
The Mitogenomic Characterization and Phylogenetic Analysis of the Plant Pathogen Phyllosticta yuccae
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling, Library Preparation and Sequenceing
2.2. Mitogenome Assembly and Annotation
2.3. Mitogenome Characteristics Analysis
2.4. Phylogenetic Analysis
3. Results
3.1. The Characteristics of P. yuccae
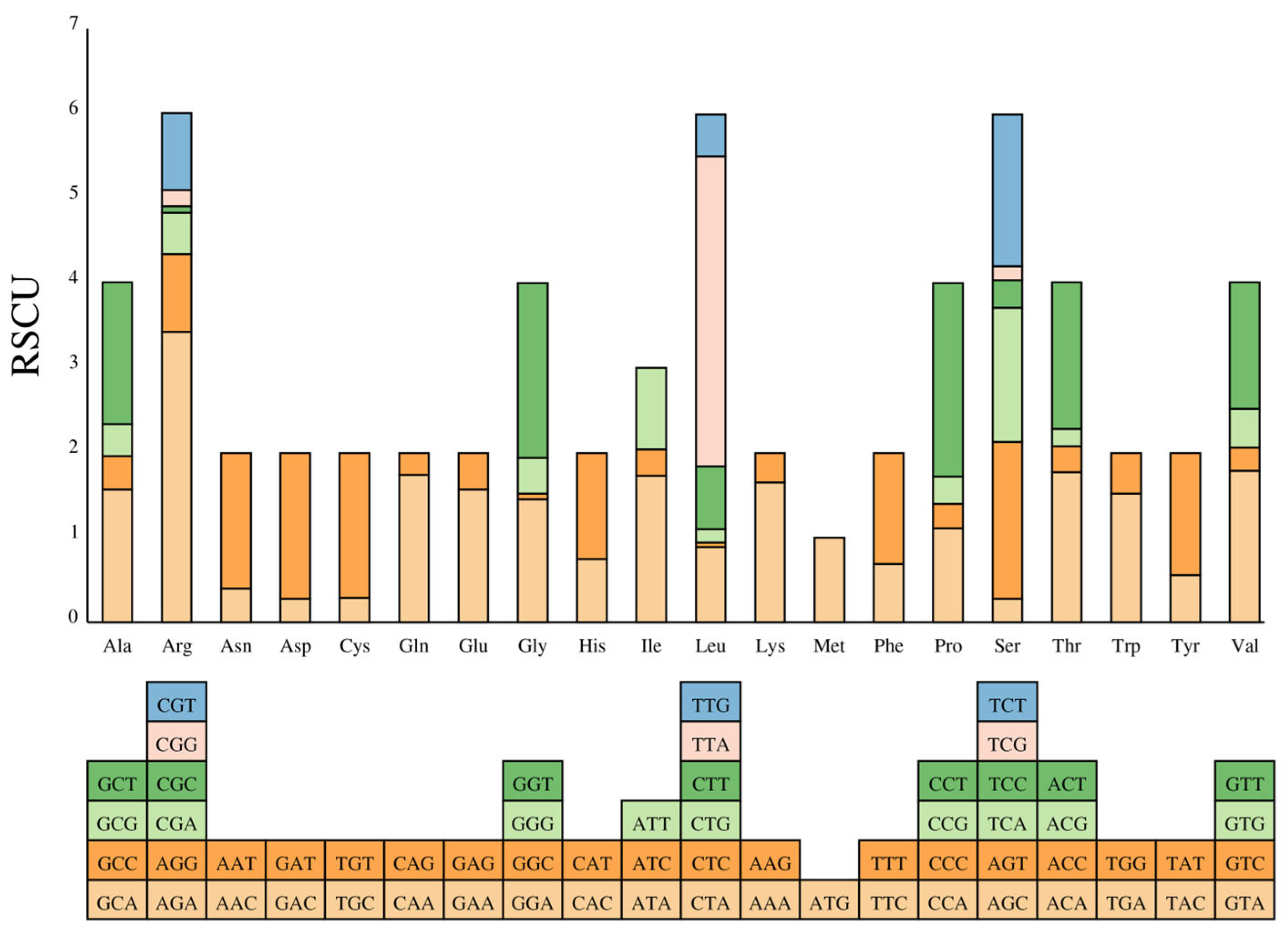
3.2. Protein-Coding Genes and Codon Usage
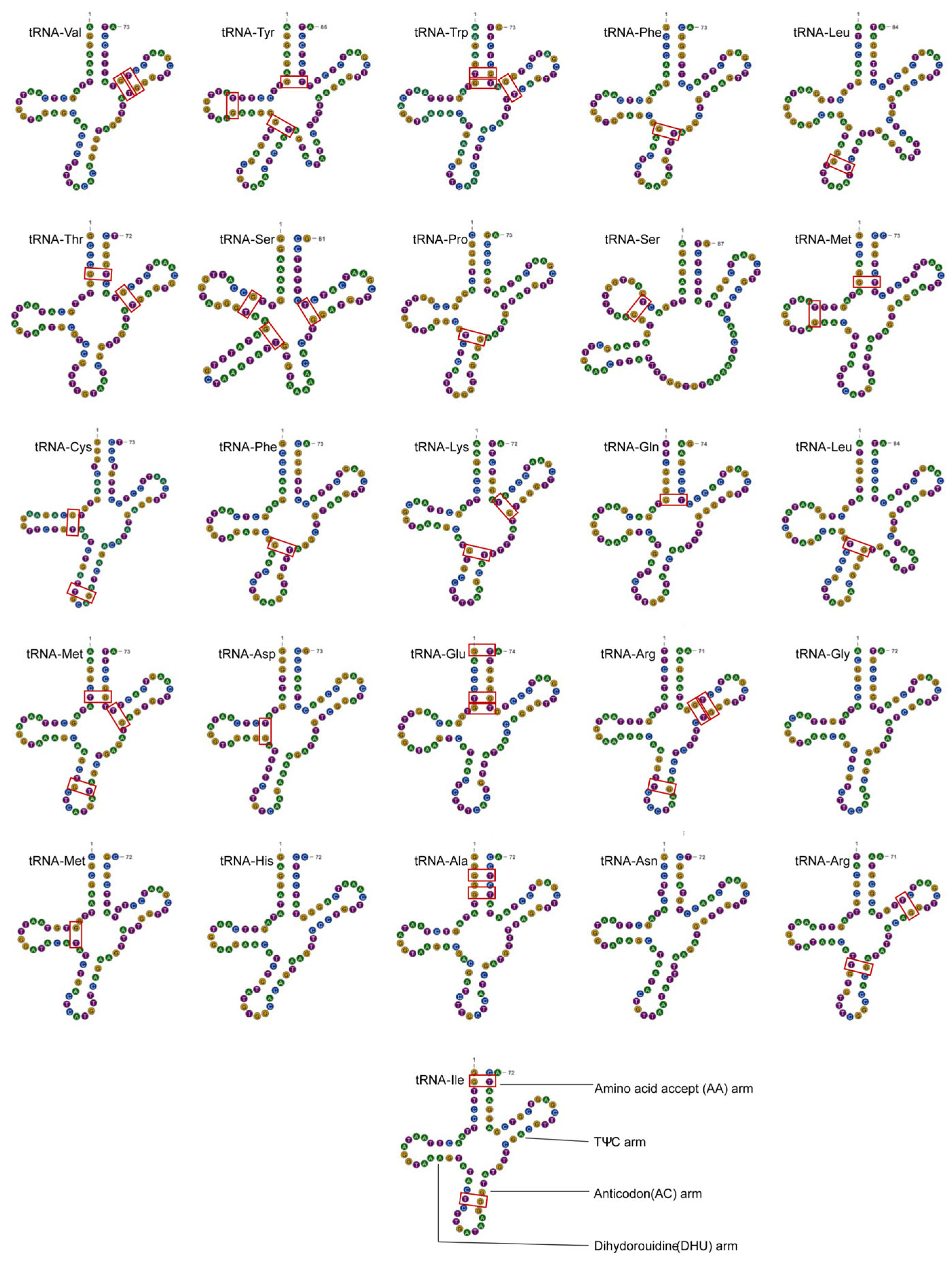
3.3. Ribosomal and Transporter RNA Genes
3.4. A Comparative Analysis of the Mitogenomes of P. yuccae and M. phaseolina
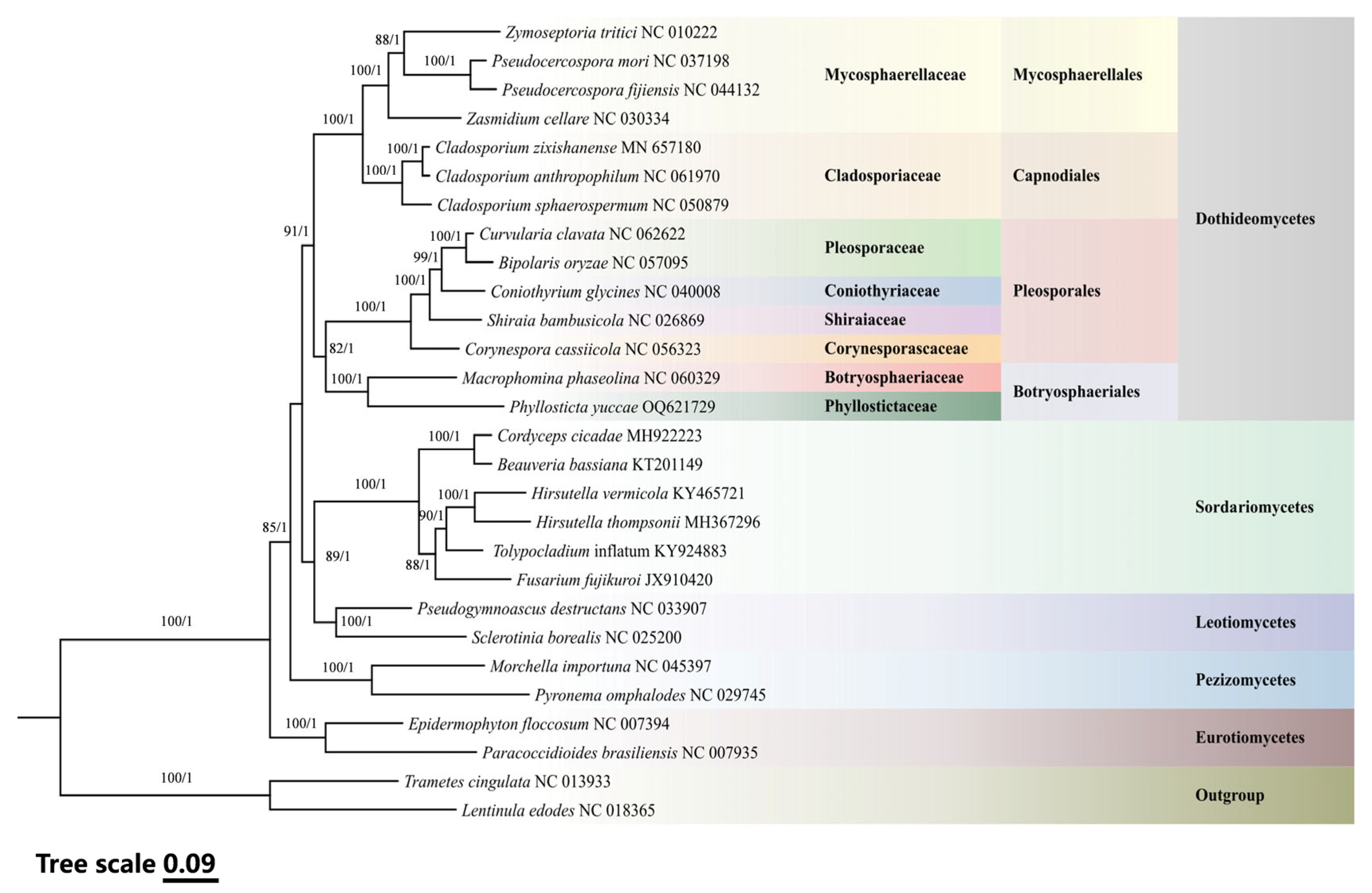
3.5. Phylogenetic Relationships
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Burger, G.; Gray, M.W.; Franz Lang, B. Mitochondrial genomes: Anything goes. Trends Genet. 2003, 19, 709–716. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar]
- Abuduaini, A.; Wang, Y.B.; Zhou, H.Y.; Kang, R.P.; Ding, M.L.; Jiang, Y.; Suo, F.Y.; Huang, L.D. The complete mitochondrial genome of Ophiocordyceps gracilis and its comparison with related species. IMA Fungus 2021, 12, 31. [Google Scholar] [CrossRef]
- Ji, X.; Tian, Y.; Liu, W.; Lin, C.; He, F.; Yang, J.; Miao, W.; Li, Z. Mitochondrial characteristics of the powdery mildew genus Erysiphe revealed an extraordinary evolution in protein-coding genes. Int. J. Biol. Macromol. 2023, 230, 123153. [Google Scholar] [CrossRef]
- Pszczolkowska, A.; Androsiuk, P.; Jastrzębski, J.P.; Paukszto, L.; Okorski, A. Rps3 as a candidate mitochondrial gene for the molecular identification of species from the Colletotrichum acutatum species complex. Genes 2020, 11, 552. [Google Scholar]
- Zhang, C.; Mao, B.; Wang, H.; Dai, L.; Huang, Y.; Chen, Z.; Huang, J. The complete mitogenomes of three grasshopper species with special notes on the phylogenetic positions of some related genera. Insects 2023, 14, 85. [Google Scholar]
