No Particle Mass Enhancement from Induced Atmospheric Ageing at a Rural Site in Northern Europe

 , , and
, , and 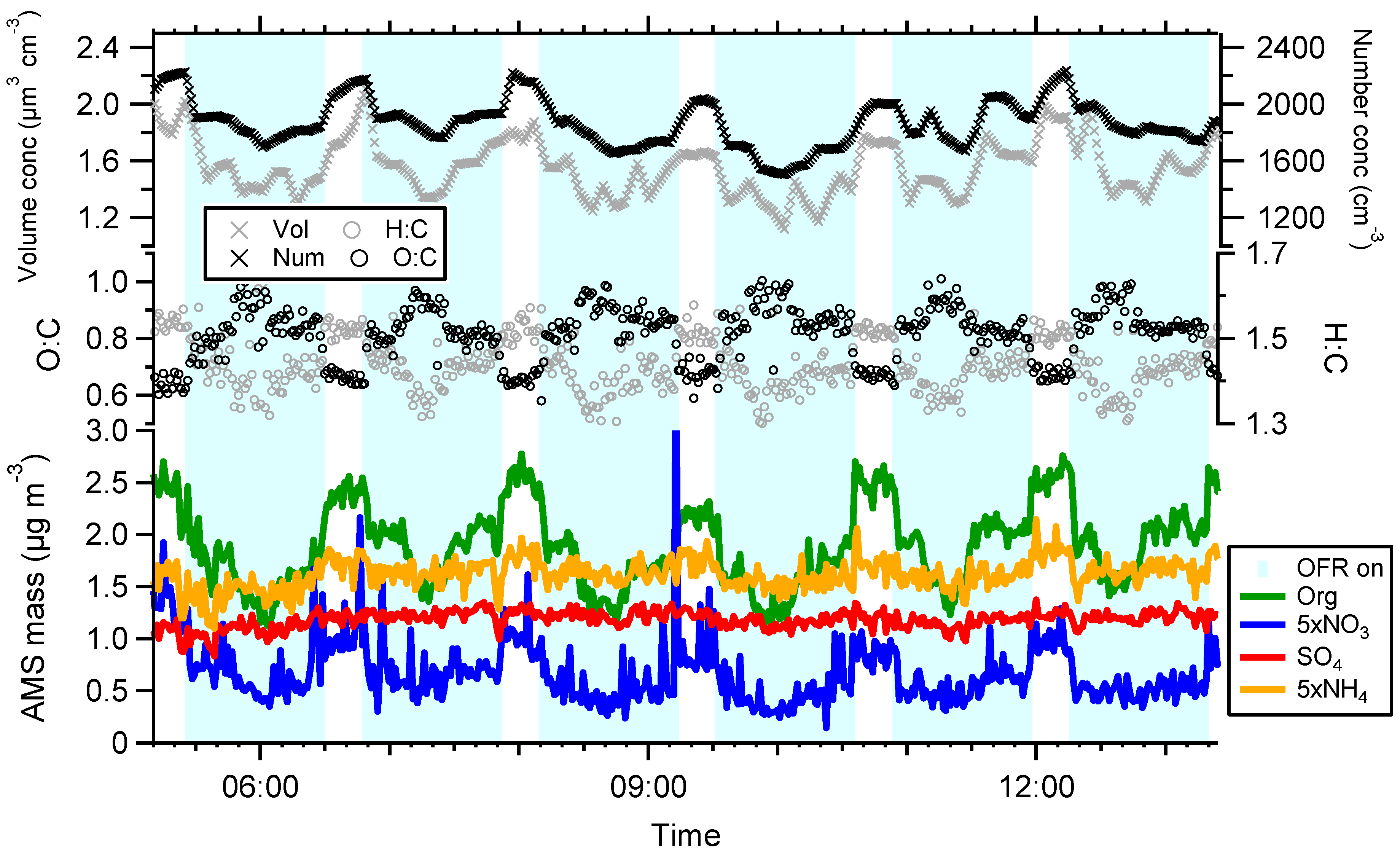{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Methods
2.1. Campaign Site and Set-Up
2.2. Instrumentation
2.3. Analysis
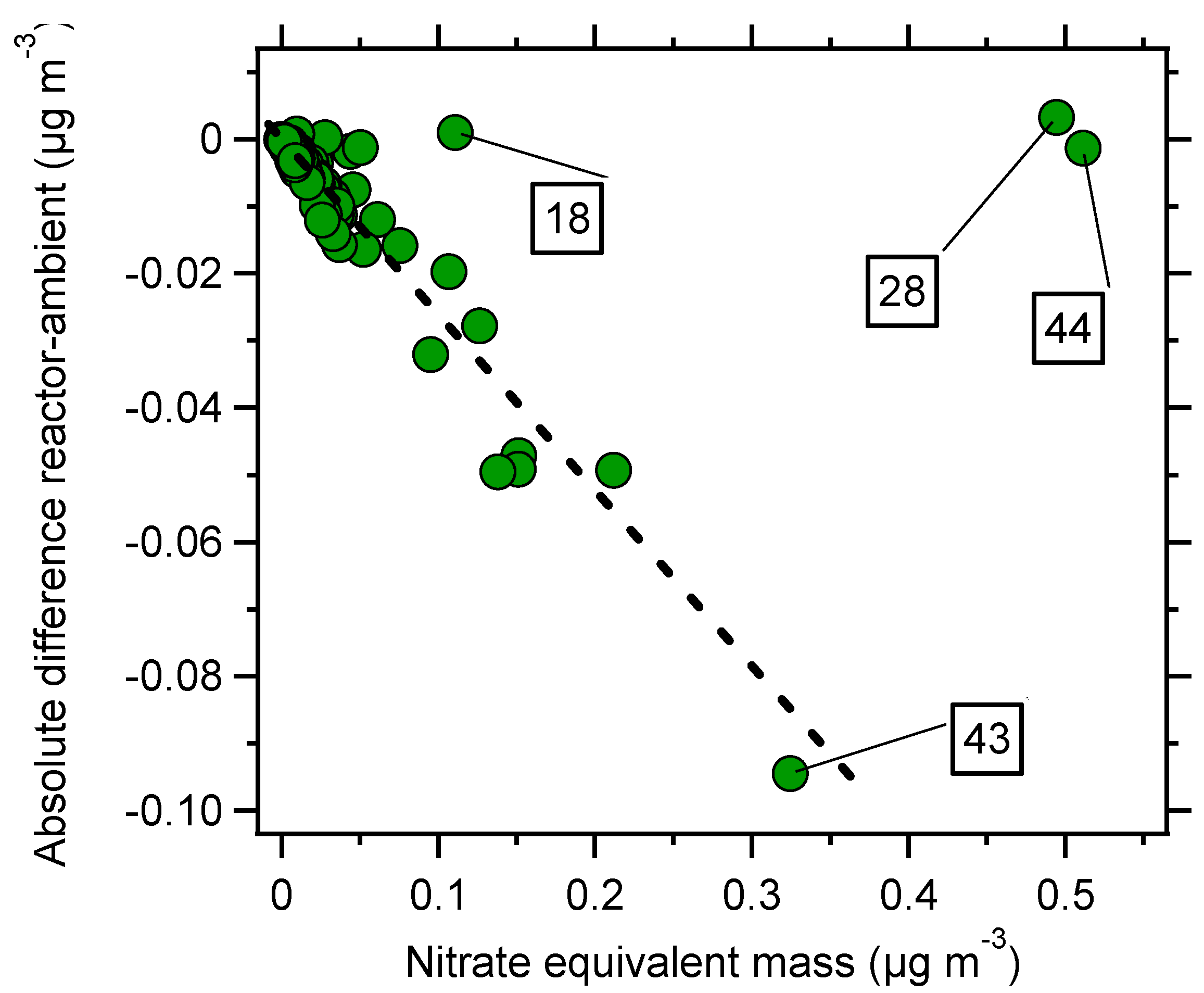
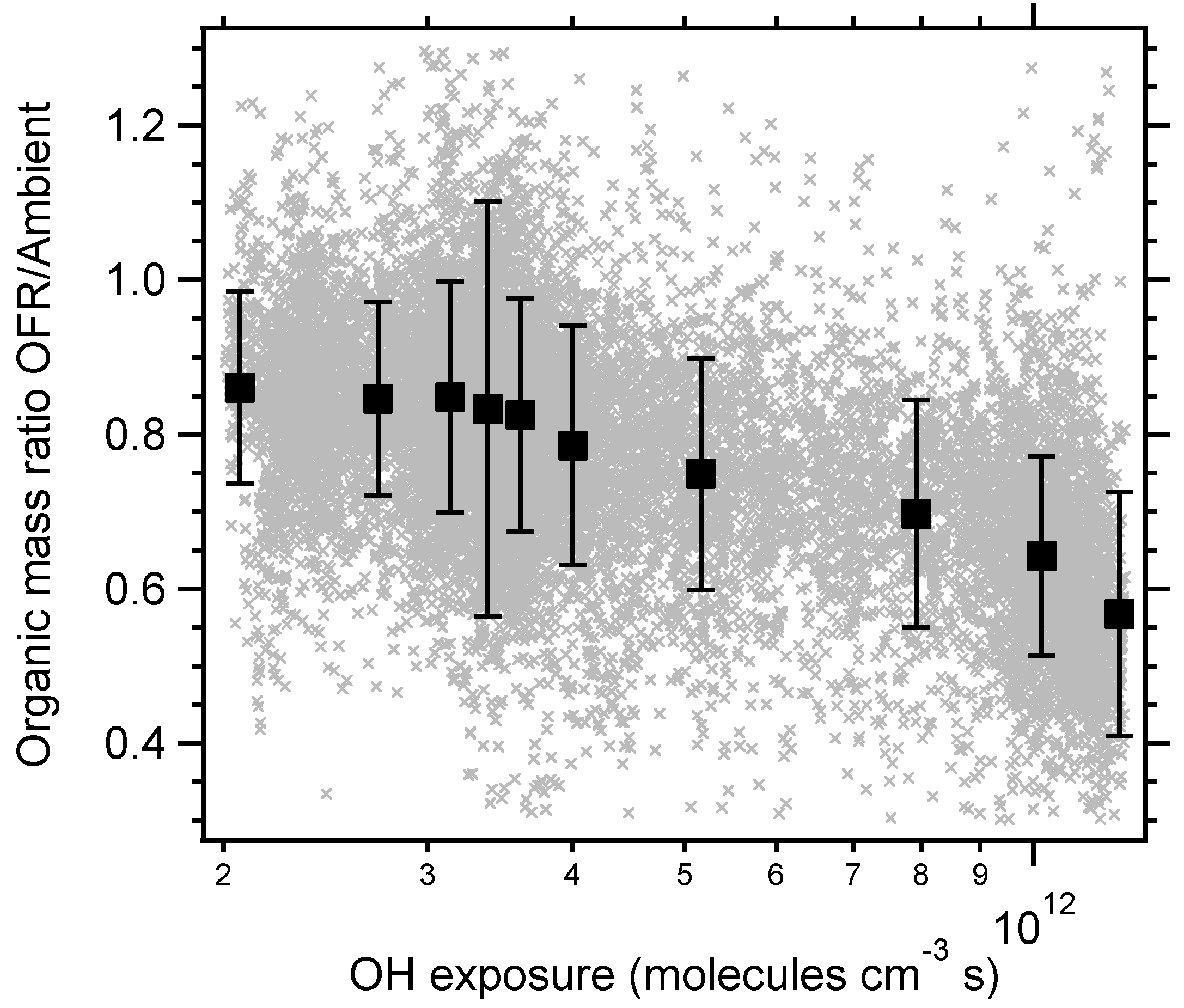
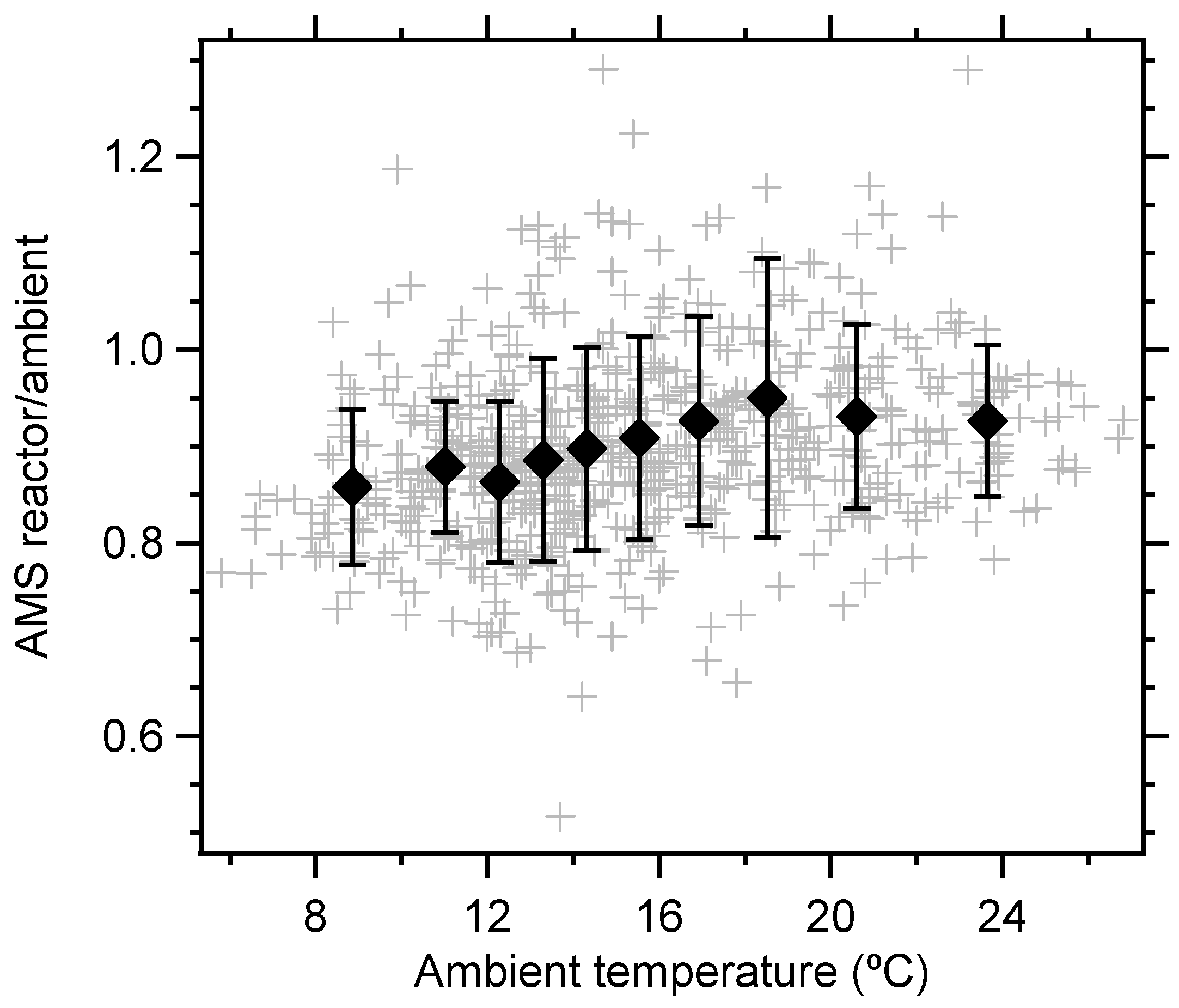
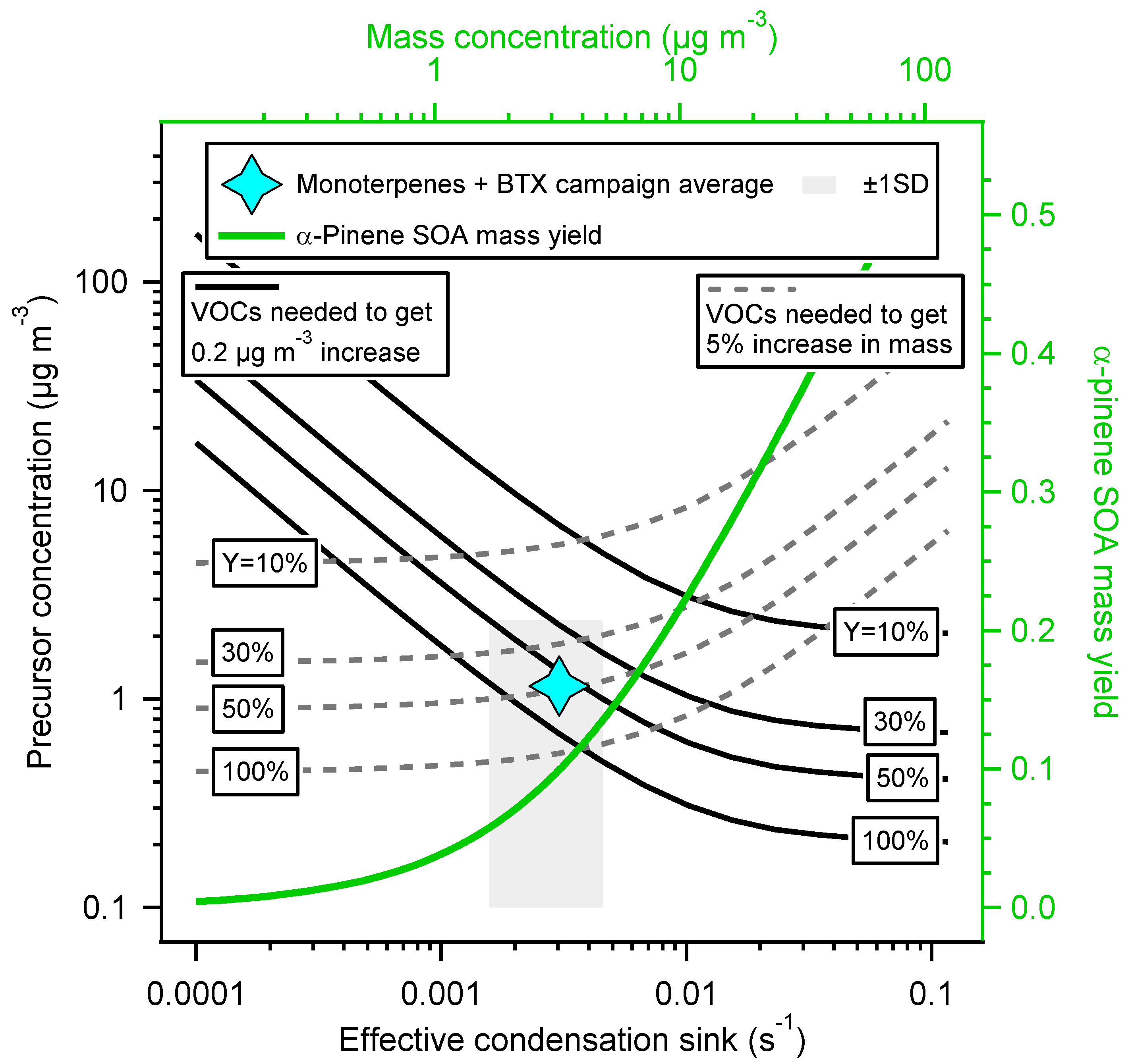
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, Q.; Jimenez, J.L.; Canagaratna, M.R.; Allan, J.D.; Coe, H.; Ulbrich, I.; Alfarra, M.R.; Takami, A.; Middlebrook, A.M.; Sun, Y.L.; et al. Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes. Geophys. Res. Lett. 2007, 34. [Google Scholar] [CrossRef] [Green Version]
- Pandis, S.N.; Skyllakou, K.; Florou, K.; Kostenidou, E.; Kaltsonoudis, C.; Hasa, E.; Presto, A.A. Urban particulate matter pollution: A tale of five cities. Faraday Discuss. 2016, 189, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Glasius, M.; Goldstein, A.H. Recent Discoveries and Future Challenges in Atmospheric Organic Chemistry. Environ. Sci. Technol. 2016, 50, 2754–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava, M.; Cappa, C.D.; Fan, J.; Goldstein, A.H.; Guenther, A.B.; Jimenez, J.L.; Kuang, C.; Laskin, A.; Martin, S.T.; Ng, N.L.; et al. Recent advances in understanding secondary organic aerosol: Implications for global climate forcing. Rev. Geophys. 