Source Apportionment of Aerosol at a Coastal Site and Relationships with Precipitation Chemistry: A Case Study over the Southeast United States

 and
and
Abstract
:1. Introduction
2. Methods
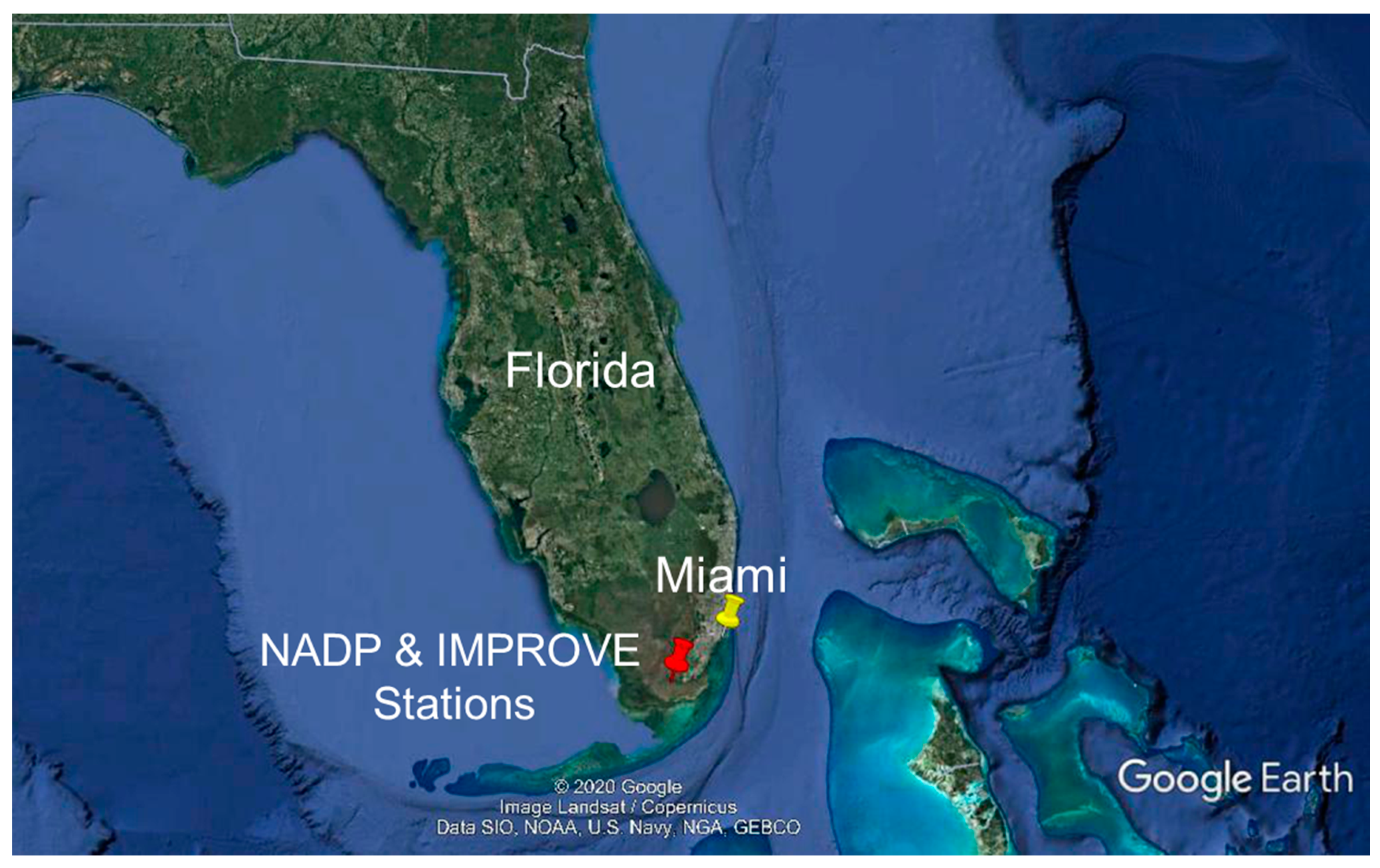
2.1. Site Description
2.2. Precipitation and Aerosol Composition
2.3. Metereological Data
2.4. Calculations
2.4.1. Positive Matrix Factorization
2.4.2. Weight Concentration Weighted Trajectory
3. Results and Discussion
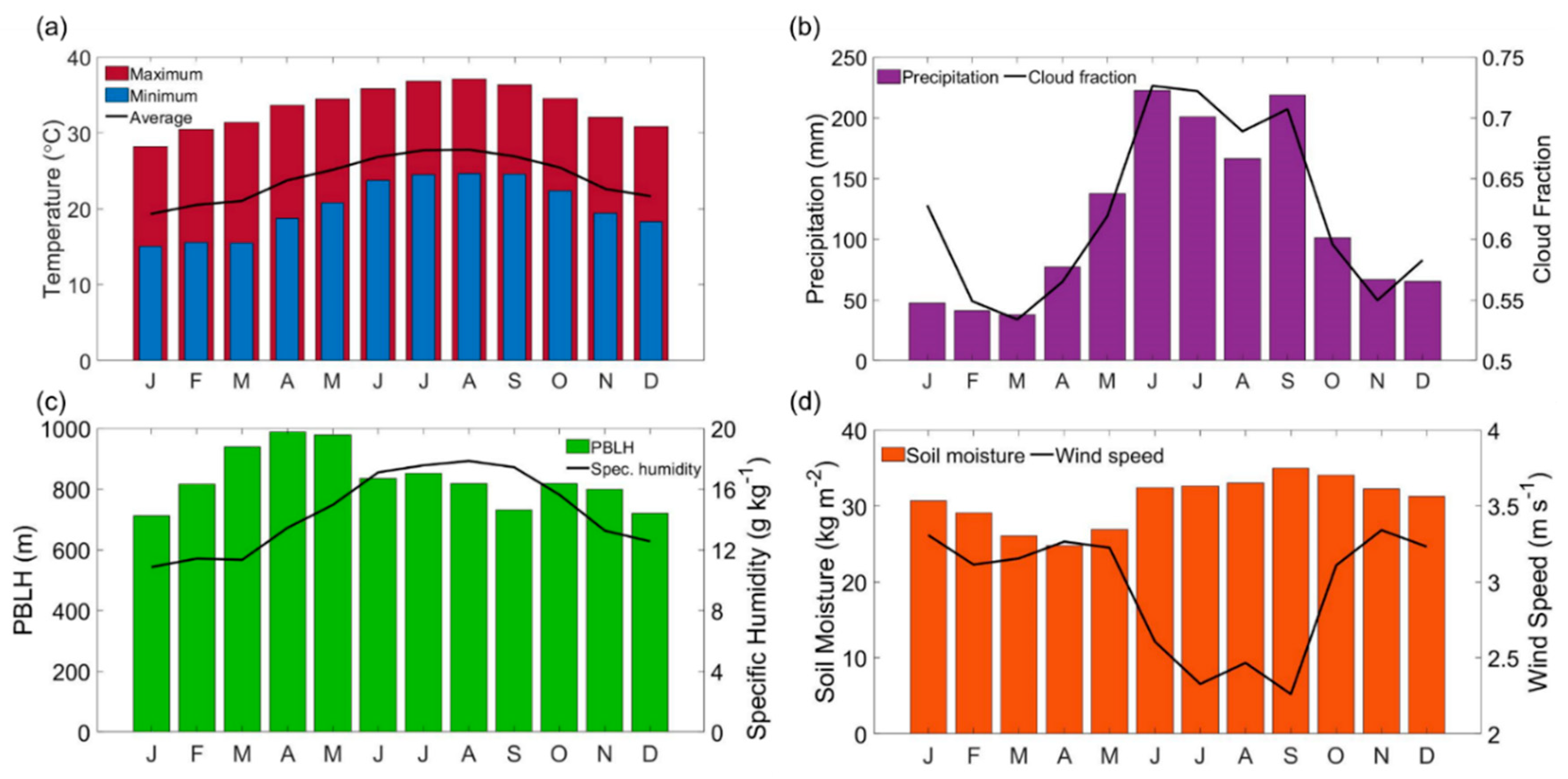
3.1. Meteorological Profile
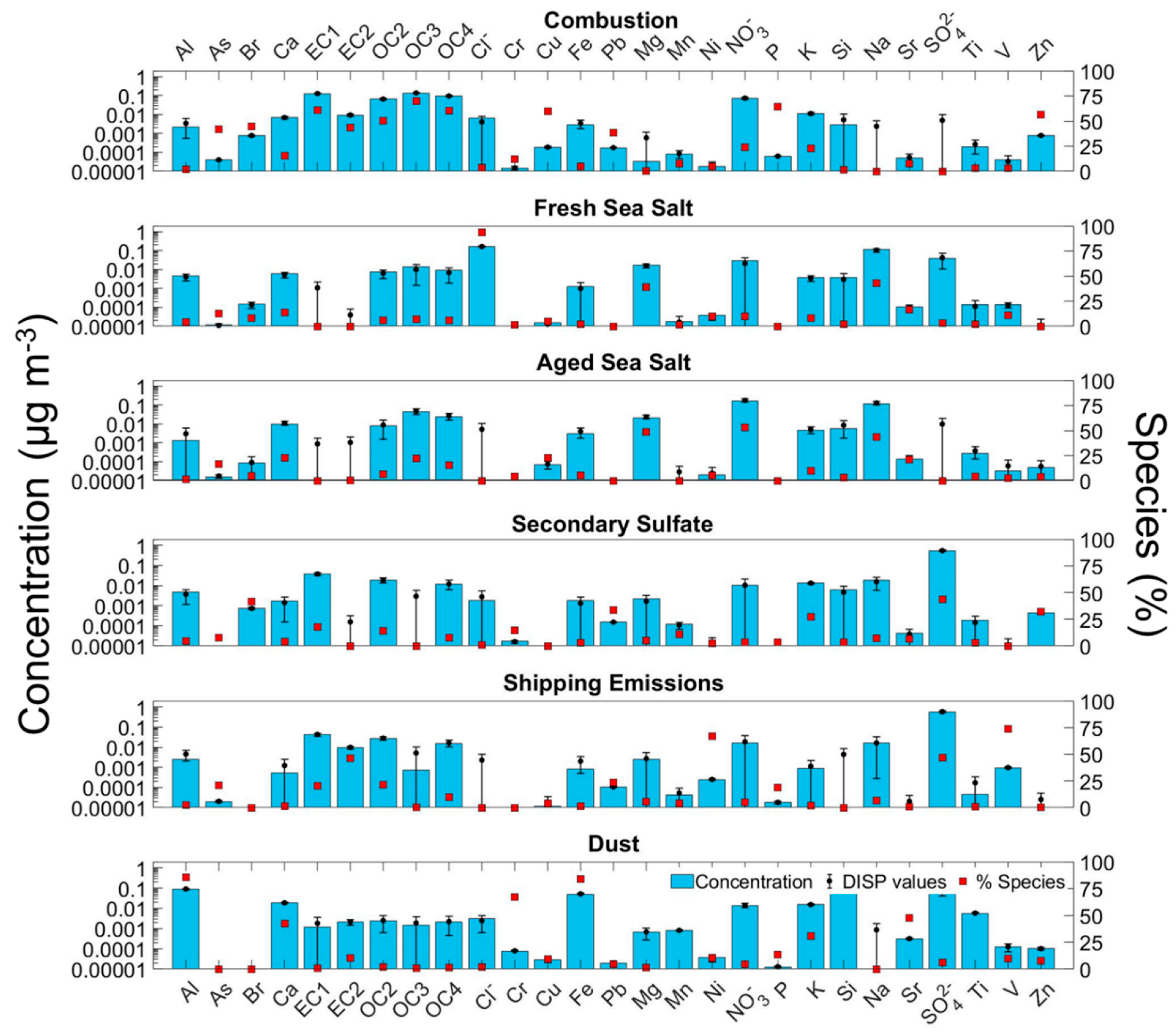
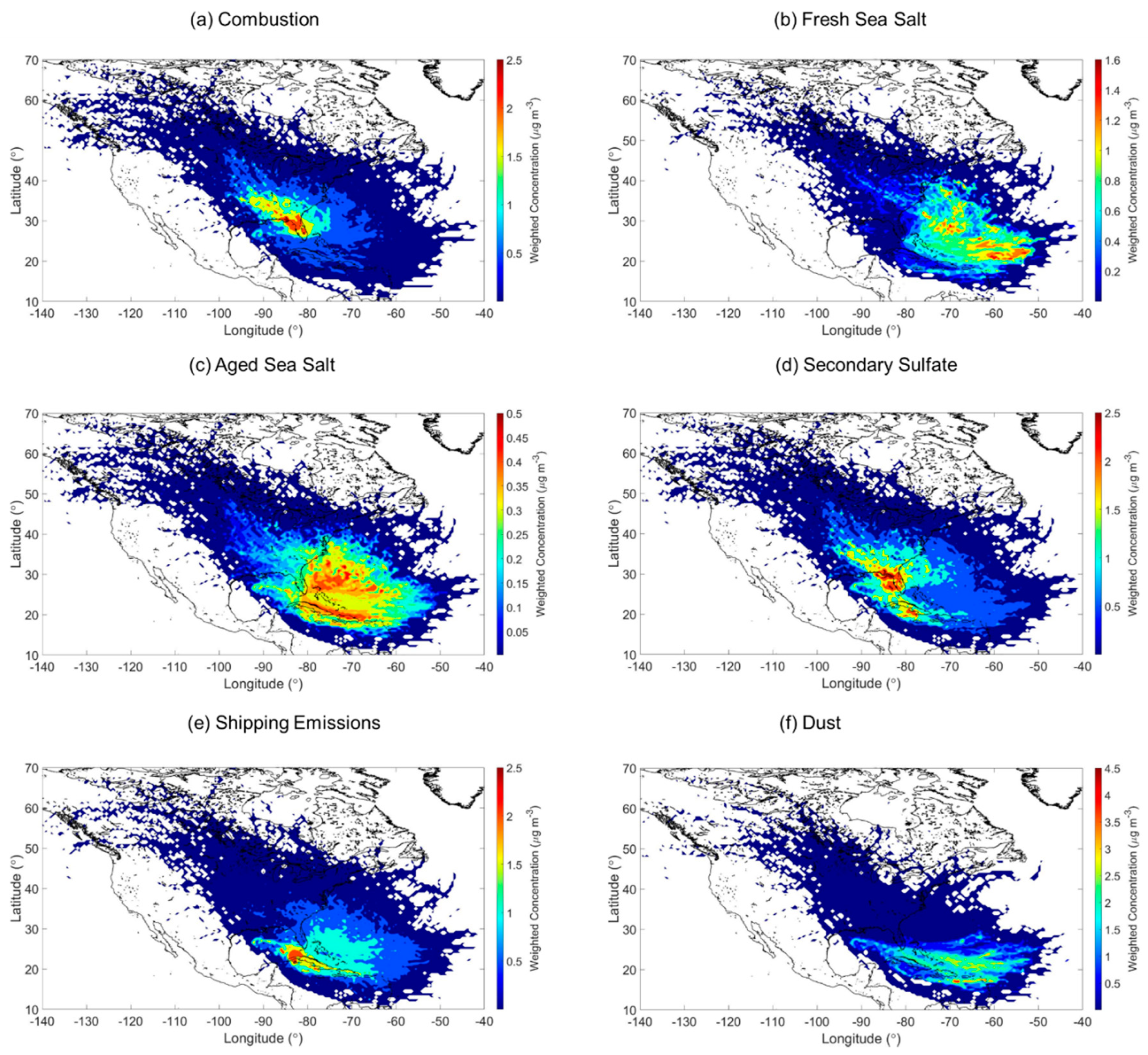
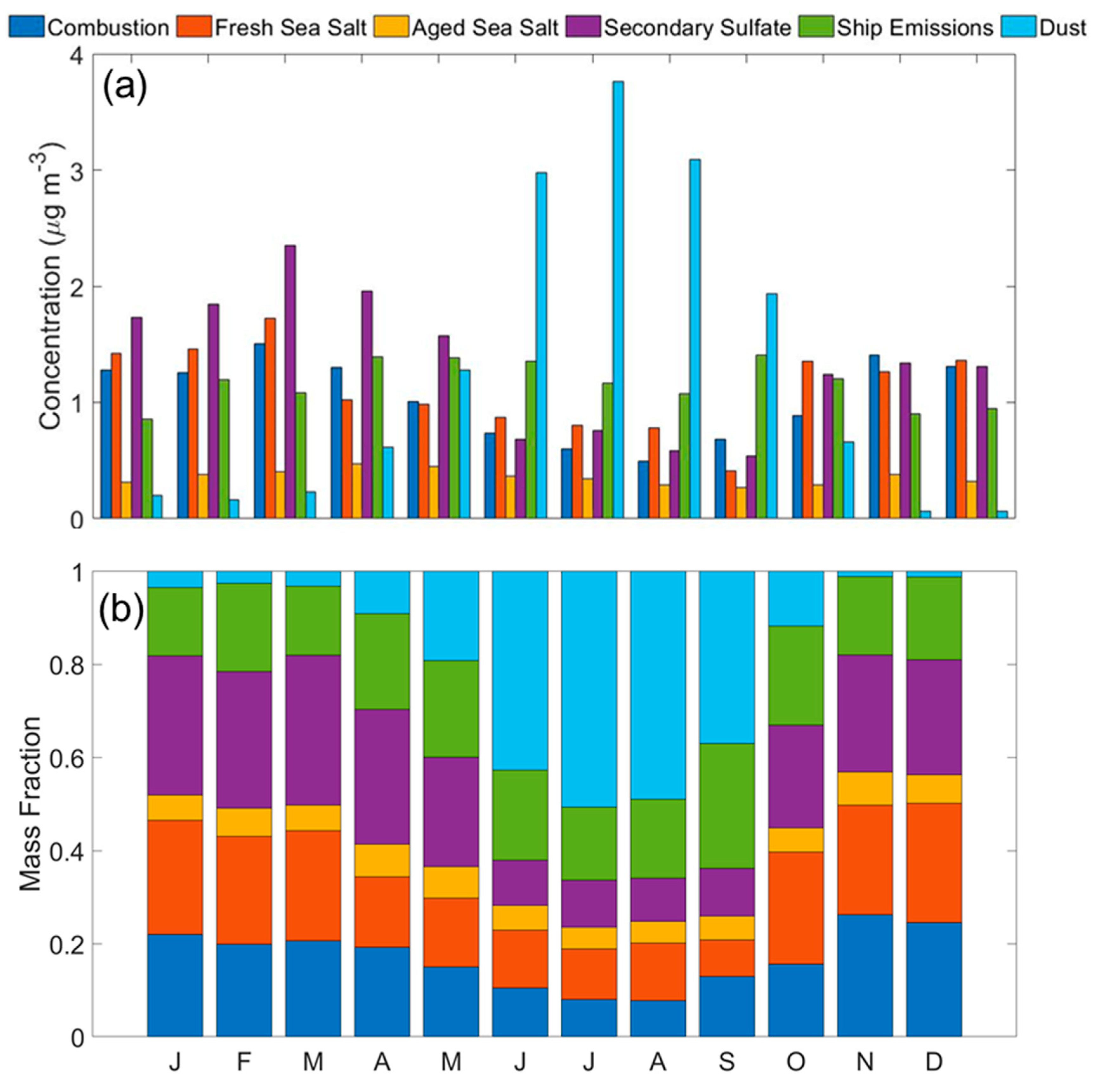
3.2. Sources of PM2.5
3.2.1. Combustion
3.2.2. Fresh and Aged Sea Salt
3.2.3. Secondary Sulfate
3.2.4. Shipping Emissions
3.2.5. Dust
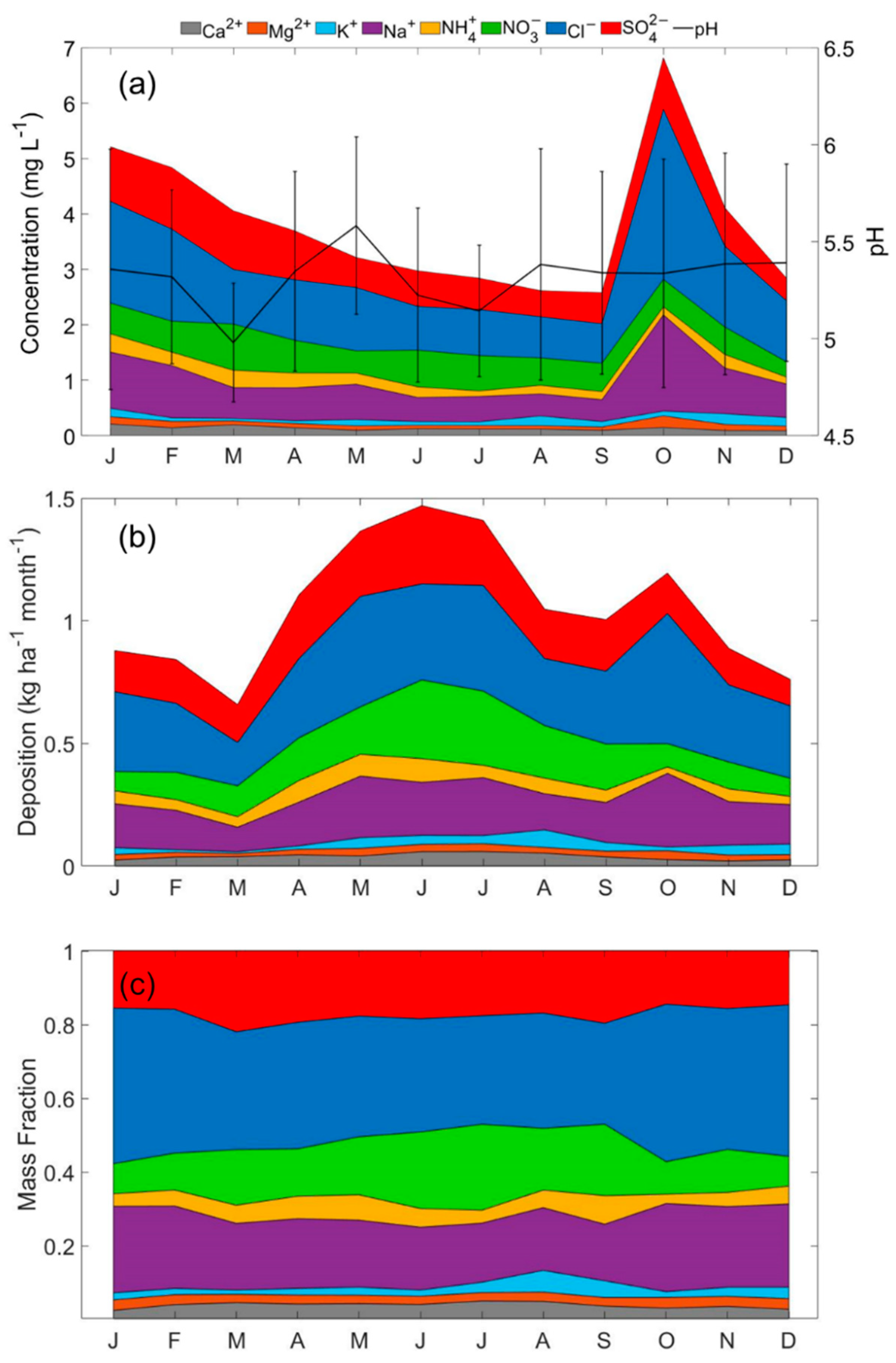
3.3. Precipiation Chemistry Profile
3.3.1. Monthly Profile
3.3.2. Interrelationships
3.4. Precipitation and Aerosol Interrelationships
4. Conclusions
- The Southern Florida coastal site was impacted by a diverse source of pollutants. The following six sources were identified in decreasing contribution to total PM2.5 (percentage contributions): (i) secondary sulfate (23.0%); (ii) dust (20.6%); (iii) shipping emissions (20.2%); (iv) combustion (17.0%); (v) fresh sea salt (12.6%); and (vi) aged sea salt (6.6%).
- Monthly mean precipitation pH ranges from 4.98 ± 0.31 (March) to 5.58 ± 0.51 (May). Values of pH were negatively related to the acidic anion SO42−, whereas they were positively related to dust presence as based on the crustal tracer species Ca2+.
- The highest mean annual wet deposition fluxes were attributed to Cl−, NO3−, SO42−, and Na+ between April and October, coinciding with months experiencing the most precipitation. Although lower in magnitude, enhanced fluxes of Ca2+, K+, and Mg2+ in summertime coincided with the main dust season.
- Fresh sea salt was the dominant component in the region’s precipitation, unlike surface PM2.5 owing partly to how the sea salt particles seeding precipitation drops likely exceed 2.5 µm. Aged sea salt was shown to be far less influential in the region’s precipitation.
- Even though dust plays a large role in PM2.5, it was much less influential in volume-weighted wet deposition concentrations and mass fractions as compared to sea salt.
- A weak positive association between precipitation Ca2+ and both aerosol NO3− and PMcoarse was linked to NO3− preferentially partitioning to coarse dust.
- Statistically significant correlations (p-value < 0.05) between related parameters indicative of dust, sea salt, and SO42− in the NADP and IMPROVE datasets demonstrated that the combined use of these long-term datasets could be useful for other regions to indirectly examine aerosol-precipitation interactions.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rosenfeld, D.; Sherwood, S.; Wood, R.; Donner, L. Atmospheric science. Climate effects of aerosol-cloud interactions. Science 2014, 343, 379–380. [Google Scholar] [CrossRef] [PubMed]
- IPCC. Climate change 2013: The physical science basis. In Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Stocker, T.F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S.K., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P.M., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2013; p. 1535. [Google Scholar]