- Van de Sande, W.W. Phylogenetic analysis of the complete mitochondrial genome of Madurella mycetomatis confirms its taxonomic position within the order Sordariales. PLoS ONE 2012, 7, e38654. [Google Scholar] [CrossRef]
- Fonseca, P.L.C.; De-Paula, R.B.; Araújo, D.S.; Tomé, L.M.R.; Mendes-Pereira, T.; Rodrigues, W.F.C.; Del-Bem, L.E.; Aguiar, E.R.G.R.; Góes-Neto, A. Global Characterization of Fungal Mitogenomes: New Insights on Genomic Diversity and Dynamism of Coding Genes and Accessory Elements. Front. Microbiol. 2021, 12, 787283. [Google Scholar] [CrossRef]
- Kulik, T.; Bilska, K.; Żelechowski, M. Promising Perspectives for Detection, Identification, and Quantification of Plant Pathogenic Fungi and Oomycetes through Targeting Mitochondrial DNA. Int. J. Mol. Sci. 2020, 21, 2645. [Google Scholar] [CrossRef]
- Kulik, T.; Van Diepeningen, A.D.; Hausner, G. Editorial: The Significance of Mitogenomics in Mycology. Front. Microbiol. 2021, 11, 628579. [Google Scholar]
- Kouvelis, V.N.; Hausner, G. Editorial: Mitochondrial genomes and mitochondrion related gene insights to fungal evolution. Front. Microbiol. 2022, 13, 897981. [Google Scholar] [CrossRef]
- Zardoya, R. Recent advances in understanding mitochondrial genome diversity. F1000Research 2020, 9, F1000. [Google Scholar] [CrossRef]
- Li, Q.; Ren, Y.; Xiang, D.; Shi, X.; Zhao, J.; Peng, L.; Zhao, G. Comparative mitogenome analysis of two ectomycorrhizal fungi (Paxillus) reveals gene rearrangement, intron dynamics, and phylogeny of basidiomycetes. IMA Fungus 2020, 11, 12. [Google Scholar] [CrossRef]
- Kong, L.; Li, Y.; Kocot, K.M.; Yang, Y.; Qi, L.; Li, Q.; Halanych, K.M. Mitogenomics reveals phylogenetic relationships of Arcoida (Mollusca, Bivalvia) and multiple independent expansions and contractions in mitochondrial genome size. Mol. Phylogenet Evol. 2020, 150, 106857. [Google Scholar]
