
Investigating Neuron Degeneration in Huntington’s Disease Using RNA-Seq Based Transcriptome Study


Abstract
:1. Introduction
2. Methods
2.1. Dataset, Pre-Processing, and Variant Analysis
2.2. Effect of Variants over Regulatory Mechanisms and Finding Essential Variant Genes
2.3. Differential Gene/Transcript Proportion Expression Analysis
2.4. Functional Enrichment and Tissue-Specific Network Interaction Analysis
3. Results
3.1. Differential Gene/Transcript Expression Profile
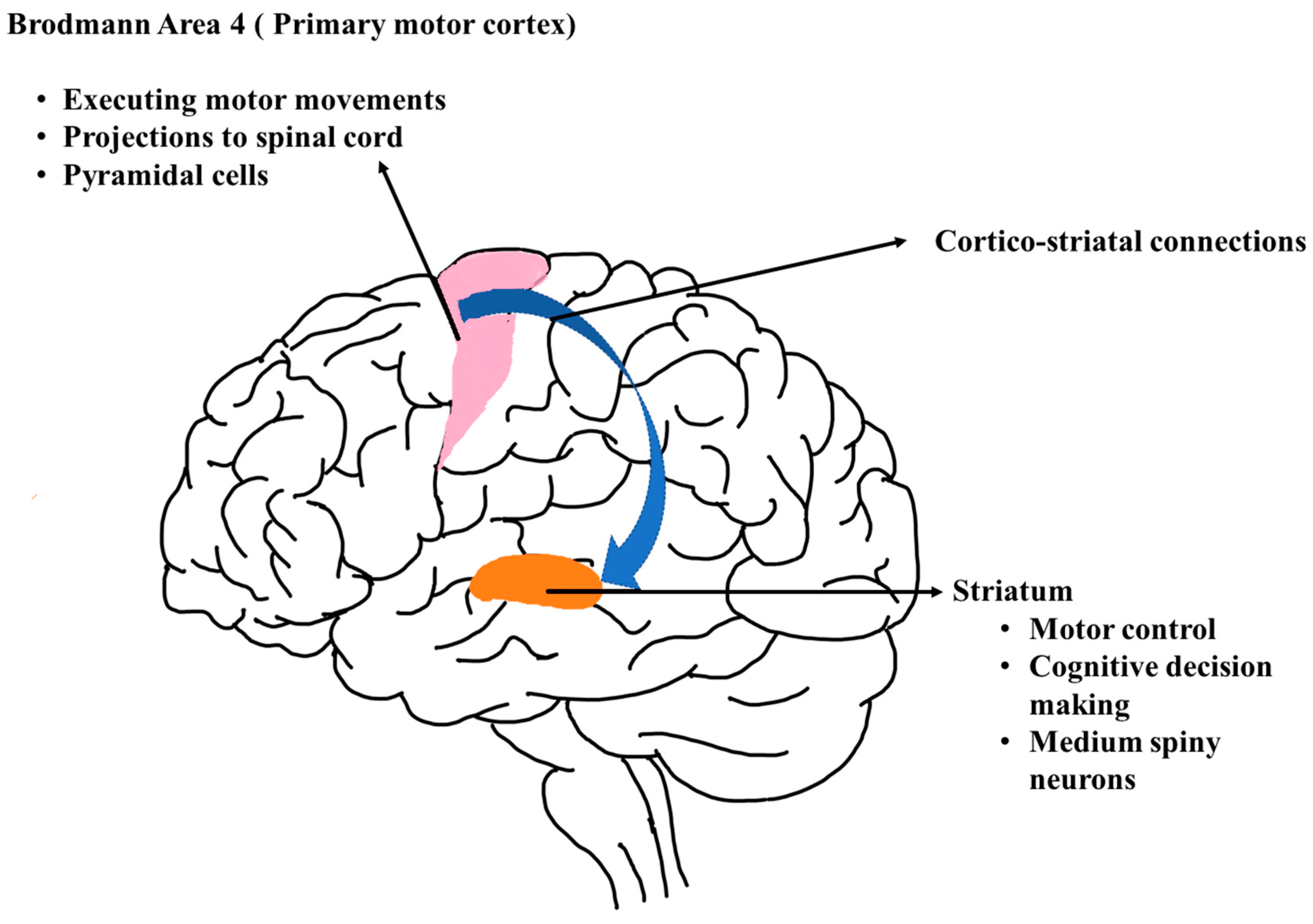
3.1.1. Novel Gene Expression Pattern in BA4
3.1.2. Differential Transcript Expression/Usage Analysis
3.1.3. Similarity of Gene Expression with Previous Bulk RNA-Seq and GTEX Datasets
3.2. Variant Analysis and Associating Variant Effect on Disease-Associated Regulatory Mechanisms
3.2.1. Role of Variants on Transcription Factor Binding
3.2.2. Role of Variants on miRNA Binding and Their Targets
3.2.3. Essential Gene Identification from Genome-Scale Metabolic Model of Variant Genes
3.3. Function Interaction Network of Differentially Expressed Genes and Transcription Factors
3.4. Differential Co-Expression Network of Genes and Function Enrichment Study
3.5. Proposed Mechanism in HD Pathology
3.6. Limitations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Nopoulos, P.C. Huntington disease: A single-gene degenerative disorder of the striatum. Dialogues Clin. Neurosci. 2022, 18, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216. [Google Scholar] [CrossRef]
- Shoulson, I.; Young, A.B. Milestones in huntington disease. Mov. Disord. 2011, 26, 1127–1133. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Leggo, J.; Coles, R.; Almqvist, E.; Biancalana, V.; Cassiman, J.J.; Chotai, K.; Connarty, M.; Craufurd, D.; Curtis, A.; et al. Phenotypic characterization of individuals with 30–40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals witinterconh 36–39 repeats. Am. J. Hum. Genet. 1996, 59, 16. [Google Scholar]
- Møllersen, L.; Rowe, A.D.; Larsen, E.; Rognes, T.; Klungland, A. Continuous and periodic expansion of CAG repeats in Huntington’s disease R6/1 mice. PLoS Genet. 2010, 6, e1001242. [Google Scholar] [CrossRef]
- Priya, S.B.; Gromiha, M.M. Structural insights into the aggregation mechanism of huntingtin exon 1 protein fragment with different polyQ-lengths. J. Cell. Biochem. 2019, 120, 10519–10529. [Google Scholar] [CrossRef]
- Videnovic, A. Treatment of huntington disease. Curr. Treat. Options Neurol. 2013, 15, 424–438. [Google Scholar] [CrossRef]
- Bonomo, R.; Elia, A.E.; Bonomo, G.; Romito, L.M.; Mariotti, C.; Devigili, G.; Cilia, R.; Giossi, R.; Eleopra, R. Deep brain stimulation in Huntington’s disease: A literature review. Neurol. Sci. 2021, 42, 4447–4457. [Google Scholar] [CrossRef]