- National Academies of Sciences, Engineering and Medicine. Thriving on Our Changing Planet: A Decadal Strategy for Earth Observation from Space; The National Academies Press: Washington, DC, USA, 2018. [Google Scholar] [CrossRef]
- Sorooshian, A.; Anderson, B.; Bauer, S.E.; Braun, R.A.; Cairns, B.; Crosbie, E.; Dadashazar, H.; Diskin, G.; Ferrare, R.; Flagan, R.C.; et al. Aerosol-cloud-meteorology interaction airborne field investigations: Using lessons learned from the U.S. West Coast in the design of ACTIVATE off the U.S. East Coast. Bull. Amer. Meteor. Soc. 2019, 100, 1511–1528. [Google Scholar] [CrossRef] [Green Version]
- Wood, R.; Mechoso, C.R.; Bretherton, C.S.; Weller, R.A.; Huebert, B.; Straneo, F.; Albrecht, B.A.; Coe, H.; Allen, G.; Vaughan, G.; et al. The VAMOS Ocean-Cloud-Atmosphere-Land Study Regional Experiment (VOCALS-REx): goals, platforms, and field operations. Atmos. Chem. Phys. 2011, 11, 627–654. [Google Scholar] [CrossRef] [Green Version]
- Sayer, A.M.; Hsu, N.C.; Lee, J.; Kim, W.V.; Burton, S.; Fenn, M.A.; Ferrare, R.A.; Kacenelenbogen, M.; LeBlanc, S.; Pistone, K.; et al. Two decades observing smoke above clouds in the south-eastern Atlantic Ocean: Deep Blue algorithm updates and validation with ORACLES field campaign data. Atmos. Meas. Tech 2019, 12, 3595–3627. [Google Scholar] [CrossRef] [Green Version]
- Redemann, J.; Wood, R.; Zuidema, P.; Doherty, S.; Luna, B.; LeBlanc, S.; Diamond, M.; Shinozuka, Y.; Gao, L.; Chang, I.; et al. An overview of the ORACLES (ObseRvations of Aerosols above CLouds and their intEractionS) project: aerosol-cloud-radiation interactions in the Southeast Atlantic basin. Atmos. Chem. Phys. 2020, 449. [Google Scholar] [CrossRef]
- Sorooshian, A.; Corral, A.F.; Braun, R.A.; Cairns, B.; Crosbie, E.; Ferrare, R.; Hair, J.; Kleb, M.M.; Mardi, A.H.; Maring, H.; et al. Atmospheric research over the Western North Atlantic Ocean region and North American East Coast: A review of past work and challenges ahead. J. Geophys. Res. Atmos. 2020, 125, e2019JD031626. [Google Scholar] [CrossRef]
- Dadashazar, H.; Ma, L.; Sorooshian, A. Sources of pollution and interrelationships between aerosol and precipitation chemistry at a central California site. Sci. Total Environ. 2019, 651, 1776–1787. [Google Scholar] [CrossRef]
- Sorooshian, A.; Shingler, T.; Harpold, A.; Feagles, C.W.; Meixner, T.; Brooks, P.D. Aerosol and precipitation chemistry in the southwestern United States: spatiotemporal trends and interrelationships. Atmos. Chem. Phys. 2013, 13, 7361–7379. [Google Scholar] [CrossRef] [Green Version]
- Mora, M.; Braun, R.A.; Shingler, T.; Sorooshian, A. Analysis of remotely sensed and surface data of aerosols and meteorology for the Mexico Megalopolis Area between 2003 and 2015. J. Geophys. Res. Atmos. 2017, 122, 8705–8723. [Google Scholar] [CrossRef] [Green Version]
- Hardy, K.A.; Akselsson, R.; Nelson, J.W.; Winchester, J.W. Elemental constituents of Miami aerosol as function of particle size. Environ. Sci. Technol. 1976, 10, 176–182. [Google Scholar] [CrossRef]
