

Sex Differences in Anderson–Fabry Cardiomyopathy: Clinical, Genetic, and Imaging Analysis in Women
 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Study Design and Population
2.2. Echocardiographic Assessment
2.3. Assessment of Clinical Outcomes
2.4. Cardiac Magnetic Resonance (CMR)
2.5. Ethical Considerations
2.6. Statistical Analysis
3. Results
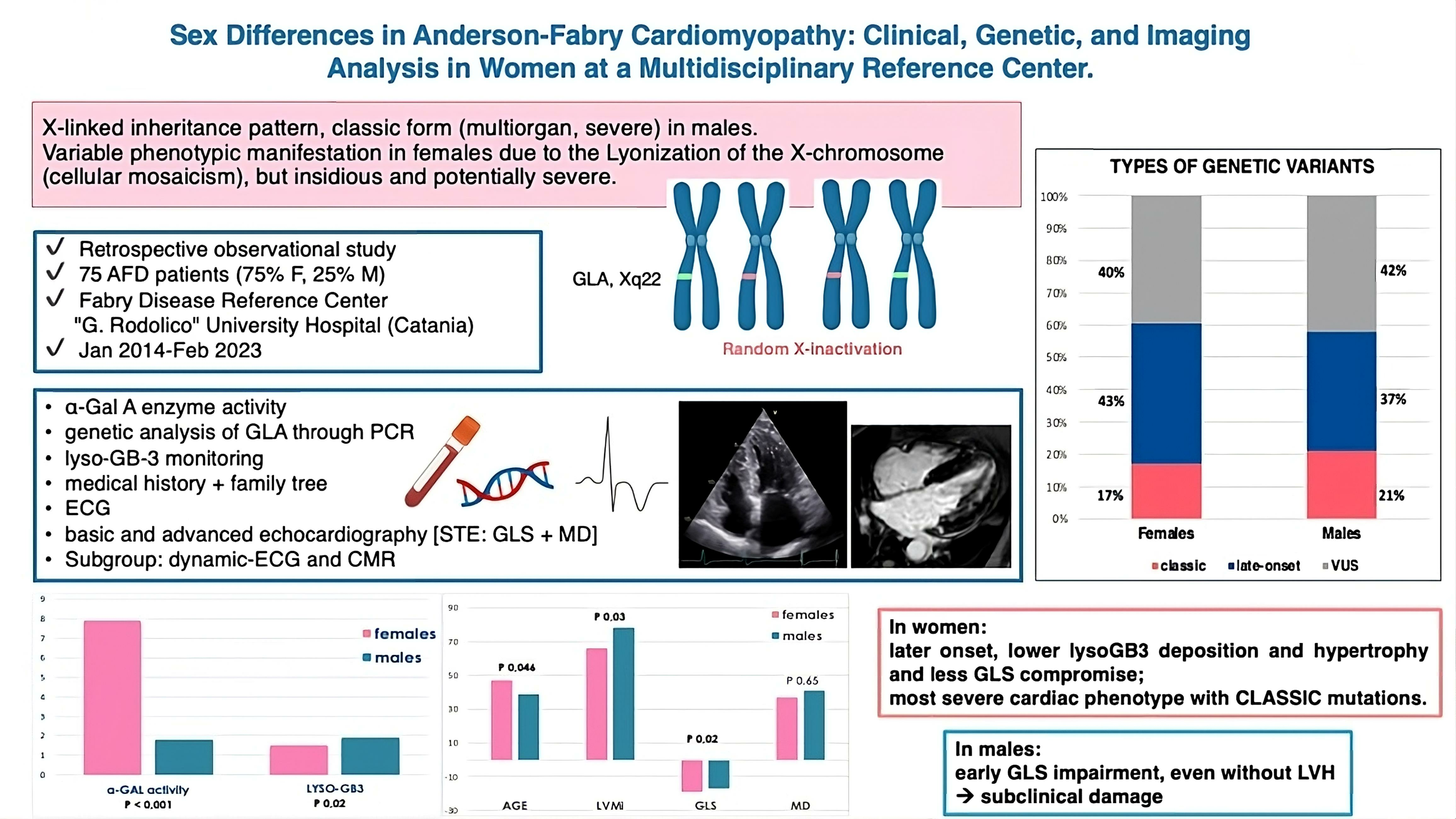
3.1. Fabry Disease Patient Population Overview
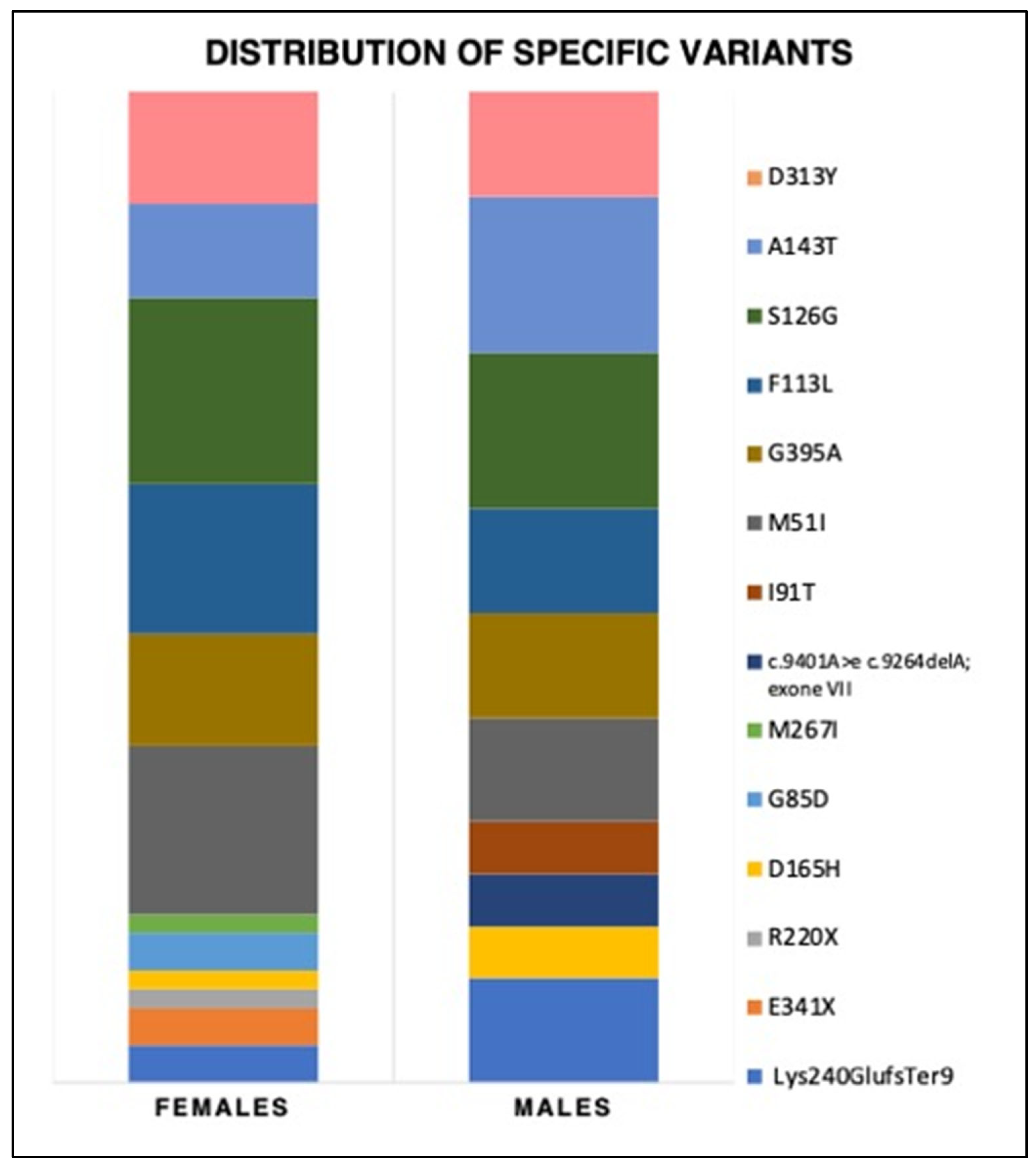
3.2. Genetic Variants, Biochemical Parameters, and Therapy
3.3. Echocardiography
3.4. Cardiac Magnetic Resonance
3.5. Analysis of Sex Differences: A Closer Examination of Female Patients with AFD
3.6. Insights from Lyso-Gb3 Values among the Groups
3.7. Subanalyses According to Therapy and Lyso-Gb3 Values


3.8. Insights from Females and Males with Peculiar Manifestations

4. Discussion
4.1. Sex Differences in AFD Cardiac Phenotype
4.2. Genetics and Specific Variants’ Contributions
4.3. Fabry Cardiomyopathy in Women: Phenotype and Genotype
4.4. Impact of Therapy (ERT and Migalastat) in the Main Historical Fabry Cohorts, and Role of Lyso-GB3
4.5. Study Limitations
4.6. Clinical Implications and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef]
- Bernardes, T.P.; Foresto, R.D.; Kirsztajn, G.M. Fabry disease: Genetics, pathology, and treatment. Rev. Assoc. Med. Bras. 2020, 66 (Suppl. 1), s10–s16. [Google Scholar] [CrossRef]
- Gal, A.; Beck, M.; Höppner, W.; Germain, D.P. Clinical utility gene card for: Fabry disease—Update 2016. Eur. J. Hum. Genet. 2017, 25, e1–e3. [Google Scholar] [CrossRef]
- Doheny, D.; Srinivasan, R.; Pagant, S.; Chen, B.; Yasuda, M.; Desnick, R.J. Fabry Disease: Prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995–2017. J. Med. Genet. 2018, 55, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Levade, T.; Hachulla, E.; Knebelmann, B.; Lacombe, D.; Seguin, V.L.; Nguyen, K.; Noël, E.; Rabès, J.P. Challenging the traditional approach for interpreting genetic variants: Lessons from Fabry disease. Clin. Genet. 2022, 101, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Altarescu, G.; Barriales-Villa, R.; Mignani, R.; Pawlaczyk, K.; Pieruzzi, F.; Terryn, W.; Vujkovac, B.; Ortiz, A. An expert consensus on practical clinical recommendations and guidance for patients with classic Fabry disease. Mol. Genet. Metab. 2022, 137, 49–61. [Google Scholar] [CrossRef]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef]
- Dobrovolny, R.; Dvorakova, L.; Ledvinova, J.; Magage, S.; Bultas, J.; Lubanda, J.C.; Elleder, M.; Karetova, D.; Pavlikova, M.; Hrebicek, M. Relationship between X-inactivation and clinical involvement in Fabry heterozygotes. Eleven novel mutations in the alpha-galactosidase A gene in the Czech and Slovak population. J. Mol. Med. 2005, 83, 647–654. [Google Scholar] [CrossRef]
- Linhart, A.; Germain, D.P.; Olivotto, I.; Akhtar, M.M.; Anastasakis, A.; Hughes, D.; Namdar, M.; Pieroni, M.; Hagège, A.; Cecchi, F.; et al. An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Eur. J. Heart Fail. 2020, 22, 1076–1096. [Google Scholar] [CrossRef]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC). Eur. Heart J. 2023, ehad194, Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Reuser, A.J.; Verheijen, F.W.; Bali, D.; van Diggelen, O.P.; Germain, D.P.; Hwu, W.L.; Lukacs, Z.; Mühl, A.; Olivova, P.; Piraud, M.; et al. The use of dried blood spot samples in the diagnosis of lysosomal storage disorders—Current status and perspectives. Mol. Genet. Metab. 2011, 104, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Rombach, S.M.; Dekker, N.; Bouwman, M.G.; Linthorst, G.E.; Zwinderman, A.H.; Wijburg, F.A.; Kuiper, S.; Vd Bergh Weerman, M.A.; Groener, J.E.; Poorthuis, B.; et al. Plasma globotriaosylsphingosine: Diagnostic value and relation to clinical manifestations of Fabry disease. Biochim. Biophys. Acta 2010, 1802, 741–748. [Google Scholar] [CrossRef]