- Morigaki, R.; Goto, S. Striatal vulnerability in Huntington’s disease: Neuroprotection versus neurotoxicity. Brain Sci. 2017, 7, 63. [Google Scholar] [CrossRef]
- Shipp, S. The functional logic of corticostriatal connections. Brain Struct. Funct. 2017, 222, 669–706. [Google Scholar] [CrossRef]
- Haber, S.N. Corticostriatal circuitry. Dialogues Clin. Neurosci. 2016, 18, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Blumenstock, S.; Dudanova, I. Cortical and striatal circuits in Huntington’s disease. Front. Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef] [PubMed]
- Agus, F.; Crespo, D.; Myers, R.H.; Labadorf, A. The caudate nucleus undergoes dramatic and unique transcriptional changes in human prodromal Huntington’s disease brain. BMC Med. Genom. 2019, 12, 137. [Google Scholar] [CrossRef] [PubMed]
- Malla, B.; Guo, X.; Senger, G.; Chasapopoulou, Z.; Yildirim, F. A systematic review of transcriptional dysregulation in Huntington’s disease studied by RNA sequencing. Front. Genet. 2021, 12, 751033. [Google Scholar] [CrossRef]
- Garcia, F.J.; Sun, N.; Lee, H.; Godlewski, B.; Mathys, H.; Galani, K.; Zhou, B.; Jiang, X.; Ng, A.P.; Mantero, J.; et al. Single-cell dissection of the human brain vasculature. Nature 2022, 603, 893–899. [Google Scholar] [CrossRef]
- Kaye, J.; Reisine, T.; Finkbeiner, S. Huntington’s disease mouse models: Unraveling the pathology caused by CAG repeat expansion. Fac. Rev. 2021, 10, 77. [Google Scholar] [CrossRef]
- Podlacha, M.; Pierzynowska, K.; Gaffke, L.; Jerzemowska, G.; Piotrowska, E.; Węgrzyn, G. Behavioral-and blood-based biomarkers for Huntington’s disease: Studies on the R6/1 mouse model with prospects for early diagnosis and monitoring of the disease. Brain Behav. Immun. Health 2022, 23, 100482. [Google Scholar] [CrossRef]
- Huang, K.L.; Marcora, E.; Pimenova, A.A.; Di Narzo, A.F.; Kapoor, M.; Jin, S.C.; Harari, O.; Bertelsen, S.; Fairfax, B.P.; Czajkowski, J.; et al. A common haplotype lowers PU.1 expression in myeloid cells and delays the onset of Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1052–1061. [Google Scholar] [CrossRef]
- Deming, Y.; Li, Z.; Kapoor, M.; Harari, O.; Del-Aguila, J.L.; Black, K.; Carrell, D.; Cai, Y.; Fernandez, M.V.; Budde, J.; et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol. 2017, 133, 839–856. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, A.; Kenna, K.P.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 2018, 97, 1268–1283. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Park, J.W.; Ramachandran, S.; Zhang, Y.; Tseng, Y.T.; Shen, S.; Waldvogel, H.J.; Curtis, M.A.; Faull, R.L.; Troncoso, J.C.; et al. Transcriptome sequencing reveals aberrant alternative splicing in Huntington’s disease. Hum. Mol. Genet. 2016, 25, 3454–3466. [Google Scholar] [CrossRef] [PubMed]