- Johansson, T.; Van Grieken, R.; Winchester, J. Marine influences on aerosol composition in the coastal zone. J. Rech. Atmos. 1974, 8, 761–776. [Google Scholar]
- Prospero, J.M. Long-range transport of mineral dust in the global atmosphere: impact of African dust on the environment of the southeastern United States. Proc. Natl. Acad. Sci. USA 1999, 96, 3396–3403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreidenweis, S.M.; Remer, L.A.; Bruintjes, R.; Dubovik, O. Smoke aerosol from biomass burning in Mexico: Hygroscopic smoke optical model. J. Geophys. Res. Atmos. 2001, 106, 4831–4844. [Google Scholar] [CrossRef] [Green Version]
- Yokelson, R.J.; Crounse, J.D.; DeCarlo, P.F.; Karl, T.; Urbanski, S.; Atlas, E.; Campos, T.; Shinozuka, Y.; Kapustin, V.; Clarke, A.D.; et al. Emissions from biomass burning in the Yucatan. Atmos. Chem. Phys. 2009, 9, 5785–5812. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, B.; Nuijens, L. Clouds at Barbados are representative of clouds across the trade wind regions in observations and climate models. Proc. Natl. Acad. Sci. USA 2016, 113, E3062–E3070. [Google Scholar] [CrossRef] [Green Version]
- Delgadillo, R.; Voss, K.J.; Zuidema, P. Characteristics of optically thin coastal Florida cumuli derived from surface-based lidar measurements. J. Geophys. Res. Atmos. 2018, 123, 10591–10605. [Google Scholar] [CrossRef]
- Court, A.; Griffiths, J. Thunderstorm climatology. In Thunderstorm Morphology and Dynamics, 2nd ed.; University of Oklahoma Press: Norman, OK, USA, 1986; pp. 9–40. [Google Scholar]
- Byers, H.R.; Rodebush, H.R. Causes of thunderstorms of the Florida peninsula. J. Meteorol. 1948, 5, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Prospero, J.M.; Landing, W.M.; Schulz, M. African dust deposition to Florida: Temporal and spatial variability and comparisons to models. J. Geophys. Res. Atmos. 2010, 115. [Google Scholar] [CrossRef] [Green Version]
- Pollman, C.; Gill, G.; Landing, W.; Guentzel, J.; Bare, D.; Porcella, D.; Zillioux, E.; Atkeson, T. Overview of the Florida atmospheric mercury study (Fams). Water Air Soil Pollut. 1995, 80, 285–290. [Google Scholar] [CrossRef]
- U.S. Census Bureau. QuickFacts Miami-Dade County, Florida; United States. 2018. Available online: https://www.census.gov/quickfacts/miamidadecountyflorida (accessed on 1 May 2020).
- Glaser, P.H.; Hansen, B.C.; Donovan, J.J.; Givnish, T.J.; Stricker, C.A.; Volin, J.C. Holocene dynamics of the Florida Everglades with respect to climate, dustfall, and tropical storms. Proc. Natl. Acad. Sci. USA 2013, 110, 17211–17216. [Google Scholar] [CrossRef] [Green Version]
- Ingebritsen, S.; McVoy, C.; Glaz, B.; Park, W. Florida Everglades. Land Subsid. USA Circa 1999, 1182, 95–120. [Google Scholar]
- USDA-NASS. Florida County Estimates Sugarcane 2017–2018. United States Department of Agriculture National Agricultural Statistics Service Southern Region. 2019. Available online: https://www.nass.usda.gov/Statistics_by_State/Florida/Publications/County_Estimates/2018/FLSugarcane2018.pdf (accessed on 1 May 2020).