2017, 55, 509–559. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, J.L. Concluding remarks: Faraday Discussion on chemistry in the urban atmosphere. Faraday Discuss. 2016, 189, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Bruns, E.A.; El Haddad, I.; Keller, A.; Klein, F.; Kumar, N.K.; Pieber, S.M.; Corbin, J.C.; Slowik, J.G.; Brune, W.H.; Baltensperger, U.; et al. Inter-comparison of laboratory smog chamber and flow reactor systems on organic aerosol yield and composition. Atmos. Meas. Tech. 2015, 8, 2315–2332. [Google Scholar] [CrossRef] [Green Version]
- Lambe, A.T.; Chhabra, P.S.; Onasch, T.B.; Brune, W.H.; Hunter, J.F.; Kroll, J.H.; Cummings, M.J.; Brogan, J.F.; Parmar, Y.; Worsnop, D.R.; et al. Effect of oxidant concentration, exposure time, and seed particles on secondary organic aerosol chemical composition and yield. Atmos. Chem. Phys. 2015, 15, 3063–3075. [Google Scholar] [CrossRef] [Green Version]
- Platt, S.M.; Haddad, I.E.; Zardini, A.A.; Clairotte, M.; Astorga, C.; Wolf, R.; Slowik, J.G. Secondary organic aerosol formation from gasoline vehicle emissions in a new mobile environmental reaction chamber. Atmos. Chem. Phys. 2013, 13, 9141–9158. [Google Scholar] [CrossRef] [Green Version]
- Ortega, A.M.; Hayes, P.L.; Peng, Z.; Palm, B.B.; Hu, W.; Day, D.A.; Li, R.; Cubison, M.J.; Brune, W.H.; Graus, M.; et al. Real-time measurements of secondary organic aerosol formation and aging from ambient air in an oxidation flow reactor in the Los Angeles area. Atmos. Chem. Phys. 2016, 16, 7411–7433. [Google Scholar] [CrossRef] [Green Version]