- Vonsattel, J.-P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P. Neuropathological Classification of Huntinqton’s Disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Jain, M. NGS QC toolkit: A toolkit for quality control of next-generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef]
- Blankenberg, D.; Gordon, A.; Von Kuster, G.; Coraor, N.; Taylor, J.; Nekrutenko, A. Manipulation of FASTQ data with galaxy. Bioinformatics 2010, 26, 1783–1785. [Google Scholar] [CrossRef]
- Krueger, F. Trim Galore: A Wrapper Tool around Cutadapt and FastQC. Trim Galore 2012. Available online: https://github.com/FelixKrueger/TrimGalore/blob/master/Docs/Trim_Galore_User_Guide.md (accessed on 20 May 2023).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Chen, K.; Wylie, T.; Larson, D.E.; McLellan, M.D.; Mardis, E.R.; Weinstock, G.M.; Wilson, R.K.; Ding, L. VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009, 25, 2283–2285. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.N.; Aleksic, J.; Butano, D.; Carr, A.; Contrino, S.; Hu, F.; Lyne, M.; Lyne, R.; Kalderimis, A.; Rutherford, K.; et al. InterMine: A flexible data warehouse system for the integration and analysis of heterogeneous biological data. Bioinformatics 2012, 28, 3163–3165. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Kumar, S.; Ambrosini, G.; Bucher, P. SNP2TFBS-a database of regulatory SNPs affecting predicted transcription factor binding site affinity. Nucleic Acids Res. 2017, 26, 728. [Google Scholar] [CrossRef]
- Han, H.; Cho, J.-W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J.; Fu, X.; Xia, M.; Zhang, Q.; Gu, Z.; Guo, A.Y. miRNASNP-v3: A comprehensive database for SNPs and disease-related variations in miRNAs and miRNA targets. Nucleic Acids Res. 2021, 49, D1276–D1281. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Ziebarth, J.D.; Cui, Y. PolymiRTS Database 3.0: Linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Res. 2014, 42, D86–D91. [Google Scholar] [CrossRef]
- Huang, H.-Y.; Lin, Y.-C.; Cui, S.; Huang, Y.; Tang, Y.; Xu, J.; Bao, J.; Li, Y.; Wen, J.; Zuo, H.; et al. miRTarBase update 2022: An informative resource for experimentally validated miRNA–target interactions. Nucleic Acids Res. 2022, 50, D222–D230. [Google Scholar] [CrossRef]
- Zheng, Z.; Huang, D.; Wang, J.; Zhao, K.; Zhou, Y.; Guo, Z.; Zhai, S.; Xu, H.; Cui, H.; Yao, H.; et al. QTLbase: An integrative resource for quantitative trait loci across multiple human molecular phenotypes. Nucleic Acids Res. 2020, 48, D983–D991. [Google Scholar] [CrossRef]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Dräger, A.; Mih, N.; Gatto, F.; Nilsson, A.; Preciat Gonzalez, G.A.; Aurich, M.K.; et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Schellenberger, J.; Que, R.; Fleming, R.M.; Thiele, I.; Orth, J.D.; Feist, A.M.; Palsson B, Ø. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox v2. 0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Robinson, M.D.; Nowicka, M. DRIMSeq: A Dirichlet-multinomial framework for multivariate count outcomes in genomics. F1000Research 2016, 5, 1356. [Google Scholar] [CrossRef]
