
Role of Telomerase in the Cardiovascular System

_Haendeler.png)

{kind=link}
{kind=link}
Abstract
:1. Introduction
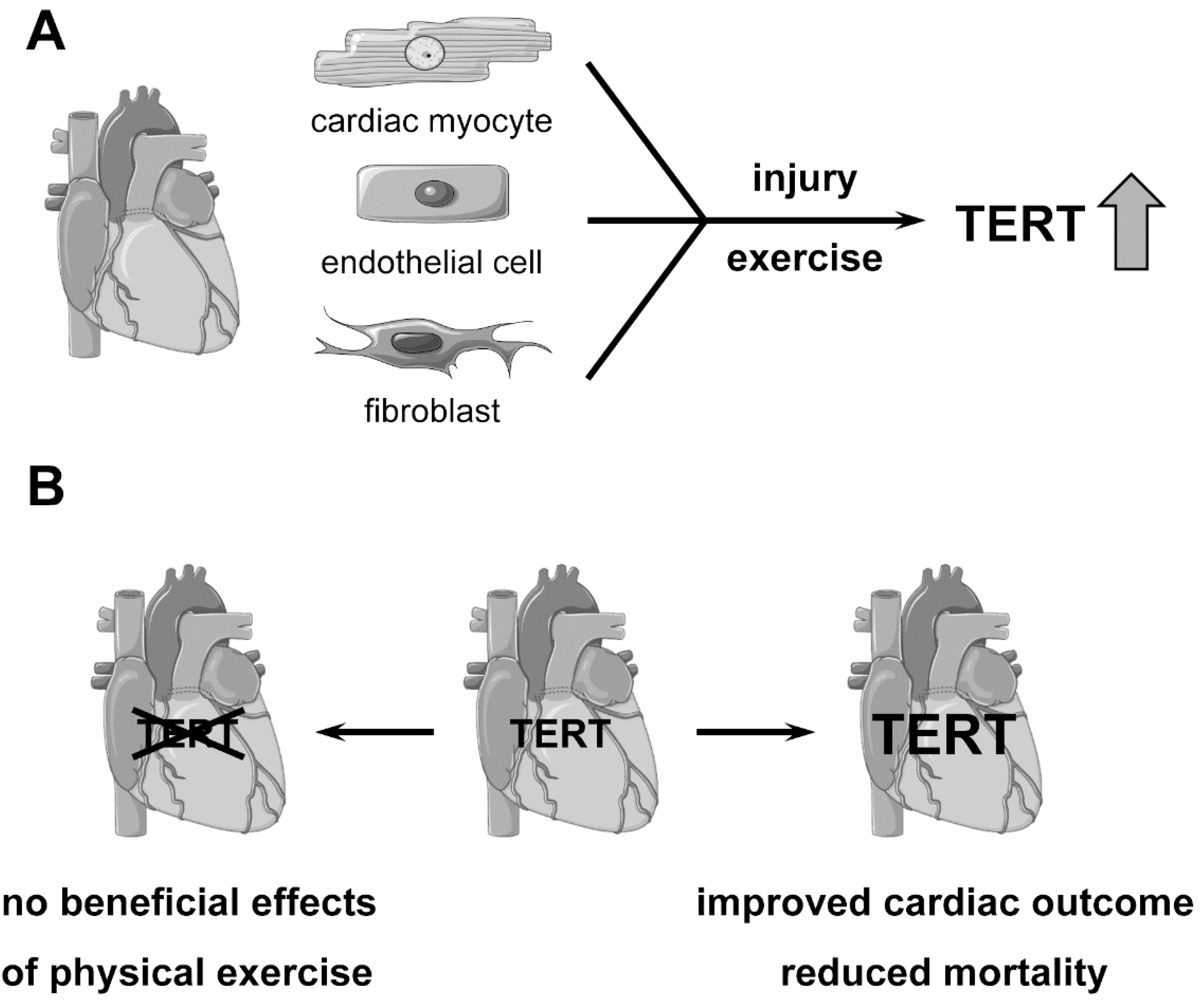
2. Role of Telomerase and TERT in the Heart
3. Role of Telomerase and TERT in Myocardial Infarction and Heart Failure
4. Physical Exercise, Telomerase and TERT in the Heart
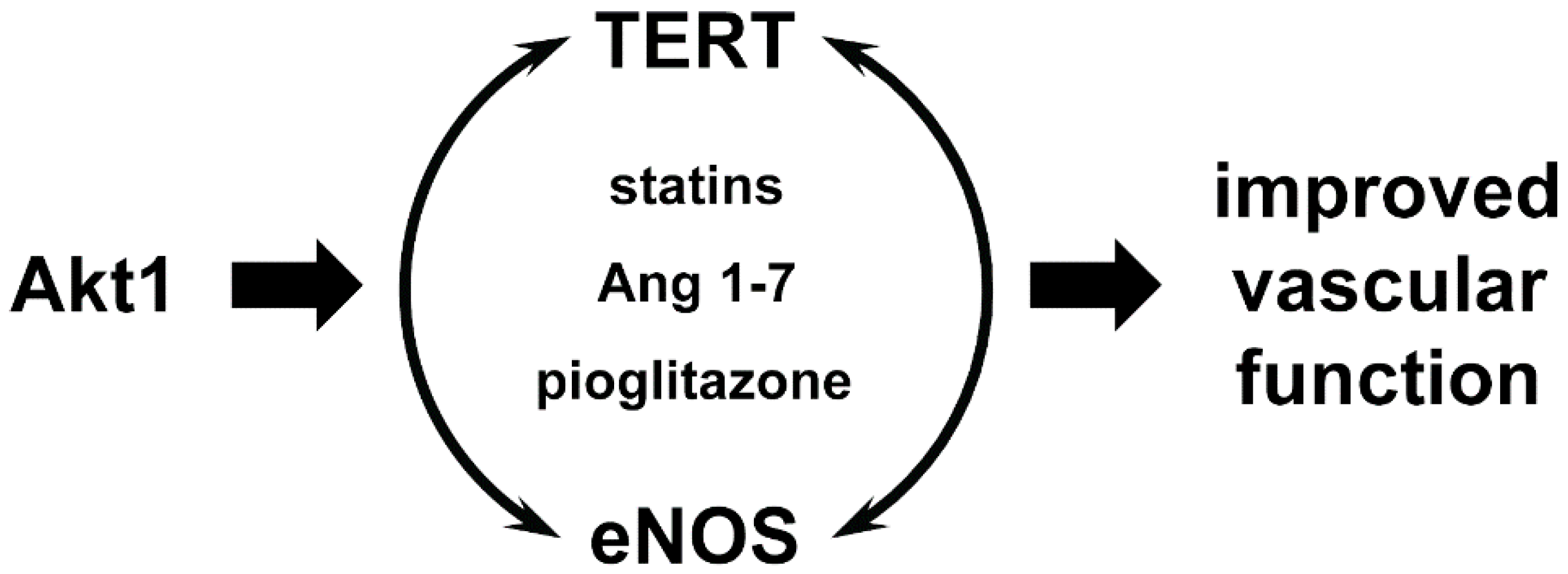
5. Role of Telomerase and TERT in the Vascular System
6. Extranuclear Functions of TERT in the Cardiovascular System
Acknowledgments
Conflicts of Interest
References
- Blasco, M.A. Telomeres and human disease: Ageing, cancer and beyond. Nat. Rev. Genet. 2005, 6, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, N.R.; Wright, W.E.; Shay, J.W. Telomerase and differentiation in multicellular organisms: Turn it off, turn it on, and turn it off again. Differentiation 2002, 69, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Kourembanas, S. Mechanisms of telomerase induction during vascular smooth muscle cell proliferation. Circ. Res. 2001, 89, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J.; Hoffmann, J.; Diehl, J.F.; Vasa, M.; Spyridopoulos, I.; Zeiher, A.M.; Dimmeler, S. Antioxidants inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ. Res. 2004, 94, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Werner, C.; Hanhoun, M.; Widmann, T.; Kazakov, A.; Semenov, A.; Pöss, J.; Bauersachs, J.; Thum, T.; Pfreundschuh, M.; Müller, P.; et al. Effects of physical exercise on myocardial telomere-regulating proteins, survival pathways, and apoptosis. J. Am. Coll. Cardiol. 2008, 52, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Werner, C.; Fürster, T.; Widmann, T.; Pöss, J.; Roggia, C.; Hanhoun, M.; Scharhag, J.; Büchner, N.; Meyer, T.; Kindermann, W.; et al. Physical exercise prevents cellular senescence in circulating leukocytes and in the vessel wall. Circulation 2009, 120, 2438–2447. [Google Scholar] [CrossRef] [PubMed]
- Richardson, G.D.; Breault, D.; Horrocks, G.; Cormack, S.; Hole, N.; Owens, W.A. Telomerase expression in the mammalian heart. FASEB J. 2012, 26, 4832–4840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasco, M.A.; Lee, H.W.; Hande, M.P.; Samper, E.; Lansdorp, P.M.; DePinho, R.A.; Greider, C.W. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 1997, 91, 25–34. [Google Scholar] [CrossRef]
- Lee, H.W.; Blasco, M.A.; Gottlieb, G.J.; Horner, J.W., 2nd; Greider, C.W.; DePinho, R.A. Essential role of mouse telomerase in highly proliferative organs. Nature 1998, 392, 569–574. [Google Scholar] [PubMed]
- Herrera, E.; Samper, E.; Martin-Caballero, J.; Flores, J.M.; Lee, H.W.; Blasco, M.A. Disease states associated with telomerase deficiency appear earlier in mice with short telomeres. EMBO J. 1999, 18, 2950–2960. [Google Scholar] [CrossRef] [PubMed]