- Aguileta, G.; de Vienne, D.M.; Ross, O.N.; Hood, M.E.; Giraud, T.; Petit, E.; Gabaldón, T. High variability of mitochondrial gene order among fungi. Genome Biol. Evol. 2014, 6, 451–465. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, S.; Zhang, G.; Liu, X.; Wang, C.; Xu, J. Comparison of mitochondrial genomes provides insights into intron dynamics and evolution in the caterpillar fungus Cordyceps militaris. Fungal Genet. Biol. 2015, 77, 95–107. [Google Scholar] [CrossRef]
- Liu, W.; Cai, Y.; Zhang, Q.; Chen, L.; Shu, F.; Ma, X.; Bian, Y. The mitochondrial genome of Morchella importuna (272.2 kb) is the largest among fungi and contains numerous introns, mitochondrial non-conserved open reading frames and repetitive sequences. Int. J. Biol. Macromol. 2020, 143, 373–381. [Google Scholar] [CrossRef]
- Bissett, J. Discochora yuccae sp. nov. with Phyllosticta and Leptodothiorella synanamorphs. Can. J. Bot. 1986, 64, 1720–1726. [Google Scholar] [CrossRef]
- Wikee, S.; Lombard, L.; Nakashima, C.; Motohashi, K.; Chukeatirote, E.; Cheewangkoon, R.; McKenzie, E.H.C.; Hyde, K.D.; Crous, P.W. A phylogenetic re-evaluation of Phyllosticta (Botryosphaeriales). Stud. Mycol. 2013, 76, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Herron, D.A.; Wingfield, M.J.; Wingfield, B.D.; Rodas, C.A.; Marincowitz, S.; Steenkamp, E.T. Novel taxa in the Fusarium fujikuroi species complex from Pinus spp. Stud. Mycol. 2015, 80, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.B.; Wang, Y.; Fang, Q.; Duan, D.E.; Zhang, G.D.; Dai, R.Q.; Dai, Y.D. Multigene phylogeny of the family Cordycipitaceae (Hypocreales): New taxa and the new systematic position of the Chinese cordycipitoid fungus Paecilomyces hepiali. Fungal Divers. 2020, 103, 1–46. [Google Scholar] [CrossRef]