- George, I.J.; Slowik, J.; Abbatt, J.P.D. Chemical aging of ambient organic aerosol from heterogeneous reaction with hydroxyl radicals. Geophys. Res. Lett. 2008, 35. [Google Scholar] [CrossRef]
- Palm, B.B.; Campuzano-Jost, P.; Ortega, A.M.; Day, D.A.; Kaser, L.; Jud, W.; Karl, T.; Hansel, A.; Hunter, J.F.; Cross, E.S.; et al. In situ secondary organic aerosol formation from ambient pine forest air using an oxidation flow reactor. Atmos. Chem. Phys. 2016, 16, 2943–2970. [Google Scholar] [CrossRef] [Green Version]
- Palm, B.B.; Campuzano-Jost, P.; Day, D.A.; Ortega, A.M.; Fry, J.L.; Brown, S.S.; Zarzana, K.J.; Dube, W.; Wagner, N.L.; Draper, D.C.; et al. Secondary organic aerosol formation from in situ OH, O-3, and NO3 oxidation of ambient forest air in an oxidation flow reactor. Atmos. Chem. Phys. 2017, 17, 5331–5354. [Google Scholar] [CrossRef]
- Slowik, J.G.; Wong, J.P.S.; Abbatt, J.P.D. Real-time, controlled OH-initiated oxidation of biogenic secondary organic aerosol. Atmos. Chem. Phys. 2012, 12, 9775–9790. [Google Scholar] [CrossRef] [Green Version]
- Palm, B.B.; Sá, S.S.D.; Day, D.A.; Campuzano-Jost, P.; Hu, W.; Seco, R.; Sjostedt, S.J.; Park, J.H.; Guenther, A.B.; Kim, S.; et al. Secondary organic aerosol formation from ambient air in an oxidation flow reactor in central Amazonia. Atmos. Chem. Phys. Discuss. 2018, 18, 467–493. [Google Scholar] [CrossRef] [Green Version]
