

Relevance of Extending FGFR3 Gene Analysis in Osteochondrodysplasia to Non-Coding Sequences: A Case Report

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Next-Generation Sequencing (NGS)
2.2. Sanger Sequencing
2.3. Minigene Assay
3. Results
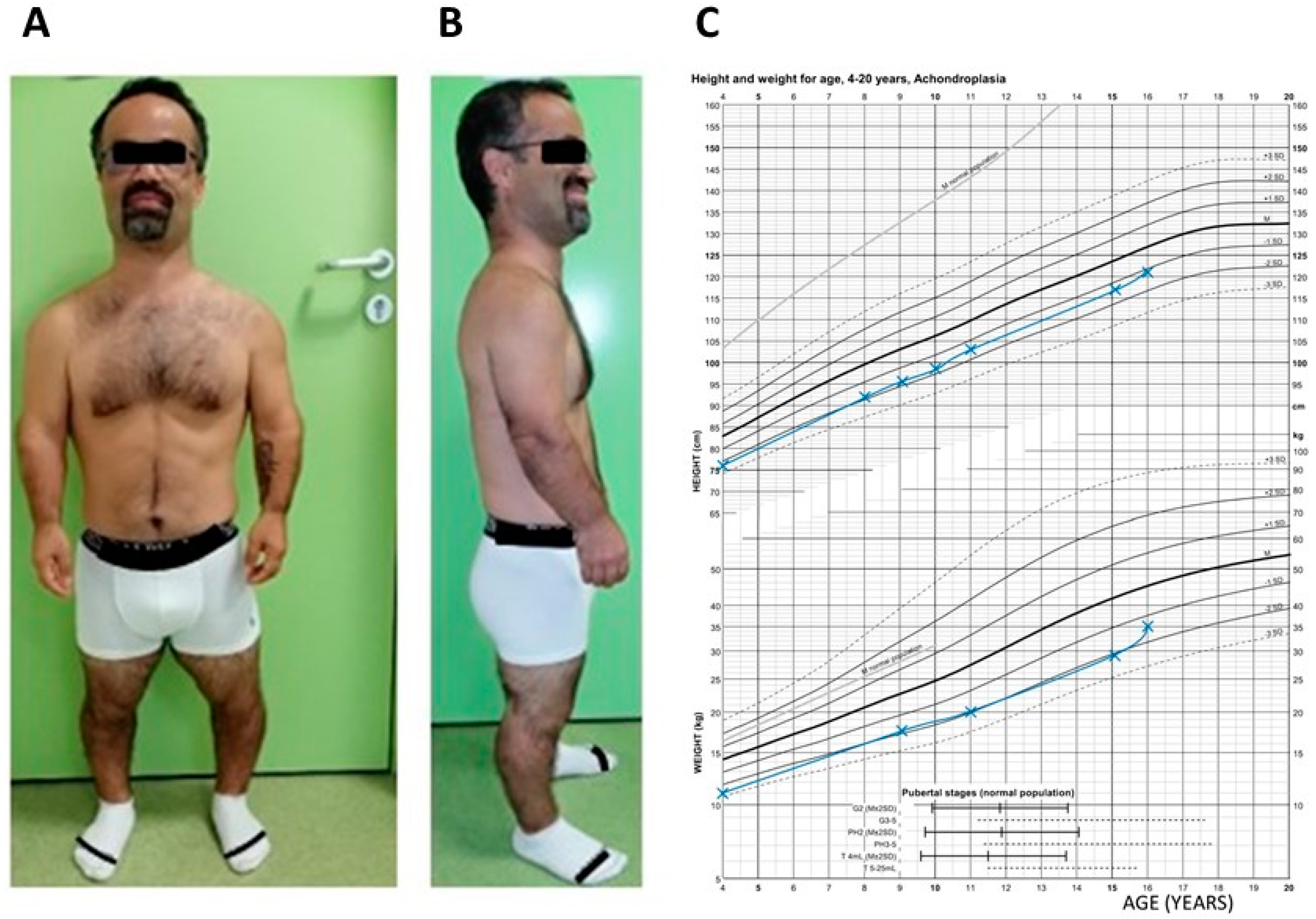
3.1. Patient Clinical History and Phenotype
3.2. Variant Discovery and Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jurcă, M.C.; Jurcă, S.I.; Mirodot, F.; Bercea, B.; Severin, E.M.; Bembea, M.; Jurcă, A.D. Changes in skeletal dysplasia nosology. Rom. J. Morphol. Embryol. 2021, 62, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Krakow, D.; Lachman, R.S.; Rimoin, D.L. Guidelines for the prenatal diagnosis of fetal skeletal dysplasias. Genet. Med. Off. J. Am. Coll. Med. Genet. 2009, 11, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Unger, S.; Ferreira, C.R.; Mortier, G.R.; Ali, H.; Bertola, D.R.; Calder, A.; Cohn, D.H.; Cormier-Daire, V.; Girisha, K.M.; Hall, C.; et al. Nosology of genetic skeletal disorders: 2023 revision. Am. J. Med. Genet. Part A 2023, 191, 1164–1209. [Google Scholar] [CrossRef] [PubMed]
- Orioli, I.M.; Castilla, E.E.; Barbosa-Neto, J.G. The birth prevalence rates for the skeletal dysplasias. J. Med. Genet. 1986, 23, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Mortier, G.R.; Cohn, D.H.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Mundlos, S.; Nishimura, G.; Robertson, S.; Sangiorgi, L.; Savarirayan, R.; et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. Part A 2019, 179, 2393–2419. [Google Scholar] [CrossRef] [PubMed]
- Makrythanasis, P.; Temtamy, S.; Aglan, M.S.; Otaify, G.A.; Hamamy, H.; Antonarakis, S.E. A novel homozygous mutation in FGFR3 causes tall stature, severe lateral tibial deviation, scoliosis, hearing impairment, camptodactyly, and arachnodactyly. Hum. Mutat. 2014, 35, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Shi, L.; Dai, W.; Gu, X.; Yu, Y.; Fan, Y. An intronic variant disrupts mRNA splicing and causes FGFR3-related skeletal dysplasia. J. Pediatr. Endocrinol. Metab. 2021, 34, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- Pauli, R.M. Achondroplasia: A comprehensive clinical review. Orphanet J. Rare Dis. 2019, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Bellus, G.A.; Hefferon, T.W.; Ortiz de Luna, R.I.; Hecht, J.T.; Horton, W.A.; Machado, M.; Kaitila, I.; McIntosh, I.; Francomano, C.A. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am. J. Hum. Genet. 1995, 56, 368–373. [Google Scholar] [PubMed]