- Mogensen, J.; van Tintelen, J.P.; Fokstuen, S.; Elliott, P.; van Langen, I.M.; Meder, B.; Richard, P.; Syrris, P.; Caforio, A.L.; Adler, Y.; et al. The current role of next-generation DNA sequencing in routine care of patients with hereditary cardiovascular conditions: A viewpoint paper of the European Society of Cardiology working group on myocardial and pericardial diseases and members of the European Society of Human Genetics. Eur. Heart J. 2015, 36, 1367–1370. [Google Scholar]
- Smid, B.E.; van der Tol, L.; Cecchi, F.; Elliott, P.M.; Hughes, D.A.; Linthorst, G.E.; Timmermans, J.; Weidemann, F.; West, M.L.; Biegstraaten, M.; et al. Uncertain diagnosis of Fabry disease: Consensus recommendation on diagnosis in adults with left ventricular hypertrophy and genetic variants of unknown significance. Int. J. Cardiol. 2014, 177, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Fouilhoux, A.; Decramer, S.; Tardieu, M.; Pillet, P.; Fila, M.; Rivera, S.; Deschênes, G.; Lacombe, D. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin. Genet. 2019, 96, 107–117. [Google Scholar] [CrossRef]
- Linhart, A.; Paleček, T. Narrative review on Morbus Fabry: Diagnosis and management of cardiac manifestations. Cardiovasc. Diagn. Ther. 2021, 11, 650–660. [Google Scholar] [CrossRef]
- Kubo, T. Fabry disease and its cardiac involvement. J. Gen. Fam. Med. 2017, 18, 225–229. [Google Scholar] [CrossRef]
- Baig, S.; Edward, N.C.; Kotecha, D.; Liu, B.; Nordin, S.; Kozor, R.; Moon, J.C.; Geberhiwot, T.; Steeds, R.P. Ventricular arrhythmia and sudden cardiac death in Fabry disease: A systematic review of risk factors in clinical practice. Europace 2018, 20, f153–f161. [Google Scholar] [CrossRef]
- Germain, D.P.; Brand, E.; Burlina, A.; Cecchi, F.; Garman, S.C.; Kempf, J.; Laney, D.A.; Linhart, A.; Maródi, L.; Nicholls, K. Phenotypic characteristics of the p.Asn215Ser (p.N215S) GLA mutation in male and female patients with Fabry disease: A multicenter Fabry Registry study. Mol. Genet. Genom. Med. 2018, 6, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Pieroni, M.; Ciabatti, M.; Graziani, F.; Camporeale, A.; Saletti, E.; Lillo, R.; Figliozzi, S.; Bolognese, L. The Heart in Fabry Disease: Mechanisms Beyond Storage and Forthcoming Therapies. Rev. Cardiovasc. Med. 2022, 23, 196. [Google Scholar] [CrossRef]
- Nordin, S.; Kozor, R.; Medina-Menacho, K.; Abdel-Gadir, A.; Baig, S.; Sado, D.M.; Lobascio, I.; Murphy, E.; Lachmann, R.H.; Mehta, A.; et al. Proposed Stages of Myocardial Phenotype Development in Fabry Disease. JACC Cardiovasc. Imaging 2019, 12, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Nordin, S.; Kozor, R.; Vijapurapu, R.; Augusto, J.B.; Knott, K.D.; Captur, G.; Treibel, T.A.; Ramaswami, U.; Tchan, M.; Geberhiwot, T.; et al. Myocardial Storage, Inflammation, and Cardiac Phenotype in Fabry Disease After One Year of Enzyme Replacement Therapy. Circ. Cardiovasc. Imaging 2019, 12, e009430. [Google Scholar] [CrossRef]
- Niemann, M.; Liu, D.; Hu, K.; Herrmann, S.; Breunig, F.; Strotmann, J.; Störk, S.; Voelker, W.; Ertl, G.; Wanner, C.; et al. Prominent papillary muscles in Fabry disease: A diagnostic marker? Ultrasound Med. Biol. 2011, 37, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Linhart, A.; Kampmann, C.; Zamorano, J.L.; Sunder-Plassmann, G.; Beck, M.; Mehta, A.; Elliott, P.M.; European FOS Investigators. Cardiac manifestations of Anderson-Fabry disease: Results from the international Fabry outcome survey. Eur. Heart J. 2007, 28, 1228–1235. [Google Scholar] [CrossRef]
- Moon, J.C.; Sheppard, M.; Reed, E.; Lee, P.; Elliott, P.M.; Pennell, D.J. The histological basis of late gadolinium enhancement cardiovascular magnetic resonance in a patient with Anderson-Fabry disease. J. Cardiovasc. Magn. Reson. 2006, 8, 479–482. [Google Scholar] [CrossRef]
- Vijapurapu, R.; Bradlow, W.; Leyva, F.; Moon, J.C.; Zegard, A.; Lewis, N.; Kotecha, D.; Jovanovic, A.; Hughes, D.A.; Woolfson, P.; et al. Cardiac device implantation and device usage in Fabry and hypertrophic cardiomyopathy. Orphanet J. Rare Dis. 2022, 17, 6. [Google Scholar] [CrossRef]
- Lillo, R.; Pieroni, M.; Camporeale, A.; Ciabatti, M.; Lombardo, A.; Massetti, M.; Graziani, F. Echocardiography in Anderson-Fabry Disease. Rev. Cardiovasc. Med. 2022, 23, 201. [Google Scholar] [CrossRef]