- Sevimoğlu, O.; Rogge, W.F. Seasonal variations of PM10—Trace elements, PAHs and levoglucosan: Rural sugarcane growing area versus coastal urban area in Southeastern Florida, USA. Part II: Elemental concentrations. Particuology 2019, 46, 99–108. [Google Scholar] [CrossRef]
- Dawson, L.; Boopathy, R. Use of post-harvest sugarcane residue for ethanol production. Bioresour. Technol. 2007, 98, 1695–1699. [Google Scholar] [CrossRef] [PubMed]
- Frias, M.; Villar-Cocina, E.; Valencia-Morales, E. Characterisation of sugar cane straw waste as pozzolanic material for construction: calcining temperature and kinetic parameters. Waste Manag. 2007, 27, 533–538. [Google Scholar] [CrossRef]
- Silva, F.S.; Cristale, J.; Andre, P.A.; Saldiva, P.H.N.; Marchi, M.R.R. PM2.5 and PM10: The influence of sugarcane burning on potential cancer risk. Atmos. Environ. 2010, 44, 5133–5138. [Google Scholar] [CrossRef]
- Sevimoglu, O.; Rogge, W.F. Seasonal size-segregated PM10 and PAH concentrations in a rural area of sugarcane agriculture versus a coastal urban area in Southeastern Florida, USA. Particuology 2016, 28, 52–59. [Google Scholar] [CrossRef]
- Malm, W.C.; Sisler, J.F.; Huffman, D.; Eldred, R.A.; Cahill, T.A. Spatial and seasonal trends in particle concentration and optical extinction in the United States. J. Geophys. Res. Atmos. 1994, 99, 1347–1370. [Google Scholar] [CrossRef]
- National Atmospheric Deposition Program (NADP). Available online: http://nadp.slh.wisc.edu/ntn/ (accessed on 1 May 2020).
- The Interagency Monitoring of Protected Visual Environments (IMPROVE). Available online: http://views.cira.colostate.edu/fed/QueryWizard/Default.aspx (accessed on 1 May 2020).
- Chow, J.C.; Watson, J.G.; Chen, L.W.; Chang, M.C.; Robinson, N.F.; Trimble, D.; Kohl, S. The IMPROVE_A temperature protocol for thermal/optical carbon analysis: maintaining consistency with a long-term database. J. Air Waste Manag. Assoc. 2007, 57, 1014–1023. [Google Scholar] [CrossRef] [Green Version]
- Chow, J.C.; Watson, J.G.; Pritchett, L.C.; Pierson, W.R.; Frazier, C.A.; Purcell, R.G. The dri thermal/optical reflectance carbon analysis system: Description, evaluation and applications in U.S. Air quality studies. Atmos. Environ. Part A Gen. Top. 1993, 27, 1185–1201. [Google Scholar] [CrossRef]
- Lim, S.; Lee, M.; Lee, G.; Kim, S.; Yoon, S.; Kang, K. Ionic and carbonaceous compositions of PM10, PM2.5 and PM1.0 at Gosan ABC Superstation and their ratios as source signature. Atmos. Chem. Phys. 2012, 12, 2007–2024. [Google Scholar] [CrossRef] [Green Version]
- Chow, J.C.; Lowenthal, D.H.; Chen, L.W.; Wang, X.; Watson, J.G. Mass reconstruction methods for PM2.5: A review. Air Qual. Atmos. Health 2015, 8, 243–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, P.A.; Crumpler, D.; Flanagan, J.B.; Jayanty, R.K.; Rickman, E.E.; McDade, C.E. U.S. national PM2.5 Chemical Speciation Monitoring Networks-CSN and IMPROVE: description of networks. J. Air Waste Manag. Assoc. 2014, 64, 1410–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- USEPA AQS. EPA AirData Air Quality System (AQS) Monitors. Available online: https://epa.maps.arcgis.com/apps/webappviewer/index.html?id=5f239fd3e72f424f98ef3d5def547eb5&extent=-146.2334,13.1913,-46.3896,56.5319 (accessed on 1 May 2020).