- Tekath, T.; Dugas, M. Differential transcript usage analysis of bulk and single-cell RNA-seq data with DTUrtle. Bioinformatics 2021, 37, 3781–3787. [Google Scholar] [CrossRef]
- Wu, G.; Dawson, E.; Duong, A.; Haw, R.; Stein, L. ReactomeFIViz: A cytoscape app for pathway and network-based data analysis. F1000Research 2014, 3, 146. [Google Scholar] [CrossRef]
- Zhou, J.; Theesfeld, C.L.; Yao, K.; Chen, K.M.; Wong, A.K.; Troyanskaya, O.G. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet. 2018, 50, 1171–1179. [Google Scholar] [CrossRef]
- Obayashi, T.; Kagaya, Y.; Aoki, Y.; Tadaka, S.; Kinoshita, K. COXPRESdb v7: A gene coexpression database for 11 animal species supported by 23 coexpression platforms for technical evaluation and evolutionary inference. Nucleic Acids Res. 2019, 47, D55–D62. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L.; et al. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37, D211–D215. [Google Scholar] [CrossRef] [PubMed]
- Labadorf, A.; Hoss, A.G.; Lagomarsino, V.; Latourelle, J.C.; Hadzi, T.C.; Bregu, J.; MacDonald, M.E.; Gusella, J.F.; Chen, J.F.; Akbarian, S.; et al. Correction: RNA sequence analysis of human huntington disease brain reveals an extensive increase in inflammatory and developmental gene expression. PLoS ONE 2016, 11, e0160295. [Google Scholar] [CrossRef] [PubMed]
- Hoss, A.G.; Kartha, V.K.; Dong, X.; Latourelle, J.C.; Dumitriu, A.; Hadzi, T.C.; MacDonald, M.E.; Gusella, J.F.; Akbarian, S.; Chen, J.-F.; et al. MicroRNAs located in the HOX gene clusters are implicated in huntington’s disease pathogenesis. PLoS Genet. 2014, 10, e1004188. [Google Scholar] [CrossRef]
- Kim, J.H.; Yoon, S.; Park, M.; Park, H.O.; Ko, J.J.; Lee, K.; Bae, J. Differential apoptotic activities of wild-type FOXL2 and the adult-type granulosa cell tumor-associated mutant FOXL2 (C134W). Oncogene 2011, 30, 1653–1663. [Google Scholar] [CrossRef]
- Zeisel, A.; Muñoz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; La Manno, G.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef]
- Gonzalez-Halphen, D.; Lindorfer, M.A.; Capaldi, R.A. Subunit arrangement in beef heart complex III. Biochemistry 1988, 27, 7021–7031. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef]
- Gu, C.; Chen, Y.; Chen, Y.; Liu, C.F.; Zhu, Z.; Wang, M. Role of G protein-coupled receptors in microglial activation: Implication in Parkinson’s disease. Front. Aging Neurosci. 2021, 13, 768156. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G. Novel therapeutic targets in neuroinflammation and neuropathic pain. Inflamm. Cell Signal. 2014, 1, e111. [Google Scholar] [PubMed]