- Kang, E.; Lee, M.; Brune, W.H. Taehyung Lee Photochemical aging of organic and inorganic ambient aerosol from the Potential Aerosol Mass (PAM) reactor experiment in East Asia. Atmos. Chem. Phys. Discuss. 2017. [Google Scholar] [CrossRef]
- Kang, E.; Root, M.J.; Toohey, D.W.; Brune, W.H. Introducing the concept of Potential Aerosol Mass (PAM). Atmos. Chem. Phys. 2007, 7, 5727–5744. [Google Scholar] [CrossRef] [Green Version]
- Lambe, A.T.; Ahern, A.T.; Williams, L.R.; Slowik, J.G.; Wong, J.P.S.; Abbatt, J.P.D.; Brune, W.H.; Ng, N.L.; Wright, J.P.; Croasdale, D.R.; et al. Characterization of aerosol photooxidation flow reactors: Heterogeneous oxidation, secondary organic aerosol formation and cloud condensation nuclei activity measurements. Atmos. Meas. Tech. 2011, 4, 445–461. [Google Scholar] [CrossRef]
- Li, R.; Palm, B.B.; Ortega, A.M.; Hlywiak, J.; Hu, W.; Peng, Z.; Day, D.A.; Knote, C.; Brune, W.H.; De Gouw, J.A.; et al. Modeling the Radical Chemistry in an Oxidation Flow Reactor: Radical Formation and Recycling, Sensitivities, and the OH Exposure Estimation Equation. J. Phys. Chem. A 2015, 119, 4418–4432. [Google Scholar] [CrossRef]
- Peng, Z.; Day, D.A.; Stark, H.; Li, R.; Lee-Taylor, J.; Palm, B.B.; Brune, W.H.; Jimenez, J.L. HOx radical chemistry in oxidation flow reactors with low-pressure mercury lamps systematically examined by modeling. Atmos. Meas. Tech. 2015, 8, 4863–4890. [Google Scholar] [CrossRef] [Green Version]