- Leri, A.; Franco, S.; Zacheo, A.; Barlucchi, L.; Chimenti, S.; Limana, F.; Nadal-Ginard, B.; Kajstura, J.; Anversa, P.; Blasco, M.A. Ablation of telomerase and telomere loss leads to cardiac dilatation and heart failure associated with p53 upregulation. EMBO J. 2003, 22, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Leri, A.; Barlucchi, L.; Limana, F.; Deptala, A.; Darzynkiewicz, Z.; Hintze, T.H.; Kajstura, J.; Nadal-Ginard, B.; Anversa, P. Telomerase expression and activity are coupled with myocyte proliferation and preservation of telomeric length in the failing heart. Proc. Natl. Acad. Sci. USA 2001, 98, 8626–8631. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Vera, E.; Bernardes de Jesus, B.; Foronda, M.; Flores, J.M.; Blasco, M.A. The rate of increase of short telomeres predicts longevity in mammals. Cell Rep. 2012, 2, 732–737. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, T.; Rietzschel, E.R.; de Buyzere, M.L.; Van Criekinge, W.; Bekaert, S. Telomere length and cardiovascular aging: The means to the ends? Ageing Res. Rev. 2011, 10, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Bernardes de Jesus, B.; Vera, E.; Schneeberger, K.; Tejera, A.M.; Ayuso, E.; Bosch, F.; Blasco, M.A. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol. Med. 2012, 4, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Taffet, G.E.; Youker, K.A.; Entman, M.L.; Overbeek, P.A.; Michael, L.H.; Schneider, M.D. Telomerase reverse transcriptase promotes cardiac muscle cell proliferation, hypertrophy, and survival. Proc. Natl. Acad. Sci. USA 2001, 98, 10308–10313. [Google Scholar] [CrossRef] [PubMed]
- Herbert, B.; Pitts, A.E.; Baker, S.I.; Hamilton, S.E.; Wright, W.E.; Shay, J.W.; Corey, D.R. Inhibition of human telomerase in immortal human cells leads to progressive telomere shortening and cell death. Proc. Natl. Acad. Sci. USA 1999, 96, 14276–14281. [Google Scholar] [CrossRef] [PubMed]
- Goytisolo, F.A.; Samper, E.; Martin-Caballero, J.; Finnon, P.; Herrera, E.; Flores, J.M.; Bouffler, S.D.; Blasco, M.A. Short telomeres result in organismal hypersensitivity to ionizing radiation in mammals. J. Exp. Med. 2000, 192, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Breault, D.T.; Min, I.M.; Carlone, D.L.; Farilla, L.G.; Ambruzs, D.M.; Henderson, D.E.; Algra, S.; Montgomery, R.K.; Wagers, A.J.; Hole, N. Generation of mTert-GFP mice as a model to identify and study tissue progenitor cells. Proc. Natl. Acad. Sci. USA 2008, 105, 10420–10425. [Google Scholar] [CrossRef] [PubMed]