- Vajo, Z.; Francomano, C.A.; Wilkin, D.J. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: The achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocr. Rev. 2000, 21, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Prinos, P.; Costa, T.; Sommer, A.; Kilpatrick, M.W.; Tsipouras, P. A common FGFR3 gene mutation in hypochondroplasia. Hum. Mol. Genet. 1995, 4, 2097–2101. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.R.; Campos-Xavier, A.B.; Medeira, A.; Cordeiro, I.; Sousa, A.B.; Lima, M.; Soares, G.; Rocha, M.; Saraiva, J.; Ramos, L.; et al. Clinical and molecular diagnosis of the skeletal dysplasias associated with mutations in the gene encoding Fibroblast Growth Factor Receptor 3 (FGFR3) in Portugal. Clin. Genet. 2009, 75, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Millat, G.; Lafont, E.; Nony, S.; Rouvet, I.; Bozon, D. Functional characterization of putative novel splicing mutations in the cardiomyopathy-causing genes. DNA Cell Biol. 2015, 34, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Baralle, D.; Baralle, M. Splicing in action: Assessing disease causing sequence changes. J. Med. Genet. 2005, 42, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Neumeyer, L.; Merker, A.; Hagenäs, L. Clinical charts for surveillance of growth and body proportion development in achondroplasia and examples of their use. Am. J. Med. Genet. Part A 2021, 185, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.N.; Modaff, P.; Wang, C.G.; Wohler, E.; Sobreira, N.L.; Donoghue, D.J.; Pauli, R.M. Typical achondroplasia secondary to a unique insertional variant of FGFR3 with in vitro demonstration of its effect on FGFR3 function. Am. J. Med. Genet. Part A 2021, 185, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Baux, D.; Van Goethem, C.; Ardouin, O.; Guignard, T.; Bergougnoux, A.; Koenig, M.; Roux, A.-F. MobiDetails: Online DNA variants interpretation. Eur. J. Hum. Genet. 2021, 29, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Fokkema, I.F.A.C.; Kroon, M.; López Hernández, J.A.; Asscheman, D.; Lugtenburg, I.; Hoogenboom, J.; den Dunnen, J.T. The LOVD3 platform: Efficient genome-wide sharing of genetic variants. Eur. J. Hum. Genet. 2021, 29, 1796–1803. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Criteria | PM2 | PS2 | PS3 |
|---|---|---|---|
| Evidence | The variant is absent from control databases, including gnomAD, despite good coverage of the genomic region in sequencing. | The variant is confirmed to be de novo using NGS and Sanger sequencing, with both maternity and paternity confirmation, in a patient with the disorder and no family history of this trouble. | The minigene in vitro functional analyses confirmed, in two independent studies, the damaging effect on FGFR3 transcript splicing. |