- Randles, C.A.; Da Silva, A.M.; Buchard, V.; Colarco, P.R.; Darmenov, A.; Govindaraju, R.; Smirnov, A.; Holben, B.; Ferrare, R.; Hair, J.; et al. The MERRA-2 aerosol reanalysis, 1980-onward, Part I: System description and data assimilation evaluation. J. Clim. 2017, 30, 6823–6850. [Google Scholar] [CrossRef] [PubMed]
- Rodell, M.; Houser, P.R.; Jambor, U.; Gottschalck, J.; Mitchell, K.; Meng, C.J.; Arsenault, K.; Cosgrove, B.; Radakovich, J.; Bosilovich, M.; et al. The global land data assimilation system. Bull. Amer. Meteor. Soc. 2004, 85, 381. [Google Scholar] [CrossRef] [Green Version]
- Acker, J.G.; Leptoukh, G. Online analysis enhances use of NASA earth science data. Eos Trans. 2007, 88, 14–17. [Google Scholar] [CrossRef]
- Hopke, P.K. Review of receptor modeling methods for source apportionment. J. Air Waste Manag. Assoc. 2016, 66, 237–259. [Google Scholar] [CrossRef]
- Brown, S.G.; Eberly, S.; Paatero, P.; Norris, G.A. Methods for estimating uncertainty in PMF solutions: examples with ambient air and water quality data and guidance on reporting PMF results. Sci. Total Environ. 2015, 518, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Reff, A.; Eberly, S.I.; Bhave, P.V. Receptor modeling of ambient particulate matter data using positive matrix factorization: review of existing methods. J. Air Waste Manag. Assoc. 2007, 57, 146–154. [Google Scholar] [CrossRef] [Green Version]
- Norris, G.; Duvall, R.; Brown, S.; Bai, S. EPA Positive Matrix Factorization (PMF) 5.0 Fundamentals and User Guide. 2014. Available online: https://www.epa.gov/sites/production/files/2015-02/documents/pmf_5.0_user_guide.pdf (accessed on 1 May 2020).
- Kim, E.; Hopke, P.K.; Edgerton, E.S. Source identification of atlanta aerosol by positive matrix factorization. J. Air Waste Manag. Assoc. 2003, 53, 731–739. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.K.; Holsen, T.M.; Hopke, P.K. Comparison of hybrid receptor models to locate PCB sources in Chicago. Atmos. Environ. 2003, 37, 545–562. [Google Scholar] [CrossRef]
- Seibert, P.; Kromp-Kolb, H.; Baltensperger, U.; Jost, D.; Schwikowski, M.; Kasper, A.; Puxbaum, H. Trajectory analysis of aerosol measurements at high alpine sites. In Air Pollution Modeling and Its Application X. NATO · Challenges of Modern Society; Gryning, S.E., Millán, M.M., Eds.; Springer: Boston, MA, USA, 1994; Volume 18, pp. 689–693. [Google Scholar]
- Sun, T.Z.; Che, H.Z.; Qi, B.; Wang, Y.Q.; Dong, Y.S.; Xia, X.G.; Wang, H.; Gui, K.; Zheng, Y.; Zhao, H.J.; et al. Aerosol optical characteristics and their vertical distributions under enhanced haze pollution events: effect of the regional transport of different aerosol types over eastern China. Atmos. Chem. Phys. 2018, 18, 2949–2971. [Google Scholar] [CrossRef] [Green Version]
- Polissar, A.V.; Hopke, P.K.; Paatero, P.; Kaufmann, Y.J.; Hall, D.K.; Bodhaine, B.A.; Dutton, E.G.; Harris, J.M. The aerosol at Barrow, Alaska: long-term trends and source locations. Atmos. Environ. 1999, 33, 2441–2458. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Zhang, X.Y.; Draxler, R.R. TrajStat: GIS-based software that uses various trajectory statistical analysis methods to identify potential sources from long-term air pollution measurement data. Environ. Model. Software 2009, 24, 938–939. [Google Scholar] [CrossRef]
- Stein, A.F.; Draxler, R.R.; Rolph, G.D.; Stunder, B.J.B.; Cohen, M.D.; Ngan, F. NOAA’s HYSPLITt atmospheric transport and dispersion modeling system. Bull. Amer. Meteor. Soc. 2015, 96, 2059–2077. [Google Scholar] [CrossRef]
- Rolph, G.; Stein, A.; Stunder, B. Real-time Environmental Applications and Display sYstem: READY. Environ. Model. Software 2017, 95, 210–228. [Google Scholar] [CrossRef]
- Masiol, M.; Hopke, P.K.; Felton, H.D.; Frank, B.P.; Rattigan, O.V.; Wurth, M.J.; LaDuke, G.H. Source apportionment of PM2.5 chemically speciated mass and particle number concentrations in New York City. Atmos. Environ. 2017, 148, 215–229. [Google Scholar] [CrossRef]
- Xin, J.Y.; Du, W.P.; Wang, Y.S.; Gao, Q.X.; Li, Z.Q.; Wang, M.X. Aerosol optical properties affected by a strong dust Storm over central and northern China. Adv. Atmos. Sci. 2010, 27, 562–574. [Google Scholar] [CrossRef]
- Zhang, M.J.; Singh, H.V.; Migliaccio, K.W.; Kisekka, I. Evaluating water table response to rainfall events in a shallow aquifer and canal system. Hydrol. Process. 2017, 31, 3907–3919. [Google Scholar] [CrossRef]
- Schlosser, J.S.; Braun, R.A.; Bradley, T.; Dadashazar, H.; MacDonald, A.B.; Aldhaif, A.A.; Aghdam, M.A.; Mardi, A.H.; Xian, P.; Sorooshian, A. Analysis of aerosol composition data for western United States wildfires between 2005 and 2015: Dust emissions, chloride depletion, and most enhanced aerosol constituents. J. Geophys. Res. Atmos. 2017, 122, 8951–8966. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Hopke, P.K. Source apportionment of fine particles in Washington, DC, utilizing temperature-resolved carbon fractions. J. Air Waste Manag. Assoc. 2004, 54, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Corbin, J.C.; Mensah, A.A.; Pieber, S.M.; Orasche, J.; Michalke, B.; Zanatta, M.; Czech, H.; Massabo, D.; Buatier de Mongeot, F.; Mennucci, C.; et al. Trace metals in soot and PM2.5 from heavy-fuel-oil combustion in a marine engine. Environ. Sci. Technol. 2018, 52, 6714–6722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linak, W.P.; Miller, C.A.; Wendt, J.O. Comparison of particle size distributions and elemental partitioning from the combustion of pulverized coal and residual fuel oil. J. Air Waste Manag. Assoc. 2000, 50, 1532–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ålander, T.; Antikainen, E.; Raunemaa, T.; Elonen, E.; Rautiola, A.; Torkkell, K. Particle emissions from a small two-stroke engine: effects of fuel, lubricating oil, and exhaust aftertreatment on particle characteristics. Aerosol Sci. Technol. 2005, 39, 151–161. [Google Scholar] [CrossRef] [Green Version]
- Pant, P.; Harrison, R.M. Estimation of the contribution of road traffic emissions to particulate matter concentrations from field measurements: A review. Atmos. Environ. 2013, 77, 78–97. [Google Scholar] [CrossRef]
- DARM. AirInfo Data Search. Florida Department of Environmental Protection Division of Air Resource Management’s. 2013. Available online: https://prodenv.dep.state.fl.us/DarmAircom/public/searchFacilityPILoad.action (accessed on 1 May 2020).