- Cianciulli, T.F.; Saccheri, M.C.; Rísolo, M.A.; Lax, J.A.; Méndez, R.J.; Morita, L.A.; Beck, M.A.; Kazelián, L.R. Mechanical dispersion in Fabry disease assessed with speckle tracking echocardiography. Echocardiography 2020, 37, 293–301. [Google Scholar] [CrossRef]
- Kawakami, H.; Nerlekar, N.; Haugaa, K.H.; Edvardsen, T.; Marwick, T.H. Prediction of Ventricular Arrhythmias With Left Ventricular Mechanical Dispersion: A Systematic Review and Meta-Analysis. JACC Cardiovasc. Imaging 2020, 13, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Faro, D.C.; Losi, V.; Rodolico, M.S.; Licciardi, S.; Monte, I.P. Speckle tracking echocardiography-derived parameters as new prognostic markers in hypertrophic cardiomyopathies. Eur. Heart J. Open 2023, 3, oead014. [Google Scholar] [CrossRef]
- Duro, G.; Zizzo, C.; Cammarata, G.; Burlina, A.; Burlina, A.; Polo, G.; Scalia, S.; Oliveri, R.; Sciarrino, S.; Francofonte, D.; et al. Mutations in the GLA Gene and LysoGb3: Is It Really Anderson-Fabry Disease? Int. J. Mol. Sci. 2018, 19, 3726. [Google Scholar] [CrossRef]
- Germain, D.P.; Oliveira, J.P.; Bichet, D.G.; Yoo, H.W.; Hopkin, R.J.; Lemay, R.; Politei, J.; Wanner, C.; Wilcox, W.R.; Warnock, D.G. Use of a rare disease registry for establishing phenotypic classification of previously unassigned GLA variants: A consensus classification system by a multispecialty Fabry disease genotype–phenotype workgroup. J. Med. Genet. 2020, 57, 542–551. [Google Scholar] [CrossRef]
- Nagueh, S.F.; Smiseth, O.A.; Appleton, C.P.; Byrd, B.F., 3rd; Dokainish, H.; Edvardsen, T.; Flachskampf, F.A.; Gillebert, T.C.; Klein, A.L.; Lancellotti, P.; et al. Recommendations for the Evaluation of Left Ventricular Diastolic Function by Echocardiography: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2016, 29, 277–314. [Google Scholar] [CrossRef] [PubMed]
- Badano, L.P.; Kolias, T.J.; Muraru, D.; Abraham, T.P.; Aurigemma, G.; Edvardsen, T.; D’Hooge, J.; Donal, E.; Fraser, A.G.; Marwick, T.; et al. Standardization of left atrial, right ventricular, and right atrial deformation imaging using two-dimensional speckle tracking echocardiography: A consensus document of the EACVI/ASE/Industry Task Force to standardize deformation imaging. Eur. Heart J. Cardiovasc. Imaging 2018, 19, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Galderisi, M.; Cosyns, B.; Edvardsen, T.; Cardim, N.; Delgado, V.; Di Salvo, G.; Donal, E.; Sade, L.E.; Ernande, L.; Garbi, M.; et al. Standardization of adult transthoracic echocardiography reporting in agreement with recent chamber quantification, diastolic function, and heart valve disease recommendations: An expert consensus document of the European Association of Cardiovascular Imaging. Eur. Heart J. Cardiovasc. Imaging 2017, 18, 1301–1310. [Google Scholar]
- Moura, B.; Aimo, A.; Al-Mohammad, A.; Keramida, K.; Ben Gal, T.; Dorbala, S.; Todiere, G.; Cameli, M.; Barison, A.; Bayes-Genis, A.; et al. Diagnosis and management of patients with left ventricular hypertrophy: Role of multimodality cardiac imaging. A scientific statement of the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2023. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Lee, B.H.; Heo, S.H.; Kim, G.H.; Kim, Y.M.; Kim, D.S.; Ko, J.M.; Sohn, Y.B.; Hong, Y.H.; Lee, D.H.; et al. Clinical characteristics and mutation spectrum of GLA in Korean patients with Fabry disease by a nationwide survey: Underdiagnosis of late-onset phenotype. Medicine 2017, 96, e7387. [Google Scholar] [CrossRef]
- Eng, C.M.; Ashley, G.A.; Burgert, T.S.; Enriquez, A.L.; D’Souza, M.; Desnick, R.J. Fabry disease: Thirty-five mutations in the alpha-galactosidase A gene in patients with classic and variant phenotypes. Mol. Med. 1997, 3, 174–182. [Google Scholar] [CrossRef]
- Sakuraba, H.; Oshima, A.; Fukuhara, Y.; Shimmoto, M.; Nagao, Y.; Bishop, D.F.; Desnick, R.J.; Suzuki, Y. Identification of point mutations in the alpha-galactosidase A gene in classical and atypical hemizygotes with Fabry disease. Am. J. Hum. Genet. 1990, 47, 784–789. [Google Scholar]