- Sun, L.; Ye, R.D. Role of G protein-coupled receptors in inflammation. Acta Pharmacol. Sin. 2012, 33, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Zulfiqar, A.; Arguelles, S.; Rasekhian, M.; Nabavi, S.F.; Silva, A.S.; Nabavi, S.M. Map kinase signaling as therapeutic target for neurodegeneration. Pharmacol. Res. 2020, 160, 105090. [Google Scholar] [CrossRef]
- Li, X.L.; Wang, B.; Yang, F.B.; Chen, L.G.; You, J. HOXA11-AS aggravates microglia-induced neuroinflammation after traumatic brain injury. Neural Regen. Res. 2022, 17, 1096. [Google Scholar]
- Zelenchuk, T.A.; Brusés, J.L. In vivo labeling of zebrafish motor neurons using an mnx1 enhancer and Gal4/UAS. Genesis 2011, 49, 546–554. [Google Scholar] [CrossRef] [PubMed]
- El-Bizri, N.; Guignabert, C.; Wang, L.; Cheng, A.; Stankunas, K.; Chang, C.P.; Mishina, Y.; Rabinovitch, M. SM22α-targeted deletion of bone morphogenetic protein receptor 1A in mice impairs cardiac and vascular development, and influences organogenesis. Development 2008, 135, 2981–2991. [Google Scholar] [CrossRef]
- Hayes, G.; Pinto, J.; Sparks, S.N.; Wang, C.; Suri, S.; Bulte, D.P. Vascular smooth muscle cell dysfunction in neurodegeneration. Front. Neurosci. 2022, 16, 1010164. [Google Scholar] [CrossRef]
- Rocha, N.P.; Ribeiro, F.M.; Furr-Stimming, E.; Teixeira, A.L. Neuroimmunology of Huntington’s disease: Revisiting evidence from human studies. Mediat. Inflamm. 2016, 2016, 8653132. [Google Scholar] [CrossRef]
- Goodwin, A.M.; d’Amore, P.A. Wnt signaling in the vasculature. Angiogenesis 2002, 5, 1–9. [Google Scholar] [CrossRef]
- Dejana, E. The role of wnt signaling in physiological and pathological angiogenesis. Circ. Res. 2010, 107, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Uchimura, E.; Ueda, T.; Nomura, T.; Fujita, S.; Matsumoto, K.; Funeriu, D.P.; Miyake, M.; Miyake, J. Identification of twinfilin-2 as a factor involved in neurite outgrowth by RNAi-based screen. Biochem. Biophys. Res. Commun. 2007, 363, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Gil-Mohapel, J.; Simpson, J.M.; Ghilan, M.; Christie, B.R. Neurogenesis in Huntington’s disease: Can studying adult neurogenesis lead to the development of new therapeutic strategies? Brain Res. 2011, 1406, 84–105. [Google Scholar] [CrossRef] [PubMed]
- Al-Dalahmah, O.; Sosunov, A.A.; Shaik, A.; Ofori, K.; Liu, Y.; Vonsattel, J.P.; Adorjan, I.; Menon, V.; Goldman, J.E. Single-nucleus RNA-Seq Identifies Huntington Disease Astrocyte States. Acta Neuropathol. Commun. 2020, 8, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.C. Approaches for establishing the function of regulatory genetic variants involved in disease. Genome Med. 2014, 6, 92. [Google Scholar] [CrossRef]