- Hwang, I.-J.; Hopke, P.K. Comparison of source apportionment of PM 2.5 using PMF2 and EPA PMF version 2. Asian J. Atmos. Environ. 2011, 5, 86–96. [Google Scholar] [CrossRef] [Green Version]
- Lang, Q.; Zhang, Q.; Jaffe, R. Organic aerosols in the Miami area, USA: temporal variability of atmospheric particles and wet/dry deposition. Chemosphere 2002, 47, 427–441. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Huey, L.; Yokelson, R.; Wang, Y.; Jimenez, J.; Campuzano-Jost, P.; Beyersdorf, A.; Blake, D.; Choi, Y. Agricultural fires in the southeastern US during SEAC4RS: Emissions of trace gases and particles and evolution of ozone, reactive nitrogen, and organic aerosol. J. Geophys. Res. Atmos. 2016, 121, 7383–7414. [Google Scholar] [CrossRef] [Green Version]
- McCarty, J.L.; Korontzi, S.; Justice, C.O.; Loboda, T. The spatial and temporal distribution of crop residue burning in the contiguous United States. Sci. Total Environ. 2009, 407, 5701–5712. [Google Scholar] [CrossRef]
- Reid, S.B.; Funk, T.H.; Sullivan, D.C.; Stiefer, P.S.; Arkinson, H.L.; Brown, S.G.; Chinkin, L.R. Research and development of emission inventories for planned burning activities for the Central State Regional Air Planning Association. In Proceedings of the 13th International Emission Inventory Conference, Clearwater, FL, USA, 8–10 June 2004. [Google Scholar]
- Wang, J.; Christopher, S.A.; Nair, U.S.; Reid, J.S.; Prins, E.M.; Szykman, J.; Hand, J.L. Mesoscale modeling of Central American smoke transport to the United States: 1. “Top-down” assessment of emission strength and diurnal variation impacts. J. Geophys. Res. Atmos. 2006, 111. [Google Scholar] [CrossRef] [Green Version]
- Kotchenruther, R.A. The effects of marine vessel fuel sulfur regulations on ambient PM2.5 at coastal and near coastal monitoring sites in the U.S. Atmos. Environ. 2017, 151, 52–61. [Google Scholar] [CrossRef]
- Adachi, K.; Buseck, P.R. Changes in shape and composition of sea-salt particles upon aging in an urban atmosphere. Atmos. Environ. 2015, 100, 1–9. [Google Scholar] [CrossRef]
- Qin, Y.; Oduyemi, K.; Chan, L.Y. Comparative testing of PMF and CFA models. Chemom. Intellig. Lab. Syst. 2002, 61, 75–87. [Google Scholar] [CrossRef]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 3rd ed; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016. [Google Scholar]
- Song, X.-H.; Polissar, A.V.; Hopke, P.K. Sources of fine particle composition in the northeastern US. Atmos. Environ. 2001, 35, 5277–5286. [Google Scholar] [CrossRef]
- Perez, N.; Pey, J.; Reche, C.; Cortes, J.; Alastuey, A.; Querol, X. Impact of harbour emissions on ambient PM10 and PM2.5 in Barcelona (Spain): Evidences of secondary aerosol formation within the urban area. Sci. Total Environ. 2016, 571, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Braun, R.A.; Dadashazar, H.; MacDonald, A.B.; Aldhaif, A.M.; Maudlin, L.C.; Crosbie, E.; Aghdam, M.A.; Hossein Mardi, A.; Sorooshian, A. Impact of wildfire emissions on chloride and bromide depletion in marine aerosol particles. Environ. Sci. Technol. 2017, 51, 9013–9021. [Google Scholar] [CrossRef] [PubMed]
- Kerminen, V.M.; Teinila, K.; Hillamo, R.; Pakkanen, T. Substitution of chloride in sea-salt particles by inorganic and organic anions. J. Aerosol Sci. 1998, 29, 929–942. [Google Scholar] [CrossRef]
- Virkkula, A.; Teinila, K.; Hillamo, R.; Matti-Kerminen, V.; Saarikoski, S.; Aurela, M.; Koponen, I.K.; Kulmala, M. Chemical size distributions of boundary layer aerosol over the Atlantic Ocean and at an Antarctic site. J. Geophys. Res. Atmos. 2006, 111. [Google Scholar] [CrossRef]
- Keene, W.C.; Khalil, M.A.K.; Erickson, D.J.; McCulloch, A.; Graedel, T.E.; Lobert, J.M.; Aucott, M.L.; Gong, S.L.; Harper, D.B.; Kleiman, G.; et al. Composite global emissions of reactive chlorine from anthropogenic and natural sources: Reactive chlorine emissions inventory. J. Geophys. Res. Atmos. 1999, 104, 8429–8440. [Google Scholar] [CrossRef] [Green Version]
- AzadiAghdam, M.; Braun, R.A.; Edwards, E.-L.; Bañaga, P.A.; Cruz, M.T.; Betito, G.; Cambaliza, M.O.; Dadashazar, H.; Lorenzo, G.R.; Ma, L.; et al. On the nature of sea salt aerosol at a coastal megacity: Insights from Manila, Philippines in Southeast Asia. Atmos. Environ. 2019, 216, 116922. [Google Scholar] [CrossRef]
- Cruz, M.T.; Banaga, P.A.; Betito, G.; Braun, R.A.; Stahl, C.; Aghdam, M.A.; Cambaliza, M.O.; Dadashazar, H.; Hilario, M.R.; Lorenzo, G.R.; et al. Size-resolved composition and morphology of particulate matter during the southwest monsoon in Metro Manila, Philippines. Atmos. Chem. Phys. 2019, 19, 10675–10696. [Google Scholar] [CrossRef] [Green Version]
- Maudlin, L.; Wang, Z.; Jonsson, H.; Sorooshian, A. Impact of wildfires on size-resolved aerosol composition at a coastal California site. Atmos. Environ. 2015, 119, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Prabhakar, G.; Ervens, B.; Wang, Z.; Maudlin, L.C.; Coggon, M.M.; Jonsson, H.H.; Seinfeld, J.H.; Sorooshian, A. Sources of nitrate in stratocumulus cloud water: Airborne measurements during the 2011 E-PEACE and 2013 NiCE studies. Atmos. Environ. 2014, 97, 166–173. [Google Scholar] [CrossRef] [Green Version]
- Murphy, D.M.; Froyd, K.D.; Bian, H.S.; Brock, C.A.; Dibb, J.E.; DiGangi, J.P.; Diskin, G.; Dollner, M.; Kupc, A.; Scheuer, E.M.; et al. The distribution of sea-salt aerosol in the global troposphere. Atmos. Chem. Phys. 2019, 19, 4093–4104. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, A.B.; Dadashazar, H.; Chuang, P.Y.; Crosbie, E.; Wang, H.; Wang, Z.; Jonsson, H.H.; Flagan, R.C.; Seinfeld, J.H.; Sorooshian, A. Characteristic vertical profiles of cloud water composition in marine stratocumulus clouds and relationships with precipitation. J. Geophys. Res. Atmos. 2018, 123, 3704–3723. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.D. Dimethyl sulfide production and marine phytoplankton: The importance of species composition and cell size. Biol. Oceanogr. 1989, 6, 375–382. [Google Scholar] [CrossRef]
- Pacyna, J.M.; Pacyna, E.G. An assessment of global and regional emissions of trace metals to the atmosphere from anthropogenic sources worldwide. Environ. Rev. 2001, 9, 269–298. [Google Scholar] [CrossRef]
- Rose, K. Source allocation of columbia gorge IMPROVE data with positive matrix factorization. Appendix E of Chemical Concentration Balance Source Apportionment of PM2.5 2004. Available online: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.424.7044&rep=rep1&type=pdf (accessed on 1 May 2020).
- Kim, E.; Hopke, P.K. Improving source apportionment of fine particles in the eastern United States utilizing temperature-resolved carbon fractions. J. Air Waste Manag. Assoc. 2005, 55, 1456–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.J.C.; Van Aswegen, S.; Webb, W.R.; Goddard, S.L. UK concentrations of chromium and chromium (VI), measured as water soluble chromium, in PM10. Atmos. Environ. 2014, 99, 385–391. [Google Scholar] [CrossRef]
- Urban, R.C.; Lima-Souza, M.; Caetano-Silva, L.; Queiroz, M.E.C.; Nogueira, R.F.P.; Allen, A.G.; Cardoso, A.A.; Held, G.; Campos, M.L.A.M. Use of levoglucosan, potassium, and water-soluble organic carbon to characterize the origins of biomass-burning aerosols. Atmos. Environ. 2012, 61, 562–569. [Google Scholar] [CrossRef]
- Barth, M.C.; Rasch, P.J.; Kiehl, J.T.; Benkovitz, C.M.; Schwartz, S.E. Sulfur chemistry in the National Center for Atmospheric Research Community Climate Model: Description, evaluation, features, and sensitivity to aqueous chemistry. J. Geophys. Res. Atmos. 2000, 105, 1387–1415. [Google Scholar] [CrossRef]
- Karanasiou, A.A.; Siskos, P.A.; Eleftheriadis, K. Assessment of source apportionment by Positive Matrix Factorization analysis on fine and coarse urban aerosol size fractions. Atmos. Environ. 2009, 43, 3385–3395. [Google Scholar] [CrossRef]
- Viana, M.; Amato, F.; Alastuey, A.; Querol, X.; Moreno, T.; Dos Santos, S.G.; Herce, M.D.; Fernandez-Patier, R. Chemical tracers of particulate emissions from commercial shipping. Environ. Sci. Technol. 2009, 43, 7472–7477. [Google Scholar] [CrossRef] [PubMed]