| Case 1 | Case 2 | Case 3 | Case 4 (Described in This Report) | |
|---|---|---|---|---|
| Gender | Male | Male | Male | Male |
| Origin | Chinese | Chinese | Chinese | European (France) |
| Prenatal findings | nd | Short lower limbs at third trimester ultrasound | nd | nd |
| Age at the beginning of genetic investigation | 5 years | 14 months | 3 years | 42 years |
| Birth length | nd | Normal (50 cm) | nd | Normal (48 cm) |
| Limb aspect at birth | nd | nd | nd | Micromelia |
| Head circumference | Mild macrocephaly | nd | Normal | Normal |
| Face morphology | Low nasal bridge | Bossing forehead, low nasal bridge, thick feet | Relatively normal facial features | Bossing forehead |
| Intelligence | Normal | Normal | Normal | Normal |
| Other clinical features | Shortening of limbs | Short limbs, continuous growth retardation | Short limbs (especially upper limbs), bowing legs | Shortening of limbs (rhizomelia and mesomelia), continuous growth retardation |
| Radiologic findings | Shortening and thickening of femora and tibia, metaphyseal flaring of distal femora and proximal tibia | Multiple skeletal abnormalities | Metaphyseal flaring of distal femora and proximal tibiae | Multiple spinal abnormalities |
| Diagnosis | Hypochondroplasia or mild achondroplasia | Achondroplasia or severe hypochondroplasia | Hypochondroplasia | Achondroplasia or severe hypochondroplasia |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouedraogo, Z.G.; Janel, C.; Janin, A.; Millat, G.; Langlais, S.; Pontier, B.; Biard, M.; Lepage, M.; Francannet, C.; Laffargue, F.; et al. Relevance of Extending FGFR3 Gene Analysis in Osteochondrodysplasia to Non-Coding Sequences: A Case Report. Genes 2024, 15, 225. https://doi.org/10.3390/genes15020225
Ouedraogo ZG, Janel C, Janin A, Millat G, Langlais S, Pontier B, Biard M, Lepage M, Francannet C, Laffargue F, et al. Relevance of Extending FGFR3 Gene Analysis in Osteochondrodysplasia to Non-Coding Sequences: A Case Report. Genes. 2024; 15(2):225. https://doi.org/10.3390/genes15020225
Chicago/Turabian StyleOuedraogo, Zangbéwendé Guy, Caroline Janel, Alexandre Janin, Gilles Millat, Sarah Langlais, Bénédicte Pontier, Marie Biard, Mathis Lepage, Christine Francannet, Fanny Laffargue, and et al. 2024. "Relevance of Extending FGFR3 Gene Analysis in Osteochondrodysplasia to Non-Coding Sequences: A Case Report" Genes 15, no. 2: 225. https://doi.org/10.3390/genes15020225