- Poss, K.D.; Wilson, L.G.; Keating, M.T. Heart regeneration in zebrafish. Science 2002, 298, 2188–2190. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, D.; González-Rosa, J.M.; Guzmán-Martínez, G.; Gutiérrez-Gutiérrez, Ó.; Aguado, T.; Sánchez-Ferrer, C.; Marques, I.J.; Galardi-Castilla, M.; de Diego, I.; Gómez, M.J.; et al. Telomerase is essential for zebrafish heart regeneration. Cell Rep. 2015, 12, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Bär, C.; Bernardes de Jesus, B.; Serrano, R.; Tejera, A.; Ayuso, E.; Jimenez, V.; Formentini, I.; Bobadilla, M.; Mizrahi, J.; de Martino, A.; et al. Telomerase expression confers cardioprotection in the adult mouse heart after acute myocardial infarction. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Leri, A.; Kajstura, J.; Anversa, P. Role of cardiac stem cells in cardiac pathophysiology: A paradigm shift in human myocardial biology. Circ. Res. 2011, 109, 941–961. [Google Scholar] [CrossRef] [PubMed]
- Senyo, S.E.; Steinhauser, M.L.; Pizzimenti, C.L.; Yang, V.K.; Cai, L.; Wang, M.; Wu, T.D.; Guerquin-Kern, J.L.; Lechene, C.P.; Lee, R.T. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 2013, 493, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Hambrecht, R.; Fiehn, E.; Weigl, C.; Gielen, S.; Hamann, C.; Kaiser, R.; Yu, J.; Adams, V.; Niebauer, J.; Schuler, G. Regular physical exercise corrects endothelial dysfunction and improves exercise capacity in patients with chronic heart failure. Circulation 1998, 98, 2709–2715. [Google Scholar] [CrossRef] [PubMed]
- Hambrecht, R.; Wolf, A.; Gielen, S.; Linke, A.; Hofer, J.; Erbs, S.; Schoene, N.; Schuler, G. Effect of exercise on coronary endothelial function in patients with coronary artery disease. N. Engl. J. Med. 2000, 342, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Van Steensel, B.; Smogorzewska, A.; de Lange, T. TRF2 protects human telomeres from end-to-end fusions. Cell 1998, 92, 401–413. [Google Scholar] [CrossRef]
- Spyridopoulos, I.; Haendeler, J.; Urbich, C.; Brummendorf, T.H.; Oh, H.; Schneider, M.D.; Zeiher, A.M.; Dimmeler, S. Statins enhance migratory capacity by upregulation of the telomere repeat-binding factor TRF2 in endothelial progenitor cells. Circulation 2004, 110, 3136–3142. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Wang, S.C.; Prahash, A.; Sano, M.; Moravec, C.S.; Taffet, G.E.; Michael, L.H.; Youker, K.A.; Entman, M.L.; Schneider, M.D. Telomere attrition and Chk2 activation in human heart failure. Proc. Natl. Acad. Sci. USA 2003, 100, 5378–5383. [Google Scholar] [CrossRef] [PubMed]