- Zhang, H.; Fu, X.; Gong, X.; Wang, Y.; Zhang, H.; Zhao, Y.; Shen, Y. Systematic dissection of key factors governing recombination outcomes by GCE-SCRaMbLE. Nat. Commun. 2022, 13, 5836. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; Depamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Putz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Valach, M.; Burger, G.; Gray, M.W.; Lang, B.F. Widespread occurrence of organelle genome-encoded 5S rRNAs including permuted molecules. Nucleic Acids Res. 2014, 42, 13764–13777. [Google Scholar] [CrossRef]
- Yu, H.; Wang, L.; Yang, F.; He, Y.; Lv, M. Complete mitochondrial genome of the important phytopathogenic fungus Macrophomina phaseolina (Botryosphaeriales, Ascomycota). Mitochondrial DNA B 2021, 6, 2972–2974. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. Organellar genome DRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 33, W54–W57. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar]
- Kurtz, S. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web a web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0 a toolkit incorporating γ-series methods and sliding window strategies. Genom. Proteom. Bioinf. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Clements, J.M.; Laz, T.M.; Sherman, F. Efficiency of translation initiation by non-AUG codons in Saccharomyces cerevisiae. Mol. Cell Biol. 1988, 8, 4533–4536. [Google Scholar]
- Ivanov, I.P.; Loughran, G.; Sachs, M.S.; Atkins, J.F. Initiation context modulates autoregulation of eukaryotic translation initiation factor 1 (elf1). Proc. Natl. Acad. Sci. USA 2010, 107, 18056–18060. [Google Scholar]
- Kolitz, S.E.; Takacs, J.E.; Lorsch, J.R. Kinetic and thermodynamic analysis of the role of start codon/anticodon base pairing during eukaryotic translation initiation. RNA 2009, 15, 138–152. [Google Scholar] [CrossRef]