- Leary, S.C.; Cobine, P.A.; Nishimura, T.; Verdijk, R.M.; de Krijger, R.; de Coo, R.; Tarnopolsky, M.A.; Winge, D.R.; Shoubridge, E.A. COX19 mediates the transduction of a mitochondrial redox signal from SCO1 that regulates ATP7A-mediated cellular copper efflux. Mol. Biol. Cell 2013, 24, 683–691. [Google Scholar] [CrossRef]
- Venkateswarlu, K.; Oatey, P.B.; Tavaré, J.M.; Jackson, T.R.; Cullen, P.J. Identification of centaurin-α1 as a potential in vivo phosphatidylinositol 3, 4, 5-trisphosphate-binding protein that is functionally homologous to the yeast ADP-ribosylation factor (ARF) GTPase-activating protein, Gcs1. Biochem. J. 1999, 340, 359–363. [Google Scholar] [CrossRef]
- Lee, J.H.; Tecedor, L.; Chen, Y.H.; Monteys, A.M.; Sowada, M.J.; Thompson, L.M.; Davidson, B.L. Reinstating aberrant mTORC1 activity in Huntington’s disease mice improves disease phenotypes. Neuron 2015, 85, 303–315. [Google Scholar] [CrossRef]
- Liakopoulos, D.; Doenges, G.; Matuschewski, K.; Jentsch, S. A novel protein modification pathway related to the ubiquitin system. EMBO J. 1998, 17, 2208–2214. [Google Scholar] [CrossRef]
- Chen, Y.; Neve, R.L.; Liu, H. Neddylation dysfunction in Alzheimer’s disease. J. Cell. Mol. Med. 2012, 16, 2583–2591. [Google Scholar] [CrossRef]
- Gizaw, S.T.; Koda, T.; Amano, M.; Kamimura, K.; Ohashi, T.; Hinou, H.; Nishimura, S.I. A comprehensive glycome profiling of Huntington’s disease transgenic mice. Biochim. Et Biophys. Acta (BBA) Gen. Subj. 2015, 1850, 1704–1718. [Google Scholar] [CrossRef]
- Walden, H.; Podgorski, M.S.; Huang, D.T.; Miller, D.W.; Howard, R.J.; Minor, D.L., Jr.; Holton, J.M.; Schulman, B.A. The structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol. Cell 2003, 12, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shen, J.K.; Hornicek, F.J.; Kan, Q.; Duan, Z. The emerging roles and therapeutic potential of cyclin-dependent kinase 11 (CDK11) in human cancer. Oncotarget 2016, 7, 40846. [Google Scholar] [CrossRef]
- Cornelis, S.; Bruynooghe, Y.; Denecker, G.; Van Huffel, S.; Tinton, S.; Beyaert, R. Identification and characterization of a novel cell cycle–regulated internal ribosome entry site. Mol. Cell 2000, 5, 597–605. [Google Scholar] [CrossRef]
- Wong, T.-S.; Li, G.; Li, S.; Gao, W.; Chen, G.; Gan, S.; Zhang, M.; Li, H.; Wu, S.; Du, Y. G protein-coupled receptors in neurodegenerative diseases and psychiatric disorders. Signal Transduct. Target. Ther. 2023, 8, 177. [Google Scholar] [CrossRef] [PubMed]
- Sileo, P.; Simonin, C.; Melnyk, P.; Chartier-Harlin, M.C.; Cotelle, P. Crosstalk between the Hippo Pathway and the Wnt Pathway in Huntington’s Disease and Other Neurodegenerative Disorders. Cells 2022, 11, 3631. [Google Scholar] [CrossRef] [PubMed]