- Ogami, M.; Ikura, Y.; Ohsawa, M.; Matsuo, T.; Kayo, S.; Yoshimi, N.; Hai, E.; Shirai, N.; Ehara, S.; Komatsu, R.; et al. Telomere shortening in human coronary artery diseases. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Hwang, J.; Saha, A.; Boo, Y.C.; Sorescu, G.P.; McNally, J.S.; Holland, S.M.; Dikalov, S.; Giddens, D.P.; Griendling, K.K.; Harrison, D.G.; et al. Oscillatory shear stress stimulates endothelial production of O2- from p47phox-dependent NAD(P)H oxidases, leading to monocyte adhesion. J. Biol. Chem. 2003, 278, 47291–47298. [Google Scholar] [CrossRef] [PubMed]
- McNally, J.S.; Davis, M.E.; Giddens, D.P.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D.G. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, 2290–2297. [Google Scholar] [CrossRef] [PubMed]
- Madamanchi, N.R.; Runge, M.S. Redox signaling in cardiovascular health and disease. Free Radic. Biol. Med. 2013, 61, 473–501. [Google Scholar] [CrossRef] [PubMed]
- Unterluggauer, H.; Hampel, B.; Zwerschke, W.; Jansen-Dürr, P. Senescence-associated cell death of human endothelial cells: The role of oxidative stress. Exp. Gerontol. 2003, 38, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Goy, C.; Czypiorski, P.; Altschmied, J.; Jakob, S.; Rabanter, L.L.; Brewer, A.C.; Ale-Agha, N.; Dyballa-Rukes, N.; Shah, A.M.; Haendeler, J. The imbalanced redox status in senescent endothelial cells is due to dysregulated Thioredoxin-1 and NADPH oxidase 4. Exp. Gerontol. 2014, 56, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Jeyapalan, J.C.; Sedivy, J.M. Cellular senescence and organismal aging. Mech. Ageing Dev. 2008, 129, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.; Haendeler, J.; Aicher, A.; Rossig, L.; Vasa, M.; Zeiher, A.M.; Dimmeler, S. Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: Important role of nitric oxide. Circ. Res. 2001, 89, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, H.; Chang, E.; Glassford, A.J.; Cooke, J.P.; Chiu, C.P.; Tsao, P.S. eNOS activity is reduced in senescent human endothelial cells: Preservation by hTERT immortalization. Circ. Res. 2001, 89, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Vasa, M.; Breitschopf, K.; Zeiher, A.M.; Dimmeler, S. Nitric oxide activates telomerase and delays endothelial cell senescence. Circ. Res. 2000, 87, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.; Gratton, J.P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [PubMed]
- Kang, S.S.; Kwon, T.; Kwon, D.Y.; Do, S.I. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J. Biol. Chem. 1999, 274, 13085–13090. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J.; Hoffmann, J.; Rahman, S.; Zeiher, A.M.; Dimmeler, S. Regulation of telomerase activity and anti-apoptotic function by protein-protein interaction and phosphorylation. FEBS Lett. 2003, 536, 180–186. [Google Scholar] [CrossRef]