- Wiedensohler, A.; Birmili, W.; Nowak, A.; Sonntag, A.; Weinhold, K.; Merkel, M.; Wehner, B.; Tuch, T.; Pfeifer, S.; Fiebig, M.; et al. Mobility particle size spectrometers: Harmonization of technical standards and data structure to facilitate high quality long-term observations of atmospheric particle number size distributions. Atmos. Meas. Tech. 2012, 5, 657–685. [Google Scholar] [CrossRef]
- DeCarlo, P.F.; Kimmel, J.R.; Trimborn, A.; Northway, M.J.; Jayne, J.T.; Aiken, A.C.; Gonin, M.; Fuhrer, K.; Horvath, T.; Docherty, K.S.; et al. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 2006, 78, 8281–8289. [Google Scholar] [CrossRef] [PubMed]
- Graus, M.; Müller, M.; Hansel, A. High Resolution PTR-TOF: Quantification and Formula Confirmation of VOC in Real Time. J. Am. Soc. Mass Spectrom. 2010, 21, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Lindinger, W.; Hansel, A.; Jordan, A. On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) - Medical applications, food control and environmental research. Int. J. Mass Spectrom. 1998, 173, 191–241. [Google Scholar] [CrossRef]
- Von der Weiden, S.L.; Drewnick, F.; Borrmann, S. Particle Loss Calculator—A new software tool for the assessment of the performance of aerosol inlet systems. Atmos. Meas. Tech. 2009, 2, 479–494. [Google Scholar] [CrossRef]
