAS-effector protein-protein interactions. eLife 2018, 7, e37122. [Google Scholar] [CrossRef]
- Bery, N.; Rabbitts, T. A cell-based screening method using an intracellular antibody for discovering small molecules targeting hard-to-drug proteins. Bio-Protocol. 2022, 12, e4324. [Google Scholar] [CrossRef]
- Canning, P.; Bataille, C.; Bery, N.; Milhas, S.; Hayes, A.; Raynaud, F.; Miller, A.; Rabbitts, T. Competitive SPR using an intracellular anti-LMO2 antibody identifies novel LMO2-interacting compounds. J. Immunol. Methods 2021, 494, 113051. [Google Scholar] [CrossRef]
- Quevedo, C.E.; Cruz-Migoni, A.; Bery, N.; Miller, A.; Tanaka, T.; Petch, D.; Bataille, C.J.R.; Lee, L.Y.W.; Fallon, P.S.; Tulmin, H.; et al. Small molecule inhibitors of RAS-effector protein interactions derived using an intracellular antibody fragment. Nat. Commun. 2018, 9, 3169. [Google Scholar] [CrossRef] [PubMed]
- Bery, N.; Rabbitts, T.H. Bioluminescence Resonance Energy Transfer 2 (BRET2)-Based RAS Biosensors to Characterize RAS Inhibitors. Curr. Protoc. Cell Biol. 2018, 83, e83. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorbara, M.; Cordelier, P.; Bery, N. Antibody-Based Approaches to Target Pancreatic Tumours. Antibodies 2022, 11, 47. https://doi.org/10.3390/antib11030047
Sorbara M, Cordelier P, Bery N. Antibody-Based Approaches to Target Pancreatic Tumours. Antibodies. 2022; 11(3):47. https://doi.org/10.3390/antib11030047
Chicago/Turabian StyleSorbara, Marie, Pierre Cordelier, and Nicolas Bery. 2022. "Antibody-Based Approaches to Target Pancreatic Tumours" Antibodies 11, no. 3: 47. https://doi.org/10.3390/antib11030047