- Viana, M.; Kuhlbusch, T.A.J.; Querol, X.; Alastuey, A.; Harrison, R.M.; Hopke, P.K.; Winiwarter, W.; Vallius, A.; Szidat, S.; Prevot, A.S.H.; et al. Source apportionment of particulate matter in Europe: A review of methods and results. J. Aerosol Sci. 2008, 39, 827–849. [Google Scholar] [CrossRef]
- Hobbs, P.V.; Garrett, T.J.; Ferek, R.J.; Strader, S.R.; Hegg, D.A.; Frick, G.M.; Hoppel, W.A.; Gasparovic, R.F.; Russell, L.M.; Johnson, D.W.; et al. Emissions from ships with respect to their effects on clouds. J. Atmos. Sci. 2000, 57, 2570–2590. [Google Scholar] [CrossRef] [Green Version]
- Isakson, J.; Persson, T.A.; Selin Lindgren, E. Identification and assessment of ship emissions and their effects in the harbour of Göteborg, Sweden. Atmos. Environ. 2001, 35, 3659–3666. [Google Scholar] [CrossRef]
- Pandolfi, M.; Gonzalez-Castanedo, Y.; Alastuey, A.; Jesus, D.; Mantilla, E.; De La Campa, A.S.; Querol, X.; Pey, J.; Amato, F.; Moreno, T. Source apportionment of PM 10 and PM 2.5 at multiple sites in the strait of Gibraltar by PMF: impact of shipping emissions. Environ. Sci. Pollut. Res. 2011, 18, 260–269. [Google Scholar] [CrossRef]
- Perry, K.D.; Cahill, T.A.; Eldred, R.A.; Dutcher, D.D.; Gill, T.E. Long-range transport of North African dust to the eastern United States. J. Geophys. Res. Atmos. 1997, 102, 11225–11238. [Google Scholar] [CrossRef]
- Flocchini, R.G.; Cahill, T.A.; Pitchford, M.L.; Eldred, R.A.; Feeney, P.J.; Ashbaugh, L.L. Characterization of particles in the arid west. Atmos. Environ. 1981, 15, 2017–2030. [Google Scholar] [CrossRef]
- Prospero, J.M.; Nees, R.T. Dust concentration in the atmosphere of the equatorial north atlantic: possible relationship to the sahelian drought. Science 1977, 196, 1196–1198. [Google Scholar] [CrossRef] [Green Version]
- Carlson, T.N.; Prospero, J.M. The large-scale movement of Saharan air outbreaks over the Northern Equatorial Atlantic. J. Appl. Meteorol. Climatol 1972, 11, 283–297. [Google Scholar] [CrossRef]
- Savoie, D.; Prospero, J. Aerosol concentration statistics for the northern tropical Atlantic. J. Geophys. Res. 1977, 82, 5954–5964. [Google Scholar] [CrossRef]
- Prospero, J.M. Assessing the impact of advected African dust on air quality and health in the eastern United States. Hum. Ecol. Risk Assess. Int. J. 1999, 5, 471–479. [Google Scholar] [CrossRef]
- Prospero, J.M. Long-term measurements of the transport of African mineral dust to the southeastern United States: Implications for regional air quality. J. Geophys. Res. Atmos. 1999, 104, 15917–15927. [Google Scholar] [CrossRef] [Green Version]
- Zuidema, P.; Alvarez, C.; Kramer, S.J.; Custals, L.; Izaguirre, M.; Sealy, P.; Prospero, J.M.; Blades, E. Is Summer African dust arriving earlier to Barbados? The updated long-term in situ dust mass concentration time series from Ragged Point, Barbados, and Miami, Florida. Bull. Amer. Meteor. Soc. 2019, 100, 1981–1986. [Google Scholar] [CrossRef]
- Kramer, S.J.; Alvarez, C.; Barkley, A.; Colarco, P.R.; Custals, L.; Delgadillo, R.; Gaston, C.; Govindaraju, R.; Zuidema, P. Apparent dust size discrepancy in aerosol reanalysis in north African dust after long-range transport. Atmos. Chem. Phys. Discuss. 2020, 2020, 1–32. [Google Scholar] [CrossRef]
- Kramer, S.J.; Kirtman, B.P.; Zuidema, P.; Ngan, F. Subseasonal variability of elevated dust concentrations over South Florida. J. Geophys. Res. Atmos. 2020, 125, e2019JD031874. [Google Scholar] [CrossRef]
- Prospero, J.M.; Bonatti, E.; Schubert, C.; Carlson, T.N. Dust in the Caribbean atmosphere traced to an African dust storm. Earth Planet. Sci. Lett. 1970, 9, 287–293. [Google Scholar] [CrossRef]
- Aldhaif, A.M.; Lopez, D.H.; Dadashazar, H.; Sorooshian, A. Sources, frequency, and chemical nature of dust events impacting the United States East Coast. Atmos. Environ. 2020, 231, 117456. [Google Scholar] [CrossRef]
- Prospero, J.M.; Collard, F.-X.; Molinié, J.; Jeannot, A. Characterizing the annual cycle of African dust transport to the Caribbean Basin and South America and its impact on the environment and air quality. Glob. Biogeochem. Cycles 2014, 28, 757–773. [Google Scholar] [CrossRef]
- Charlson, R.J.; Rodhe, H. Factors controlling the acidity of natural rainwater. Nature 1982, 295, 683. [Google Scholar] [CrossRef]
- Grimshaw, H.J.; Dolske, D.A. Rainfall concentrations and wet atmospheric deposition of phosphorus and other constituents in Florida, USA. Water Air Soil Pollut. 2002, 137, 117–140. [Google Scholar] [CrossRef]
- Satyanarayana, J.; Reddy, L.A.K.; Kulshrestha, M.J.; Rao, R.N.; Kulshrestha, U.C. Chemical composition of rain water and influence of airmass trajectories at a rural site in an ecological sensitive area of Western Ghats (India). J. Atmos. Chem. 2011, 66, 101–116. [Google Scholar] [CrossRef]
- de Mello, W.Z.; de Almeida, M.D. Rainwater chemistry at the summit and southern flank of the Itatiaia massif, Southeastern Brazil. Environ. Pollut. 2004, 129, 63–68. [Google Scholar] [CrossRef]
- Cao, Y.Z.; Wang, S.Y.; Zhang, G.; Luo, J.Y.; Lu, S.Y. Chemical characteristics of wet precipitation at an urban site of Guangzhou, South China. Atmos. Res. 2009, 94, 462–469. [Google Scholar] [CrossRef]
- Zhang, N.N.; Cao, J.J.; He, Y.Q.; Xiao, S. Chemical composition of rainwater at Lijiang on the Southeast Tibetan Plateau: influences from various air mass sources. J. Atmos. Chem. 2014, 71, 157–174. [Google Scholar] [CrossRef]
- Sanusi, A.; Wortham, H.; Millet, M.; Mirabel, P. Chemical composition of rainwater in eastern France. Atmos. Environ. 1996, 30, 59–71. [Google Scholar] [CrossRef]
- Rastegari Mehr, M.; Keshavarzi, B.; Sorooshian, A. Influence of natural and urban emissions on rainwater chemistry at a southwestern Iran coastal site. Sci. Total Environ. 2019, 668, 1213–1221. [Google Scholar] [CrossRef]
- Ahmady-Birgani, H.; Ravan, P.; Schlosser, J.S.; Cuevas-Robles, A.; AzadiAghdam, M.; Sorooshian, A. On the chemical nature of wet deposition over a major desiccated lake: Case study for Lake Urmia basin. Atmos. Res. 2020, 234, 104762. [Google Scholar] [CrossRef]
- Bisht, D.S.; Srivastava, A.K.; Joshi, H.; Ram, K.; Singh, N.; Naja, M.; Srivastava, M.K.; Tiwari, S. Chemical characterization of rainwater at a high-altitude site “Nainital” in the central Himalayas, India. Environ. Sci. Pollut. Res. Int. 2017, 24, 3959–3969. [Google Scholar] [CrossRef]
- Harrison, R.M.; Hester, R.E. Chemistry in the Marine Environment; Royal Society of Chemistry: Cambridge, UK, 2000. [Google Scholar]
- Zieger, P.; Vaisanen, O.; Corbin, J.C.; Partridge, D.G.; Bastelberger, S.; Mousavi-Fard, M.; Rosati, B.; Gysel, M.; Krieger, U.K.; Leck, C.; et al. Revising the hygroscopicity of inorganic sea salt particles. Nat. Commun. 2017, 8, 15883. [Google Scholar] [CrossRef]
- Jung, E.; Albrecht, B.A.; Jonsson, H.H.; Chen, Y.C.; Seinfeld, J.H.; Sorooshian, A.; Metcalf, A.R.; Song, S.; Fang, M.; Russell, L.M. Precipitation effects of giant cloud condensation nuclei artificially introduced into stratocumulus clouds. Atmos. Chem. Phys. 2015, 15, 5645–5658. [Google Scholar] [CrossRef] [Green Version]
- Sofiev, M.; Soares, J.; Prank, M.; de Leeuw, G.; Kukkonen, J. A regional-to-global model of emission and transport of sea salt particles in the atmosphere. J. Geophys. Res. Atmos. 2011, 116. [Google Scholar] [CrossRef] [Green Version]
- Engling, G.; Lee, J.J.; Tsai, Y.W.; Lung, S.C.C.; Chou, C.C.K.; Chan, C.Y. Size-resolved anhydrosugar composition in smoke aerosol from controlled field burning of rice straw. Aerosol Sci. Technol. 2009, 43, 662–672. [Google Scholar] [CrossRef]
- Zhang, Z.S.; Gao, J.; Engling, G.; Tao, J.; Chai, F.H.; Zhang, L.M.; Zhang, R.J.; Sang, X.F.; Chan, C.Y.; Lin, Z.J.; et al. Characteristics and applications of size-segregated biomass burning tracers in China’s Pearl River Delta region. Atmos. Environ. 2015, 102, 290–301. [Google Scholar] [CrossRef]
- Fourtziou, L.; Liakakou, E.; Stavroulas, I.; Theodosi, C.; Zarmpas, P.; Psiloglou, B.; Sciare, J.; Maggos, T.; Bairachtari, K.; Bougiatioti, A.; et al. Multi-tracer approach to characterize domestic wood burning in Athens (Greece) during wintertime. Atmos. Environ. 2017, 148, 89–101. [Google Scholar] [CrossRef]