- Chung, J.; Khadka, P.; Chung, I.K. Nuclear import of hTERT requires a bipartite nuclear localization signal and Akt-mediated phosphorylation. J. Cell Sci. 2012, 125, 2684–2697. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Chen, W.; Kim, R.H.; Kang, M.K.; Park, N.H. Regulation of the hTERT promoter activity by MSH2, the hnRNPs K and D, and GRHL2 in human oral squamous cell carcinoma cells. Oncogene 2009, 28, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Dong, Q.; Shin, K.H.; Kim, R.H.; Oh, J.E.; Park, N.H.; Kang, M.K. Grainyhead-like 2 enhances the human telomerase reverse transcriptase gene expression by inhibiting DNA methylation at the 5′-CpG island in normal human keratinocytes. J. Biol. Chem. 2010, 285, 40852–40863. [Google Scholar] [CrossRef] [PubMed]
- Ting, S.B.; Wilanowski, T.; Cerruti, L.; Zhao, L.L.; Cunningham, J.M.; Jane, S.M. The identification and characterization of human Sister-of-Mammalian Grainyhead (SOM) expands the grainyhead-like family of developmental transcription factors. Biochem. J. 2003, 370, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Boglev, Y.; Wilanowski, T.; Caddy, J.; Parekh, V.; Auden, A.; Darido, C.; Hislop, N.R.; Cangkrama, M.; Ting, S.B.; Jane, S.M. The unique and cooperative roles of the Grainy head-like transcription factors in epidermal development reflect unexpected target gene specificity. Dev. Biol. 2011, 349, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Guardiola-Serrano, F.; Haendeler, J.; Lukosz, M.; Sturm, K.; von Melchner, H.; Altschmied, J. Gene trapping identifies a putative tumor suppressor and a new inducer of cell migration. Biochem. Biophys. Res. Commun. 2008, 376, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Lukosz, M.; Mlynek, A.; Czypiorski, P.; Altschmied, J.; Haendeler, J. The transcription factor Grainyhead like 3 (GRHL3) affects endothelial cell apoptosis and migration in a NO-dependent manner. Biochem. Biophys. Res. Commun. 2011, 412, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J.; Mlynek, A.; Büchner, N.; Lukosz, M.; Graf, M.; Guettler, C.; Jakob, S.; Farrokh, S.; Kunze, K.; Goy, C.; et al. Two isoforms of Sister-Of-Mammalian Grainyhead have opposing functions in endothelial cells and in vivo. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Polytarchou, C.; Iliopoulos, D.; Hatziapostolou, M.; Kottakis, F.; Maroulakou, I.; Struhl, K.; Tsichlis, P.N. Akt2 regulates all Akt isoforms and promotes resistance to hypoxia through induction of miR-21 upon oxygen deprivation. Cancer Res. 2011, 71, 4720–4731. [Google Scholar] [CrossRef] [PubMed]
- Dormandy, J.A.; Charbonnel, B.; Eckland, D.J.; Erdmann, E.; Massi-Benedetti, M.; Moules, I.K.; Skene, A.M.; Tan, M.H.; Lefebvre, P.J.; Murray, G.D.; et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): A randomised controlled trial. Lancet 2005, 366, 1279–1289. [Google Scholar] [CrossRef]