- Middlebrook, A.M.; Bahreini, R.; Jimenez, J.L.; Canagaratna, M.R. Evaluation of Composition-Dependent Collection Efficiencies for the Aerodyne Aerosol Mass Spectrometer using Field Data. Aerosol Sci. Technol. 2012, 46, 258–271. [Google Scholar] [CrossRef]
- Farmer, D.K.; Matsunaga, A.; Docherty, K.S.; Surratt, J.D.; Seinfeld, J.H.; Ziemann, P.J.; Jimenez, J.L. Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry. Proc. Natl. Acad. Sci. USA 2010, 107, 6670–6675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canagaratna, M.R.; Jimenez, J.L.; Kroll, J.H.; Chen, Q.; Kessler, S.H.; Massoli, P.; Hildebrandt Ruiz, L.; Fortner, E.; Williams, L.R.; Wilson, K.R.; et al. Worsnop: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: Characterization, improved calibration, and implications. Atmos. Chem. Phys. 2015, 15, 253–272. [Google Scholar] [CrossRef]
- Holzinger, R. PTRwid: A new widget tool for processing PTR-TOF-MS data. Atmos. Meas. Tech. 2015, 8, 3903–3922. [Google Scholar] [CrossRef] [Green Version]
- Holzinger, R.; Kasper-Giebl, A.; Staudinger, M.; Schauer, G.; Röckmann, T. Analysis of the chemical composition of organic aerosol at the Mt. Sonnblick observatory using a novel high mass resolution thermal-desorption proton-transfer-reaction mass-spectrometer (hr-TD-PTR-MS). Atmos. Chem. Phys. 2010, 10, 10111–10128. [Google Scholar] [CrossRef] [Green Version]