- Sánchez, R.; Ripoll-Vera, T.; López-Mendoza, M.; de Juan-Ribera, J.; Gimeno, J.R.; Hermida, Á.; Ruz-Zafra, M.A.; Torregrosa, J.V.; Mora, A.; García-Pinilla, J.M.; et al. The Spanish Fabry women study: A retrospective observational study describing the phenotype of females with GLA variants. Orphanet J. Rare Dis. 2023, 18, 8. [Google Scholar] [CrossRef]
- El Sayed, M.; Hirsch, A.; Boekholdt, M.; van Dussen, L.; Datema, M.; Hollak, C.; Langeveld, M. Influence of sex and phenotype on cardiac outcomes in patients with Fabry disease. Heart 2021, 107, 1889–1897. [Google Scholar] [CrossRef] [PubMed]
- Niemann, M.; Herrmann, S.; Hu, K.; Breunig, F.; Strotmann, J.; Beer, M.; Machann, W.; Voelker, W.; Ertl, G.; Wanner, C.; et al. Differences in Fabry cardiomyopathy between female and male patients: Consequences for diagnostic assessment. JACC Cardiovasc. Imaging 2011, 4, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Beer, M.; Kralewski, M.; Siwy, J.; Kampmann, C. Early detection of organ involvement in Fabry disease by biomarker assessment in conjunction with LGE cardiac MRI: Results from the SOPHIA study. Mol. Genet. Metab. 2019, 126, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, O.; Gaspar, P.; Sá Miranda, C.; Cunha, D.; Medeiros, R.; Lourenço, A. Left ventricular noncompaction in a patient with fabry disease: Overdiagnosis, morphological manifestation of fabry disease or two unrelated rare conditions in the same patient? Cardiology 2011, 119, 155–159. [Google Scholar] [CrossRef]
- Azevedo, O.; Marques, N.; Craveiro, N.; Pereira, A.R.; Antunes, H.; Reis, L.; Guerreiro, R.A.; Pontes Dos Santos, R.; Miltenberger-Miltenyi, G.; Sousa, N.; et al. Screening for Fabry disease in patients with left ventricular noncompaction. Rev. Port. Cardiol. 2019, 38, 709–716. [Google Scholar] [CrossRef]
- Krämer, J.; Niemann, M.; Störk, S.; Frantz, S.; Beer, M.; Ertl, G.; Wanner, C.; Weidemann, F. Relation of burden of myocardial fibrosis to malignant ventricular arrhythmias and outcomes in Fabry disease. Am. J. Cardiol. 2014, 114, 895–900. [Google Scholar] [CrossRef]
- Biagini, E. “Effetto donna” nelle cardiomiopatie. G. Ital. Cardiol. 2012, 13, 8. [Google Scholar]
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef]
- Stiles, A.R.; Zhang, H.; Dai, J.; McCaw, P.; Beasley, J.; Rehder, C.; Koeberl, D.D.; McDonald, M.; Bali, D.S.; Young, S.P. A comprehensive testing algorithm for the diagnosis of Fabry disease in males and females. Mol. Genet. Metab. 2020, 130, 209–214. [Google Scholar] [CrossRef]
- Avanesov, M.; Asgari, A.; Muschol, N.; Köhn, A.F.; Tahir, E.; Adam, G.; Kirchhof, P.; Lund, G.; Cavus, E.; Patten, M. Comparison of classical Fabry and its p.D313Y and p.A143T variants by cardiac T1 mapping, LGE and feature tracking myocardial strain. Sci. Rep. 2023, 13, 5809. [Google Scholar] [CrossRef] [PubMed]
- Vijapurapu, R.; Nordin, S.; Baig, S.; Liu, B.; Rosmini, S.; Augusto, J.; Tchan, M.; Hughes, D.A.; Geberhiwot, T.; Moon, J.C.; et al. Global longitudinal strain, myocardial storage and hypertrophy in Fabry disease. Heart 2019, 105, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, O.; Gago, M.F.; Miltenberger-Miltenyi, G.; Robles, A.R.; Costa, M.A.; Pereira, O.; Vide, A.T.; Castelo Branco, G.; Simões, S.; Guimarães, M.J.; et al. Natural history of the late-onset phenotype of Fabry disease due to the p.F113L mutation. Mol. Genet. Metab. Rep. 2020, 22, 100565. [Google Scholar] [CrossRef] [PubMed]
- Maki, N.; Komatsuda, A.; Wakui, H.; Oyama, Y.; Kodama, T.; Ohtani, H.; Kigawa, A.; Aiba, N.; Imai, H.; Motegi, M.; et al. A nonsense mutation (R220X) in the alpha-galactosidase A gene causes typical Fabry disease in both genders. Clin. Nephrol. 2004, 61, 185–190. [Google Scholar] [CrossRef]
- Monte, M.A.; Veroux, M.; Rodolico, M.S.; Losi, V.; Di Pino, L.; Bella, R.; Lanza, G.; Monte, I.P. Fabry’s Disease: The Utility of a Multidisciplinary Screening Approach. Life 2022, 12, 623. [Google Scholar] [CrossRef] [PubMed]
- Zizzo, C.; Monte, I.; Pisani, A.; Fatuzzo, P.; Riccio, E.; Rodolico, M.S.; Colomba, P.; Uva, M.; Cammarata, G.; Alessandro, R.; et al. Molecular and clinical studies in five index cases with novel mutations in the GLA gene. Gene 2016, 578, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Iemolo, F.; Pizzo, F.; Albeggiani, G.; Zizzo, C.; Colomba, P.; Scalia, S.; Bartolotta, C.; Duro, G. De novo mutation in a male patient with Fabry disease: A case report. BMC Res. Notes 2014, 7, 11. [Google Scholar] [CrossRef]