- Wei, J.; Zhang, Y.; Ivanov, I.P.; Sachs, M.S. The stringency of start codon selection in the filamentous fungus Neurospora crassa. J. Biol. Chem. 2013, 288, 9549–9562. [Google Scholar]
- James, T.Y.; Pelin, A.; Bonen, L.; Ahrendt, S.; Sain, D.; Corradi, N.; Stajich, J.E. Shared signatures of parasitism and phylogenomics unite Cryptomycota and Microsporidia. Curr. Biol. 2013, 23, 1548–1553. [Google Scholar]
- Zaccaron, A.Z.; Stergiopoulos, I. Characterization of the mitochondrial genomes of three powdery mildew pathogens reveals remarkable variation in size and nucleotide composition. Microbiol. Genomics 2021, 7, 000720. [Google Scholar]
- Li, Q.; Wu, P.; Li, L.; Feng, H.; Tu, W.; Bao, Z.; Xiong, C.; Gui, M.; Huang, W. The first eleven mitochondrial genomes from the ectomycorrhizal fungal genus (Boletus) reveal intron loss and gene rearrangement. Int. J. Biol. Macromol. 2021, 172, 560–572. [Google Scholar]
- Zhong, L.; Wang, M.; Li, D.; Tang, S.; Zhang, T.; Bian, W.; Chen, X. Complete mitochondrial genome of Odontobutis haifengensis (Perciformes, Odontobutiae): A unique rearrangement of tRNAs and additional non-coding regions identified in the genus Odontobutis. Genomics 2018, 110, 382–388. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, J.; Rouse, G.W.; Wiklund, H.; Pleijel, F.; Watanabe, H.K.; Chen, C.; Qian, P.Y.; Qiu, J.W. Phylogeny, evolution and mitochondrial gene order rearrangement in scale worms (Aphroditiformia, Annelida). Mol. Phylogenet Evol. 2018, 125, 220–231. [Google Scholar] [CrossRef]
- Li, Q.; Yang, L.; Xiang, D.; Wan, Y.; Wu, Q.; Huang, W.; Zhao, G. The complete mitochondrial genomes of two model ectomycorrhizal fungi (Laccaria): Features, intron dynamics and phylogenetic implications. Int. J. Biol. Macromol. 2020, 145, 974–984. [Google Scholar]