- Cappellin, L.; Biasioli, F.; Schuhfried, E.; Soukoulis, C.; Märk, T.D.; Gasperi, F. Extending the dynamic range of proton transfer reaction time-of-flight mass spectrometers by a novel dead time correction. Rapid Commun. Mass Spectrom. 2011, 25, 179–183. [Google Scholar] [CrossRef]
- Ahlberg, E.; Eriksson, A.; Brune, W.H.; Roldin, P.; Svenningsson, B. Effect of salt seed particle surface area, composition and phase on secondary organic aerosol mass yields in oxidation flow reactors. Atmos. Chem. Phys. 2019, 19, 2701–2712. [Google Scholar] [CrossRef] [Green Version]
- Hinds, W.C. Aerosol Technology: Properties, Behavior, and Measurement of Airborne Particles; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Lugg, G.A. Diffusion Coefficients of Some Organic and Other Vapors in Air. Anal. Chem. 1968, 40, 1072–1077. [Google Scholar] [CrossRef]
- Tang, M.J.; Shiraiwa, M.; Pöschl, U.; Cox, R.A.; Kalberer, M. Compilation and evaluation of gas phase diffusion coefficients of reactive trace gases in the atmosphere: Volume 2. Diffusivities of organic compounds, pressure-normalised mean free paths, and average Knudsen numbers for gas uptake calculations. Atmos. Chem. Phys. 2015, 15, 5585–5598. [Google Scholar] [CrossRef] [Green Version]
- Mcmurry, P.H.; Grosjean, D. Gas and Aerosol Wall Losses in Teflon Film Smog Chambers. Environ. Sci. Technol. 1985, 19, 1176–1182. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, A.; Ziemann, P.J. Gas-Wall Partitioning of Organic Compounds in a Teflon Film Chamber and Potential Effects on Reaction Product and Aerosol Yield Measurements. Aerosol Sci. Technol. 2010, 44, 881–892. [Google Scholar] [CrossRef]