- Cammarata, G.; Fatuzzo, P.; Rodolico, M.S.; Colomba, P.; Sicurella, L.; Iemolo, F.; Zizzo, C.; Alessandro, R.; Bartolotta, C.; Duro, G.; et al. High Variability of Fabry Disease Manifestations in an Extended Italian Family. BioMed Res. Int. 2015, 2015, 504784. [Google Scholar] [CrossRef]
- Schiffmann, R.; Fuller, M.; Clarke, L.A.; Aerts, J.M. Is it Fabry disease? Genet. Med. 2016, 18, 1181–1185. [Google Scholar] [CrossRef]
- Yeniçerioğlu, Y.; Akdam, H.; Dursun, B.; Alp, A.; Sağlam Eyiler, F.; Akın, D.; Gün, Y.; Hüddam, B.; Batmazoğlu, M.; Gibyeli Genek, D.; et al. Screening Fabry’s disease in chronic kidney disease patients not on dialysis: A multicenter study. Ren. Fail. 2017, 39, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Giese, A.K.; Markoff, A.; Grittner, U.; Kolodny, E.; Mascher, H.; Lackner, K.J.; Meyer, W.; Wree, P.; Saviouk, V.; et al. Functional Characterisation of alpha-Galactosidase A Mutations as a Basis for a New Classification System in Fabry Disease. PLoS Genet. 2013, 9, e1003632. [Google Scholar] [CrossRef] [PubMed]
- Koulousios, K.; Stylianou, K.; Pateinakis, P.; Zamanakou, M.; Loules, G.; Manou, E.; Kyriklidou, P.; Katsinas, C.; Ouzouni, A.; Kyriazis, J.; et al. Fabry disease due to D313Y and novel GLA mutations. BMJ Open 2017, 7, e017098. [Google Scholar] [CrossRef] [PubMed]
- Froissart, R.; Guffon, N.; Vanier, M.T.; Desnick, R.J.; Maire, I. Fabry disease: D313Y is an α-galactosidase A sequence variant that causes pseudodeficient activity in plasma. Mol. Genet. Metab. 2003, 80, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Desnick, R.J.; Brady, R.; Barranger, J.; Collins, A.J.; Germain, D.P.; Goldman, M.; Grabowski, G.; Packman, S.; Wilcox, W.R. Fabry disease, an under-recognized multisystemic disorder: Expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann. Intern. Med. 2003, 138, 338–346. [Google Scholar] [CrossRef]
- Wang, R.Y.; Lelis, A.; Mirocha, J.; Wilcox, W.R. Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet. Med. 2007, 9, 34–45. [Google Scholar] [CrossRef]
- Wagenhäuser, L.; Rickert, V.; Sommer, C.; Wanner, C.; Nordbeck, P.; Rost, S.; Üçeyler, N. X-chromosomal inactivation patterns in women with Fabry disease. Mol. Genet. Genom. Med. 2022, 10, e2029. [Google Scholar] [CrossRef] [PubMed]
- De Riso, G.; Cuomo, M.; Di Risi, T.; Della Monica, R.; Buonaiuto, M.; Costabile, D.; Pisani, A.; Cocozza, S.; Chiariotti, L. Ultra-Deep DNA Methylation Analysis of X-Linked Genes: GLA and AR as Model Genes. Genes 2020, 11, 620. [Google Scholar] [CrossRef] [PubMed]
- Di Risi, T.; Vinciguerra, R.; Cuomo, M.; Della Monica, R.; Riccio, E.; Cocozza, S.; Imbriaco, M.; Duro, G.; Pisani, A.; Chiariotti, L. DNA methylation impact on Fabry disease. Clin. Epigenetics 2021, 13, 24. [Google Scholar] [CrossRef]
- Wanner, C.; Feldt-Rasmussen, U.; Jovanovic, A.; Linhart, A.; Yang, M.; Ponce, E.; Brand, E.; Germain, D.P.; Hughes, D.A.; Jefferies, J.L.; et al. Cardiomyopathy and kidney function in agalsidase beta-treated female Fabry patients: A pre-treatment vs. post-treatment analysis. ESC Heart Fail. 2020, 7, 825–834. [Google Scholar] [CrossRef]
- Alharbi, F.J.; Baig, S.; Auray-Blais, C.; Boutin, M.; Ward, D.G.; Wheeldon, N.; Steed, R.; Dawson, C.; Hughes, D.; Geberhiwot, T. Globotriaosylsphingosine (Lyso-Gb(3)) as a biomarker for cardiac variant (N215S) Fabry disease. J. Inherit. Metab. Dis. 2018, 41, 239–247. [Google Scholar] [CrossRef]
- van Breemen, M.J.; Rombach, S.M.; Dekker, N.; Poorthuis, B.J.; Linthorst, G.E.; Zwinderman, A.H.; Breunig, F.; Wanner, C.; Aerts, J.M.; Hollak, C.E. Reduction of elevated plasma globotriaosylsphingosine in patients with classic Fabry disease following enzyme replacement therapy. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2011, 1812, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.; Mechtler, T.; Kasper, D.C.; Desnick, R.J. Correlation of Lyso-Gb3 levels in dried blood spots and sera from patients with classic and Later-Onset Fabry disease. Mol. Genet. Metab. 2017, 121, 320–324. [Google Scholar] [CrossRef]