- Johnston, I.G.; Williams, B.P. Evolutionary inference across eukaryotes identifies specific pressures favoring mitochondrial gene retention. Cell Syst. 2016, 2, 101–111. [Google Scholar] [CrossRef]
- Bogenhagen, D.F.; Clayton, D.A. The mitochondrial DNA replication bubble has not burst. Trends Biochem. Sci. 2003, 28, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.A.; Cecconi, C.; Tkachuk, A.N.; Bustamante, C.; Clayton, D.A. Replication of mitochondrial DNA occurs by strand displacement with alternative light-strand origins, not via a strand-coupled mechanism. Genes Dev. 2005, 19, 2466–2476. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | M. phaseolina | P. yuccae | |
|---|---|---|---|
| Accession No. | NC_060329 | OQ621729 | |
| Genome size (bp) | 101,198 | 178,540 | |
| GC content (%) | 29.8% | 31.13% | |
| PCGs | Number | 14 | 14 |
| Length (bp) | 15,741 | 16,476 | |
| Ratio (%) | 15.55% | 9.23% | |
| tRNA | Number | 26 | 26 |
| Length (bp) | 1940 | 1944 | |
| Ratio (%) | 1.92% | 1.09% | |
| rRNA | Number | 2 | 2 |
| Length (bp) | 4330 | 3576 | |
| Ratio (%) | 4.28% | 2.00% | |
| ORF | Number | 38 | 22 |
| Length (bp) | 37,683 | 24,168 | |
| Ratio (%) | 37.24% | 13.54% | |
| Intron | Number | 21 | 39 |
| Length (bp) | 33,788 | 120,240 | |
| Ratio (%) | 33.39% | 64.35% | |
| SSR | Number | 34 | 74 |
| Length (bp) | 427 | 789 | |
| Ratio (%) | 0.42% | 0.44% | |
| Tandem | Number | 16 | 14 |
| Length (bp) | 778 | 848 | |
| Ratio (%) | 0.77% | 0.47% | |
| Interspersed | Number | 157 | 310 |
| Length (bp) | 11,210 | 25,890 | |
| Ratio (%) | 11.08% | 14.75% | |
| Intergenic regions | Length (bp) | 25,863 | 33,964 |
| Ratio (%) | 25.56 | 19.02 | |
| Gene Name | Type | Position | Length | Codons | Intron Number | Introns Length | Strand | ||
|---|---|---|---|---|---|---|---|---|---|
| From | To | Start | Stop | ||||||