- Werner, C.; Gensch, C.; Poss, J.; Haendeler, J.; Böhm, M.; Laufs, U. Pioglitazone activates aortic telomerase and prevents stress-induced endothelial apoptosis. Atherosclerosis 2011, 216, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, D.; Nomiyama, T.; Nakamachi, T.; Heywood, E.B.; Stone, J.F.; Berger, J.P.; Law, R.E.; Bruemmer, D. Activation of peroxisome proliferator-activated receptor gamma suppresses telomerase activity in vascular smooth muscle cells. Circ. Res. 2006, 98, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Durand, M.J.; Zinkevich, N.S.; Riedel, M.; Gutterman, D.D.; Nasci, V.L.; Salato, V.K.; Hijjawi, J.B.; Reuben, C.F.; North, P.E.; Beyer, A.M. Vascular actions of angiotensin 1-7 in the human microcirculation: Novel role for telomerase. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Assmus, B.; Urbich, C.; Aicher, A.; Hofmann, W.K.; Haendeler, J.; Rossig, L.; Spyridopoulos, I.; Zeiher, A.M.; Dimmeler, S. HMG-CoA reductase inhibitors reduce senescence and increase proliferation of endothelial progenitor cells via regulation of cell cycle regulatory genes. Circ. Res. 2003, 92, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, T.; Hano, T.; Sawamura, T.; Nishio, I. Oxidized low-density lipoprotein induces endothelial progenitor cell senescence, leading to cellular dysfunction. Clin. Exp. Pharmacol. Physiol. 2004, 31, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Boccardi, V.; Barbieri, M.; Rizzo, M.R.; Marfella, R.; Esposito, A.; Marano, L.; Paolisso, G. A new pleiotropic effect of statins in elderly: Modulation of telomerase activity. FASEB J. 2013, 27, 3879–3885. [Google Scholar] [CrossRef] [PubMed]
- Bennaceur, K.; Atwill, M.; Al Zhrany, N.; Hoffmann, J.; Keavney, B.; Breault, D.; Richardson, G.; von Zglinicki, T.; Saretzki, G.; Spyridopoulos, I. Atorvastatin induces T cell proliferation by a telomerase reverse transcriptase (TERT) mediated mechanism. Atherosclerosis 2014, 236, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N. Regulatory T cells in atherosclerosis and strategies to induce the endogenous atheroprotective immune response. Immunol Lett. 2013, 151, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Poch, E.; Carbonell, P.; Franco, S.; Diez-Juan, A.; Blasco, M.A.; Andres, V. Short telomeres protect from diet-induced atherosclerosis in apolipoprotein E-null mice. FASEB J. 2004, 18, 418–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.H.; Meyer, J.N.; Skorvaga, M.; Annab, L.A.; Van Houten, B. Mitochondrial hTERT exacerbates free-radical-mediated mtDNA damage. Aging Cell 2004, 3, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.H.; Meyer, J.N.; Van Houten, B. Mitochondrial localization of telomerase as a determinant for hydrogen peroxide-induced mitochondrial DNA damage and apoptosis. Hum. Mol. Genet. 