- Auray-Blais, C.; Lavoie, P.; Boutin, M.; Ntwari, A.; Hsu, T.R.; Huang, C.K.; Niu, D.M. Biomarkers associated with clinical manifestations in Fabry disease patients with a late-onset cardiac variant mutation. Clin. Chim. Acta 2017, 466, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Burlina, A.; Brand, E.; Hughes, D.; Kantola, I.; Krämer, J.; Nowak, A.; Tøndel, C.; Wanner, C.; Spada, M. An expert consensus on the recommendations for the use of biomarkers in Fabry disease. Mol. Genet. Metab. 2023, 139, 107585. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Abiose, A.; Bichet, D.G.; Cabrera, G.; Charrow, J.; Germain, D.P.; Hopkin, R.J.; Jovanovic, A.; Linhart, A.; Maruti, S.S.; et al. Time to treatment benefit for adult patients with Fabry disease receiving agalsidase β: Data from the Fabry Registry. J. Med. Genet. 2016, 53, 495–502. [Google Scholar] [CrossRef]
- Nowak, A.; Beuschlein, F.; Sivasubramaniam, V.; Kasper, D.; Warnock, D.G. Lyso-Gb3 associates with adverse long-term outcome in patients with Fabry disease. J. Med. Genet. 2022, 59, 287–293. [Google Scholar] [CrossRef]
- Maruyama, H.; Miyata, K.; Mikame, M.; Taguchi, A.; Guili, C.; Shimura, M.; Murayama, K.; Inoue, T.; Yamamoto, S.; Sugimura, K.; et al. Effectiveness of plasma lyso-Gb3 as a biomarker for selecting high-risk patients with Fabry disease from multispecialty clinics for genetic analysis. Genet. Med. 2019, 21, 44–52. [Google Scholar] [CrossRef]
- Hsu, T.R.; Chang, F.P.; Chu, T.H.; Sung, S.H.; Bizjajeva, S.; Yu, W.C.; Niu, D.M. Correlations between Endomyocardial Biopsies and Cardiac Manifestations in Taiwanese Patients with the Chinese Hotspot IVS4+919G>A Mutation: Data from the Fabry Outcome Survey. Int. J. Mol. Sci. 2017, 18, 119. [Google Scholar] [CrossRef]
- Kampmann, C.; Perrin, A.; Beck, M. Effectiveness of agalsidase alfa enzyme replacement in Fabry disease: Cardiac outcomes after 10 years’ treatment. Orphanet J. Rare Dis. 2015, 10, 125. [Google Scholar] [CrossRef]
- Ramaswami, U.; Beck, M.; Hughes, D.; Kampmann, C.; Botha, J.; Pintos-Morell, G.; West, M.L.; Niu, D.M.; Nicholls, K.; Giugliani, R.; et al. Cardio- Renal Outcomes With Long- Term Agalsidase Alfa Enzyme Replacement Therapy: A 10- Year Fabry Outcome Survey (FOS) Analysis. Drug Des. Devel. Ther. 2019, 13, 3705–3715. [Google Scholar]
- Lenders, M.; Nordbeck, P.; Kurschat, C.; Karabul, N.; Kaufeld, J.; Hennermann, J.B.; Patten, M.; Cybulla, M.; Müntze, J.; Üçeyler, N.; et al. Treatment of Fabry’s Disease With Migalastat: Outcome From a Prospective Observational Multicenter Study (FAMOUS). Clin. Pharmacol. Ther. 2020, 108, 326–337. [Google Scholar] [CrossRef]
- Hughes, D.A.; Barba Romero, M.Á.; Hollak, C.E.; Giugliani, R.; Deegan, P.B. Response of women with Fabry disease to enzyme replacement therapy: Comparison with men, using data from FOS—the Fabry Outcome Survey. Mol. Genet. Metab. 2011, 103, 207–214. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AFD Tot (N = 72) | AFD-LVH (N = 12) | AFD-N (N = 60) | NH (N = 40) | |
|---|---|---|---|---|
| Age, yo | 45 ± 16.1 | 60.7 ± 7.2 | 41.8 ± 15.6 | 44.1 ± 13.9 |
| M | 19 (26.4%) | 3 (25%) | 16 (26.7%) | 26 (65%) |
| F | 53 (73.6%) | 9 (75%) | 44 (73.3%) | 14 (35%) |
| Hypertension | 24 (33.3%) | 8 (66.7%) | 16 (26.7%) | |
| Diabetes | 9 (12.5%) | 2 (16.7%) | 7 (11.7%) | |
| Smoke | 14 (19.4%) | 2 (16.7%) | 12 (20%) | |
| Dyslipidemia | 9 (12.5%) | 1 (8.3%) | 8 (13.3%) | |
| CKD | 9 (12.5%) | 5 (41.7%) | 4 (6.7%) | |
| Stroke/TIA | 6 (8.3%) | 2 (16.7%) | 4 (6.7%) | |
| Syncope | 6 (8.3%) | 2 (16.7%) | 4 (6.7%) | |
| Fam. history SCD | 1 (1.4%) | 1 (8.3%) | 0 | |
| Fam. history AFD | 41 (56.9%) | 8 (66.7%) | 33 (55%) | |
| Kidney transplant | 6 (8%) | 4 (30.8%) | 2 (3.2%) | |
| α-Gal A (nmol/mL/h) | 5.0 [2.6–9.4] | 6.4 [0.3–9.6] | 4.9 [2.6–9.8] | |
| Lyso-Gb3 (nmol/L) | 1.5 [1.3–2.7] | 9.2 [6.3–16.1] | 1.5 [1.2–1.7] | |
| CV Death | 2 (2.8%) | 2 (16.7%) | 0 | |
| NYHA 1 | 53 (73.6%) | 4 (33.3%) | 49 (81.7%) | |