- Wonaschütz, A.; Hersey, S.; Sorooshian, A.; Craven, J.; Metcalf, A.; Flagan, R.; Seinfeld, J. Impact of a large wildfire on water-soluble organic aerosol in a major urban area: the 2009 Station Fire in Los Angeles County. Atmos. Chem. Phys. 2011, 11, 8257–8270. [Google Scholar] [CrossRef] [Green Version]
- Reid, J.; Koppmann, R.; Eck, T.; Eleuterio, D. A review of biomass burning emissions part II: intensive physical properties of biomass burning particles. Atmos. Chem. Phys. 2005, 5, 799–825. [Google Scholar] [CrossRef] [Green Version]
- Aherne, J.; Mongeon, A.; Watmough, S.A. Temporal and spatial trends in precipitation chemistry in the Georgia Basin, British Columbia. J. Limnol. 2010, 69, 4–10. [Google Scholar] [CrossRef]
- Wetherbee, G.A.; Latysh, N.E.; Gordon, J.D. Spatial and temporal variability of the overall error of National Atmospheric Deposition Program measurements determined by the USGS collocated-sampler program, water years 1989-2001. Environ. Pollut. 2005, 135, 407–418. [Google Scholar] [CrossRef]
- Knipping, E.M.; Dabdub, D. Impact of chlorine emissions from sea-salt aerosol on coastal urban ozone. Environ. Sci. Technol. 2003, 37, 275–284. [Google Scholar] [CrossRef]
- Martens, C.S.; Wesolowski, J.J.; Harriss, R.C.; Kaifer, R. Chlorine loss from Puerto Rican and San Francisco Bay area marine aerosols. J. Geophys. Res. 1973, 78, 8778–8792. [Google Scholar] [CrossRef]
- Chesselet, R.; Buatmena, P.; Morelli, J. Variations in ionic ratios between reference sea-water and marine aerosols. J. Geophys. Res. 1972, 77, 5116. [Google Scholar] [CrossRef]
- Graedel, T.E.; Keene, W.C. Tropospheric budget of reactive chlorine. Glob. Biogeochem. Cycles 1995, 9, 47–77. [Google Scholar] [CrossRef]
- Keene, W.C.; Pszenny, A.A.P.; Galloway, J.N.; Hawley, M.E. Sea-Salt Corrections and Interpretation of Constituent Ratios in Marine Precipitation. J. Geophys. Res. Atmos. 1986, 91, 6647–6658. [Google Scholar] [CrossRef]
- Keene, W.C.; Long, M.S.; Pszenny, A.A.P.; Sander, R.; Maben, J.R.; Wall, A.J.; O’Halloran, T.L.; Kerkweg, A.; Fischer, E.V.; Schrems, O. Latitudinal variation in the multiphase chemical processing of inorganic halogens and related species over the eastern North and South Atlantic Oceans. Atmos. Chem. Phys. 2009, 9, 7361–7385. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, J.B.; Simpson, H.J.; Griffin, K.L.; Schuster, W.S.F. Precipitation chloride at West Point, NY: Seasonal patterns and possible contributions from non-seawater sources. Atmos. Environ. 2007, 41, 2240–2254. [Google Scholar] [CrossRef]
- Junge, C.E.; Werby, R.T. The concentration of chloride, sodium, potassium, calcium, and sulfate in rain water over the United States. J. Meteorol. 1958, 15, 417–425. [Google Scholar] [CrossRef] [Green Version]
- van der Swaluw, E.; Asman, W.A.; van Jaarsveld, H.; Hoogerbrugge, R. Wet deposition of ammonium, nitrate and sulfate in the Netherlands over the period 1992–2008. Atmos. Environ. 2011, 45, 3819–3826. [Google Scholar] [CrossRef]
- Cooksey, R.W. Correlational Statistics for Characterising Relationships. In Illustrating Statistical Procedures: Finding Meaning in Quantitative Data; Springer Publishing: New York, NY, USA, 2020; pp. 141–239. [Google Scholar]
- Prospero, J.M.; Nees, R.T.; Uematsu, M. Deposition rate of particulate and dissolved aluminum derived from Saharan dust in precipitation at Miami, Florida. J. Geophys. Res. Atmos. 1987, 92, 14723–14731. [Google Scholar] [CrossRef]
- Munger, J.W. Chemistry of atmospheric precipitation in the north-central United States: Influence of sulfate, nitrate, ammonia and calcareous soil particulates. Atmos. Environ. 1982, 16, 1633–1645. [Google Scholar] [CrossRef]
- Samara, C.; Tsitouridou, R. Fine and coarse ionic aerosol components in relation to wet and dry deposition. Water Air Soil Pollut. 2000, 120, 71–88. [Google Scholar] [CrossRef]
- Ma, L.; Dadashazar, H.; Braun, R.A.; MacDonald, A.B.; Aghdam, M.A.; Maudlin, L.C.; Sorooshian, A. Size-resolved characteristics of water-soluble particulate elements in a coastal area: Source identification, influence of wildfires, and diurnal variability. Atmos. Environ. 2019, 206, 72–84. [Google Scholar] [CrossRef]
- Savoie, D.L.; Prospero, J.M. Particle-size distribution of nitrate and sulfate in the marine atmosphere. Geophys. Res. Lett. 1982, 9, 1207–1210. [Google Scholar] [CrossRef] [Green Version]
- Savoie, D.L.; Prospero, J.M.; Saltzman, E.S. Non-sea-salt sulfate and nitrate in trade-wind aerosols at Barbados - Evidence for long-range transport. J. Geophys. Res. Atmos. 1989, 94, 5069–5080. [Google Scholar] [CrossRef]
- Zhang, D.; Shi, G.-Y.; Iwasaka, Y.; Hu, M. Mixture of sulfate and nitrate in coastal atmospheric aerosols: individual particle studies in Qingdao (36°04′ N, 120°21′ E), China. Atmos. Environ. 2000, 34, 2669–2679. [Google Scholar] [CrossRef]
- Hayden, K.L.; Macdonald, A.M.; Gong, W.; Toom-Sauntry, D.; Anlauf, K.G.; Leithead, A.; Li, S.M.; Leaitch, W.R.; Noone, K. Cloud processing of nitrate. J. Geophys. Res. Atmos. 2008, 113. [Google Scholar] [CrossRef] [Green Version]
- Barnes, I.; Hjorth, J.; Mihalopoulos, N. Dimethyl sulfide and dimethyl sulfoxide and their oxidation in the atmosphere. Chem. Rev. 2006, 106, 940–975. [Google Scholar] [CrossRef]
- Grahame, T.; Schlesinger, R. Evaluating the health risk from secondary sulfates in eastern North American regional ambient air particulate matter. Inhal. Toxicol. 2005, 17, 15–27. [Google Scholar] [CrossRef]
- Milford, J.B.; Davidson, C.I. The sizes of particulate sulfate aed nitrate in the Atmosphere—A-Review. JAPCA 1987, 37, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Hossein Mardi, A.; Dadashazar, H.; MacDonald, A.B.; Crosbie, E.; Coggon, M.M.; Azadi Aghdam, M.; Woods, R.K.; Jonsson, H.H.; Flagan, R.C.; Seinfeld, J.H. Effects of biomass burning on stratocumulus droplet characteristics, drizzle rate, and composition. J. Geophys. Res. Atmos. 2019, 124, 12301–12318. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | pH | Ca2+ | Mg2+ | K+ | Na+ | NH4+ | NO3− | Cl− | SO42− |
|---|---|---|---|---|---|---|---|---|---|
| pH | 1 | ||||||||
| Ca2+ | 0.31 | 1 | |||||||
| Mg2+ | 0.33 | 1 | |||||||
| K+ | 0.45 | 0.16 | 0.35 | 1 | |||||
| Na+ | 0.31 | 0.99 | 0.26 | 1 | |||||
| NH4+ | 0.51 | 0.48 | 0.22 | 0.42 | 0.18 | 1 | |||
| NO3− | 0.07 | 0.89 | 0.18 | 0.09 | 0.16 | 0.47 | 1 | ||
| Cl− | 0.29 | 0.99 | 0.26 | 1.00 | 0.17 | 0.15 | 1 | ||
| SO42− | −0.29 | 0.41 | 0.66 | 0.20 | 0.66 | 0.40 | 0.40 | 0.64 | 1 |
| Aerosol Parameters | Wet Deposition | |||||||
|---|---|---|---|---|---|---|---|---|
| Ca2+ | Mg2+ | K+ | Na+ | NH4+ | NO3− | Cl− | SO42− | |
| Combustion | 0.15 | 0.16 | 0.14 | |||||
| Fresh Sea Salt | 0.32 | 0.33 | 0.33 | 0.30 | ||||
| Aged Sea Salt | ||||||||
| Secondary Sulfate | −0.14 | 0.15 | 0.23 | |||||
| Shipping Emissions | −0.17 | −0.17 | −0.17 | |||||
| Dust | −0.16 | −0.18 | ||||||
| Mg | 0.23 | 0.24 | 0.24 | |||||
| Na | 0.32 | 0.33 | 0.33 | 0.20 | ||||
| NO3− | 0.17 | |||||||
| Cl− | 0.35 | 0.36 | 0.35 | 0.17 | ||||
| PMcoarse | 0.18 | |||||||
| Fine Soil | −0.18 | −0.19 | −0.16 | −0.19 | −0.21 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corral, A.F.; Dadashazar, H.; Stahl, C.; Edwards, E.-L.; Zuidema, P.; Sorooshian, A. Source Apportionment of Aerosol at a Coastal Site and Relationships with Precipitation Chemistry: A Case Study over the Southeast United States. Atmosphere 2020, 11, 1212. https://doi.org/10.3390/atmos11111212
Corral AF, Dadashazar H, Stahl C, Edwards E-L, Zuidema P, Sorooshian A. Source Apportionment of Aerosol at a Coastal Site and Relationships with Precipitation Chemistry: A Case Study over the Southeast United States. Atmosphere. 2020; 11(11):1212. https://doi.org/10.3390/atmos11111212
Chicago/Turabian StyleCorral, Andrea F., Hossein Dadashazar, Connor Stahl, Eva-Lou Edwards, Paquita Zuidema, and Armin Sorooshian. 2020. "Source Apportionment of Aerosol at a Coastal Site and Relationships with Precipitation Chemistry: A Case Study over the Southeast United States" Atmosphere 11, no. 11: 1212. https://doi.org/10.3390/atmos11111212