2006, 15, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Passos, J.F.; Birket, M.J.; Beckmann, T.; Brings, S.; Peters, H.; Birch-Machin, M.A.; von Zglinicki, T.; Saretzki, G. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J. Cell Sci. 2008, 121, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J.; Dröse, S.; Büchner, N.; Jakob, S.; Altschmied, J.; Goy, C.; Spyridopoulos, I.; Zeiher, A.M.; Brandt, U.; Dimmeler, S. Mitochondrial telomerase reverse transcriptase binds to and protects mitochondrial DNA and function from damage. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.K.; Reyes, A.; Green, P.; Caron, M.J.; Bonini, M.G.; Gordon, D.M.; Holt, I.J.; Santos, J.H. Human telomerase acts as a hTR-independent reverse transcriptase in mitochondria. Nucleic Acids Res. 2012, 40, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Maida, Y.; Yasukawa, M.; Furuuchi, M.; Lassmann, T.; Possemato, R.; Okamoto, N.; Kasim, V.; Hayashizaki, Y.; Hahn, W.C.; Masutomi, K. An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature 2009, 461, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Büchner, N.; Zschauer, T.C.; Lukosz, M.; Altschmied, J.; Haendeler, J. Downregulation of mitochondrial telomerase reverse transcriptase induced by H2O2 is Src kinase dependent. Exp. Gerontol. 2010, 45, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J.; Hoffmann, J.; Brandes, R.P.; Zeiher, A.M.; Dimmeler, S. Hydrogen peroxide triggers nuclear export of telomerase reverse transcriptase via Src kinase family-dependent phosphorylation of tyrosine 707. Mol. Cell. Biol. 2003, 23, 4598–4610. [Google Scholar] [CrossRef] [PubMed]
- Büchner, N.; Ale-Agha, N.; Jakob, S.; Sydlik, U.; Kunze, K.; Unfried, K.; Altschmied, J.; Haendeler, J. Unhealthy diet and ultrafine carbon black particles induce senescence and disease associated phenotypic changes. Exp. Gerontol. 2013, 48, 8–16. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zurek, M.; Altschmied, J.; Kohlgrüber, S.; Ale-Agha, N.; Haendeler, J. Role of Telomerase in the Cardiovascular System. Genes 2016, 7, 29. https://doi.org/10.3390/genes7060029
Zurek M, Altschmied J, Kohlgrüber S, Ale-Agha N, Haendeler J. Role of Telomerase in the Cardiovascular System. Genes. 2016; 7(6):29. https://doi.org/10.3390/genes7060029
Chicago/Turabian StyleZurek, Mark, Joachim Altschmied, Stefanie Kohlgrüber, Niloofar Ale-Agha, and Judith Haendeler. 2016. "Role of Telomerase in the Cardiovascular System" Genes 7, no. 6: 29. https://doi.org/10.3390/genes7060029