| NYHA 2 | 18 (25%) | 7 (58.3%) | 11 (18.3%) | |
| NYHA 3 | 1 (1.4%) | 1 (8.3%) | 0 | |
| NYHA 4 | 0 | 0 | 0 | |
| BP-sys (mmHg) | 125 [120–135] | 137.5 [126.2–143.7] | 120 [115–130] | 130 [120–140] |
| BP-dia (mmHg) | 80 [70–80] | 80 [71.2–80] | 75 [70–80] | 80 [70–80] |
| HR (bpm) | 69.5 ± 8.8 | 64.7 ± 11.1 | 70.5 ± 8 | 66 ± 12.8 |
| ERT | 18 (25%) | 7 (58.3%) | 11 (18.3%) | |
| Migalastat | 8 (11.1%) | 1 (8.3%) | 7 (11.7%) | |
| ICD | 1 (1.4%) | 1 (8.3%) | 0 | |
| PM | 1 (1.4%) | 1 (8.3%) | 0 | |
| GLA variant type: | ||||
| 13 (18.1%) | 5 (41.7%) | 8 (13.4%) | |
| 30 (41.6%) | 4 (33.3%) | 26 (43.3%) | |
| 29 (40.3%) | 3 (25%) | 26 (43.3%) |
| AFD (tot) (N = 72) | AFD-LVH (N = 12) | AFD-N (N = 60) | |
|---|---|---|---|
| RBBB | 4 (5.5%) | 1 (8.3%) | 3 (4.84%) |
| AVB-III | 1 (1.33%) | 1 (7.7%) | 0 |
| Short PR | 3 (4.2%) | 3 (25%) | 0 |
| D-ECG done | 12 (16.7%) | 8 (66.7%) | 4 (6.7%) |
| VT/NSVT | 6 (8.3%) | 5 (41.7%) | 1 (1.7%) |
| AF | 7 (9.7%) | 7 (58.3%) | 0 |
| PSVT | 7 (9.7%) | 6 (50%) | 1 (1.7%) |
| CMR done | 14 (19.4%) | 6 (50%) | 8 (13.3%) |
| LGE+ (% tot) | 9 (12.5%) | 5 (41.7%) | 4 (6.7%) |
| LGE+ (% pts with CMR) | 9/14 (64.2%) | 5/6 (83.3%) | 4/8 (50%) |
| N° segments LGE | 2.5 [2–5] | 3 [2–8.5] | 2 [1.5–4.5] |
| Panel A | Type | Variant | tot | N° Families | Males | Females | |
|---|---|---|---|---|---|---|---|
| Pathogenic/ likely pathogenic | CLASSIC | Lys240GlufsTer9 | 4 | 1 | 2 | 2 | |
| E341X | 3 | 1 | 1 | 2 | |||
| R220X | 1 | 1 | 0 | 1 | |||
| D165H | 2 | 1 | 1 | 1 | |||
| G85D | 4 | 1 | 1 | 3 | |||
| M267I | 2 | 1 | 0 | 2 | |||
| c.9401A > e c.9264delA; exone VII | 1 | 1 | 1 | 0 | |||
| LATE-ONSET | I91T | 3 | 1 | 1 | 2 | ||
| M51I | 17 (11.8%) | 3 | 5 | 12 | |||
| G395A | 12 (8.3%) | 4 | 3 | 9 | |||
| F113L | 11 (7.6%) | 2 | 2 | 9 | |||
| Panel B | type | variant | N° tot | AFD-LVH | AFD-N | Males | Females |
| Pathogenic/ likely pathogenic | CLASSIC | Lys240GlufsTer9 | 4 | 1 F | 2M 1F | 2 | 2 |
| E341X | 2 | 1 F | 1 F | - | 2 | ||
| R220X | 1 | 1 F | - | - | 1 | ||
| D165H | 2 | 1 M | 1 F | 1 | 1 | ||
| G85D | 2 | 1 F | 1 F | - | 2 | ||
| M267I | 1 | - | 1 F | - | 1 | ||
| c.9401A > e c.9264delA; exone VII | 1 | - | 1 M | 1 | - | ||
| LATE-ONSET | I91T | 1 | 1 M | - | 1 | - | |
| M51I | 11 (15.3%) | 1 F | 10 | 2 (10.5%) | 9 (17%) | ||
| G395A | 8 (11.1%) | - | 8 | 2 (10.5%) | 6 (11.3%) | ||
| F113L | 10 (13.9%) | 1F 1M | 8 | 2 (10.5%) | 8 (15.1%) | ||
| AFD (tot) (N = 72) | AFD-LVH (N = 12) | AFD-N (N = 60) | NH (N = 40) | p Value (All Groups) | AFD-LVH vs. AFD-N | AFD-LVH vs. NH | AFD-N vs. NH | |
|---|---|---|---|---|---|---|---|---|
| IVS (mm) | 8.4 [7.0–9.2] | 13 [12.1–17.3] | 8 [6.5–9] | 8.7 [7.1–9.4] | <0.001 | <0.001 | <0.001 | 0.10 |
| PW (mm) | 8.0 [7.0–9.3] | 12 [10.2–14] | 8 [7–9] | 8.9 [7–10] | <0.001 | <0.001 | <0.001 | 0.05 |
| LVMi (g/sqm) | 71.5 [57.2–86.2] | 116 [103.2–180.7] | 67 [54–78] | 76.8 [60.2–89.7] | <0.001 | <0.001 | <0.001 | 0.01 |
| EF (%) | 65 [62–67.7] | 65 [62.2–69.2] | 65 [61.2–67.7] | 64.5 [62–66.7] | 0.66 | 0.79 | 0.44 | 0.45 |
| LAVi (ml/sqm) | 24 [17–31] | 42.5 [33.2–54] | 22 [17–27] | 19 [14.7–27] | <0.001 | <0.001 | <0.001 | 0.25 |
| E/e’ | 7 [6–10] | 11.5 [9.2–16.7] | 7 [5.6–9] | 7 [6–8] | 0.001 | 0.001 | <0.001 | 0.71 |
| TR-Vmax (m/s) | 2.3 [1.9–2.4] | 2.3 [2.1–2.4] | 2.2 [1.8–2.4] | 2.1 [1.9–2.3] | 0.28 | 0.16 | 0.11 | 0.83 |
| LV-GLS (-%) | −18 [−16/−21] | −11.5 [−9.2/−16.7] | −19 [−17/−22] | −20 [−18/−21] | <0.001 | <0.001 | <0.001 | 0.24 |
| LV-MD (ms) | 39 [29–60] | 74 [59.2–90] | 36 [29–46] | 31 [27–41] | <0.001 | <0.001 | <0.001 | 0.13 |
| Papill Hypertr. | 4 (5.6%) | 1 (8.3%) | 3 (5%) | 0 |
| AFD Tot F (N = 53) | AFD Tot M (N = 19) | p-Value | |
|---|---|---|---|
| AGE, yo | 47.2 ± 16.2 | 38.8 ± 14.6 | 0.046 |
| CLASSIC variant | 9 (17%) | 4 (21%) | 0.73 |
| LATE-ONSET | 23 (43.4%) | 7 (36.9%) | 0.78 |
| VUS | 21 (39.6%) | 8 (42.1%) | 1 |
| α-Gal A (nmol/mL/h) | 7.9 [4.6–11.3] | 1.8 [0.3–3.35] | <0.001 |
| LYSO-GB3 (nmol/L) | 1.5 [1.1–1.7] | 1.9 [1.5–17.3] | 0.02 |
| ERT | 14 (26.4%) | 4 (21.1%) | 0.76 |
| Migalastat | 6 (11.3%) | 2 (10.5%) | 1 |
| V-AR | 4 (7.5%) | 2 (10.5%) | 0.65 |
| AF | 5 (9.4%) | 2 (10.5%) | 1 |