| atp6 | CDS | 1 | 3268 | 765 | GTG | TAG | 1 | 2503 | + |
| nad3 | CDS | 5707 | 6210 | 504 | GTG | TAA | - | - | + |
| rns | rRNA | 7032 | 14,366 | 1608 | - | - | 2 | 5727 | + |
| nad6 | CDS | 19,372 | 19,947 | 576 | ATG | TAA | - | - | + |
| rnl | rRNA | 23,680 | 39,033 | 1968 | - | - | 7 | 13,386 | + |
| cox3 | CDS | 48,055 | 62,352 | 810 | ATG | TAG | 7 | 13,488 | + |
| atp9 | CDS | 63,244 | 63,468 | 225 | ATG | TAA | - | - | + |
| cox1 | CDS | 65,320 | 96,273 | 936 | ATG | TAG | 5 | 30,018 | + |
| cox2 | CDS | 96,830 | 97,837 | 1008 | ATG | TAG | - | - | + |
| nad4L | CDS | 99,943 | 100,431 | 489 | ATG | TAA | - | - | + |
| nad5 | CDS | 101,949 | 118,788 | 2139 | ATG | TAA | 6 | 14,701 | + |
| rps3 | CDS | 109,790 | 111,196 | 1407 | GTG | TAA | - | - | + |
| nad2 | CDS | 123,965 | 135,750 | 1896 | ATG | TAG | 4 | 9890 | + |
| nad4 | CDS | 138,561 | 143,823 | 3633 | ATG | TAG | 1 | 1630 | + |
| nad1 | CDS | 144,594 | 150,225 | 1095 | ATG | TAA | 2 | 4537 | + |
| cob | CDS | 151,541 | 176,893 | 993 | ATG | TAA | 4 | 24,360 | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, H.; Zhu, Z.; Tian, Z.; Wei, C.; Fan, Q.; Wang, Y.; Shen, S.; Deng, G.; Ding, M. The Mitogenomic Characterization and Phylogenetic Analysis of the Plant Pathogen Phyllosticta yuccae. Genes 2024, 15, 111. https://doi.org/10.3390/genes15010111
Xu H, Zhu Z, Tian Z, Wei C, Fan Q, Wang Y, Shen S, Deng G, Ding M. The Mitogenomic Characterization and Phylogenetic Analysis of the Plant Pathogen Phyllosticta yuccae. Genes. 2024; 15(1):111. https://doi.org/10.3390/genes15010111
Chicago/Turabian StyleXu, Hui, Ziyi Zhu, Zeyuan Tian, Cuiyuan Wei, Qi Fan, Yuanbing Wang, Shikang Shen, Gang Deng, and Mingliang Ding. 2024. "The Mitogenomic Characterization and Phylogenetic Analysis of the Plant Pathogen Phyllosticta yuccae" Genes 15, no. 1: 111. https://doi.org/10.3390/genes15010111