- Pagonis, D.; Krechmer, J.E.; de Gouw, J.; Jimenez, J.L.; Ziemann, P.J. Effects of Gas-Wall Partitioning in Teflon Tubing and Instrumentation on Time-Resolved Measurements of Gas-Phase Organic Compounds. Atmos. Meas. Tech. Discuss. 2017, 10, 4687–4696. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahlberg, E.; Ausmeel, S.; Eriksson, A.; Holst, T.; Karlsson, T.; Brune, W.H.; Frank, G.; Roldin, P.; Kristensson, A.; Svenningsson, B. No Particle Mass Enhancement from Induced Atmospheric Ageing at a Rural Site in Northern Europe. Atmosphere 2019, 10, 408. https://doi.org/10.3390/atmos10070408
Ahlberg E, Ausmeel S, Eriksson A, Holst T, Karlsson T, Brune WH, Frank G, Roldin P, Kristensson A, Svenningsson B. No Particle Mass Enhancement from Induced Atmospheric Ageing at a Rural Site in Northern Europe. Atmosphere. 2019; 10(7):408. https://doi.org/10.3390/atmos10070408
Chicago/Turabian StyleAhlberg, Erik, Stina Ausmeel, Axel Eriksson, Thomas Holst, Tomas Karlsson, William H. Brune, Göran Frank, Pontus Roldin, Adam Kristensson, and Birgitta Svenningsson. 2019. "No Particle Mass Enhancement from Induced Atmospheric Ageing at a Rural Site in Northern Europe" Atmosphere 10, no. 7: 408. https://doi.org/10.3390/atmos10070408