| Pts with LVH | 9 (17%) | 3 (15.8%) | 1 |
| LVMi (g/sqm) | 66 [54–85.5] | 78 [73–96] | 0.03 |
| EF, (%) | 65 [62–67.5] | 64 [60–68] | 0.38 |
| E/e’ | 8 [6–10] | 7 [5–10] | 0.22 |
| LV-GLS (-%) | −19 [−17, −22] | −17 [−14, −20] | 0.02 |
| MD (ms) | 37 [29.2–49.7] | 41 [29–60] | 0.65 |
| LGE+ | 4 [57% of CRM] | 7 [87.5%] | 0.001 |
| LGE segments, n° | 3 [1–3] | 2 [2–5] | 0.003 |
| AFD-LVH F (N = 9) | AFD-LVH M (N = 3) | p Value | AFD-N F (N = 44) | AFD-N M (N = 16) | p Value | |
|---|---|---|---|---|---|---|
| AGE, yo | 62.5 ± 3.9 | 55.3 ± 13 | 0.44 | 44 ± 16 | 35.7 ± 12.9 | 0.048 |
| CLASSIC MUTATION | 4 (40%) | 1 (33.3%) | 1 | 5 (11.4%) | 3 (18.7%) | 0.43 |
| LATE-ONSET | 2 (20%) | 2 (66.7%) | 0.24 | 21 (47.7%) | 5 (31.3%) | 0.38 |
| VUS | 3 (30%) | 0 | 0.50 | 18 (40.9%) | 6 (37.5%) | 1 |
| α-Gal A (nmol/mL/h) | 8.5 [4.8–10.9] | 0.3 [0–0.3] | 0.017 | 6.8 [4.5–13.8] | 2.75 [0.57–4.05] | 0.003 |
| LYSO-GB3 (nmol/L) | 8.5 [2.8–9.3] | 22.9 [9.4–22.9] | 0.13 | 1.5 [1.1–1.7] | 1.65 [1.4–2.7] | 0.04 |
| ERT | 5 (55.6%) | 2 (66.7%) | 1 | 9 (20.5%) | 2 (12.5%) | 0.71 |
| Migalastat | 0 | 1 | 0.25 | 6 (13.6%) | 1 (6.3%) | 0.66 |
| V-AR | 3 (33.3%) | 2 (66.7%) | 0.52 | 1 (2.3%) | 0 | 1 |
| AF | 5 (55.6%) | 2 (66.7%) | 1 | 0 | 0 | 1 |
| LVMi (g/sqm) | 115 [102–140] | 227 [116–227] | 0.06 | 61.5 [53–74.2] | 77.5 [69.2–80] | 0.008 |
| EF (%) | 65 [62–72] | 65 [62–65] | 0.86 | 65.5 [62–67.25] | 64 [60–68.75] | 0.42 |
| E/e’ | 13 [7.5–15] | 10 [7.7–17] | 0.72 | 7 [6–9] | 6.5 [5–7.75] | 0.09 |
| LV-GLS (-%) | −12.6 [−10/−18.5] | −10 [−9/−10] | 0.20 | −20 [−17/−22] | −17 [−16/−20] | 0.01 |
| MD (ms) | 70 [48–90.5] | 87 [57–87] | 0.72 | 35 [29–45] | 38 [28.2–58.2] | 0.78 |
| AFD-LVH Females (N = 9) | AFD-N Females (N = 44) | p Value | |
|---|---|---|---|
| Age (yo) | 62.5 ± 3.9 | 44 ± 16 | 0.001 |
| α-Gal A (nmol/mL/h) | 8.5 [4.8–10.9] | 6.8 [4.5–13.8] | 0.85 |
| Lyso-Gb3 (nmol/L) | 8.5 [2.8–9.3] | 1.5 [1.1–1.7] | 0.048 |
| Classic mutation | 4 (40%) | 5 (11.4%) | 0.03 |
| Late-onset | 2 (20%) | 21 (47.7%) | 0.27 |
| VUS | 3 (30%) | 18 (40.9%) | 1 |
| V-AR | 3 (33.3%) | 1 (2.3%) | 0.01 |
| AF | 5 (55.6%) | 0 | <0.001 |
| LVMi (g/sqm) | 115 [102–140] | 61.5 [53–74.2] | <0.001 |
| EF (%) | 65 [62–72] | 65.5 [62–67.25] | 0.88 |
| E/e’ | 13 [7.5–15] | 7 [6–9] | 0.009 |
| GLS (-%) | −12.6 [−10/−18.5] | −20 [−17/−22] | 0.001 |
| MD (ms) | 70 [48–90.5] | 35 [29–45] | 0.002 |
| AFD-N Females N = 44 | NH Females N = 14 | p Value | AFD-N Males N = 16 | NH Males N = 26 | p Value | |
|---|---|---|---|---|---|---|
| Age, yo | 44 ± 16 | 48.7 ± 11.5 | 0.30 | 35.7 ± 12.9 | 41.6 ±14.6 | 0.20 |
| LVMi (g/sqm) | 61.5 [53–74.2] | 64 [50.5–78.5] | 0.89 | 77.5 [69.2–80] | 83 [72–81.7] | 0.11 |
| EF (%) | 65.5 [62–67.25] | 65 [62.7–66.25] | 0.66 | 64 [60–68.7] | 63.5 [60.7–67.2] | 0.87 |
| LAVi (ml/sqm) | 22 [17–27] | 25.7 [17.7–31.2] | 0.35 | 24 [17–26] | 17.5 [14–19.7] | 0.14 |
| E/e’ | 7 [6–9] | 7.15 [5.9–8.3] | 0.52 | 6.5 [5–7.7] | 7 [5.9–8.1] | 0.29 |
| TR-Vmax (m/s) | 2.2 [1.8–2.4] | 2.2 [2–2.4] | 0.72 | 2 [1.8–2.4] | 2 [1.9–2.3] | 0.84 |
| LV-GLS (-%) | −20 [−17/−22] | −20 [−16.8/−21.5] | 0.71 | −17 [−16/−20] | −20 [−19/−21.2] | 0.004 |
| LV-MD (ms) | 35 [29–45] | 29.6 [24.5–38.3] | 0.08 | 38 [28.2–58.2] | 34 [27.5–42.5] | 0.50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faro, D.C.; Losi, V.; Rodolico, M.S.; Torrisi, E.M.; Colomba, P.; Duro, G.; Monte, I.P. Sex Differences in Anderson–Fabry Cardiomyopathy: Clinical, Genetic, and Imaging Analysis in Women. Genes 2023, 14, 1804. https://doi.org/10.3390/genes14091804
Faro DC, Losi V, Rodolico MS, Torrisi EM, Colomba P, Duro G, Monte IP. Sex Differences in Anderson–Fabry Cardiomyopathy: Clinical, Genetic, and Imaging Analysis in Women. Genes. 2023; 14(9):1804. https://doi.org/10.3390/genes14091804
Chicago/Turabian StyleFaro, Denise Cristiana, Valentina Losi, Margherita Stefania Rodolico, Elvira Mariateresa Torrisi, Paolo Colomba, Giovanni Duro, and Ines Paola Monte. 2023. "Sex Differences in Anderson–Fabry Cardiomyopathy: Clinical, Genetic, and Imaging Analysis in Women" Genes 14, no. 9: 1804. https://doi.org/10.3390/genes14091804