- Kasubuchi, M.; Watanabe, K.; Hirano, K.; Inoue, D.; Li, X.; Terasawa, K.; Konishi, M.; Itoh, N.; Kimura, I. Membrane progesterone receptor β (mPRβ/Paqr8) promotes progesterone-dependent neurite outgrowth in PC12 neuronal cells via non-G protein-coupled receptor (GPCR) signaling. Sci. Rep. 2017, 7, 5168. [Google Scholar] [CrossRef]
- De Carvalho, J.B.; De Morais, G.L.; Vieira, T.C.D.S.; Rabelo, N.C.; Llerena, J.C., Jr.; Gonzalez, S.M.D.C.; De Vasconcelos, A.T.R. miRNA genetic variants alter their secondary structure and expression in patients with RASopathies syndromes. Front. Genet. 2019, 10, 1144. [Google Scholar] [CrossRef]
- Catanesi, M.; d’Angelo, M.; Tupone, M.G.; Benedetti, E.; Giordano, A.; Castelli, V.; Cimini, A. MicroRNAs dysregulation and mitochondrial dysfunction in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 5986. [Google Scholar] [CrossRef]
- Shi, L.; Fisslthaler, B.; Zippel, N.; Frömel, T.; Hu, J.; Elgheznawy, A.; Heide, H.; Popp, R.; Fleming, I. MicroRNA-223 antagonizes angiogenesis by targeting β1 integrin and preventing growth factor signaling in endothelial cells. Circ. Res. 2013, 113, 1320–1330. [Google Scholar] [CrossRef]
- Lee, S.G.; Su, Z.Z.; Emdad, L.; Sarkar, D.; Franke, T.F.; Fisher, P.B. Astrocyte elevated gene-1 activates cell survival pathways through PI3K-Akt signaling. Oncogene 2008, 27, 1114–1121. [Google Scholar] [CrossRef]
- Kimple, A.J.; Soundararajan, M.; Hutsell, S.Q.; Roos, A.K.; Urban, D.J.; Setola, V.; Temple, B.R.; Roth, B.L.; Knapp, S.; Willard, F.S.; et al. Structural determinants of G-protein α subunit selectivity by regulator of G-protein signaling 2 (RGS2). J. Biol. Chem. 2009, 284, 19402–19411. [Google Scholar] [CrossRef]
- Pattingre, S.; De Vries, L.; Bauvy, C.; Chantret, I.; Cluzeaud, F.; Ogier-Denis, E.; Vandewalle, A.; Codogno, P. The G-protein regulator AGS3 controls an early event during macroautophagy in human intestinal HT-29 cells. J. Biol. Chem. 2003, 278, 20995–21002. [Google Scholar] [CrossRef]
- Emdad, L.; Lee, S.G.; Su, Z.Z.; Jeon, H.Y.; Boukerche, H.; Sarkar, D.; Fisher, P.B. Astrocyte elevated gene-1 (AEG-1) functions as an oncogene and regulates angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21300–21305. [Google Scholar] [CrossRef] [PubMed]
- Emdad, L.; Sarkar, D.; Su, Z.Z.; Randolph, A.; Boukerche, H.; Valerie, K.; Fisher, P.B. Activation of the nuclear factor κB pathway by astrocyte elevated gene-1: Implications for tumor progression and metastasis. Cancer Res. 2006, 66, 1509–1516. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, S.K.; Kegelman, T.P.; Das, S.K.; Azab, B.; Su, Z.Z.; Lee, S.G.; Sarkar, D.; Fisher, P.B. Astrocyte elevated gene-1 induces protective autophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 22243–22248. [Google Scholar] [CrossRef]
- Wang, J.; Gao, Q.S.; Wang, Y.; Lafyatis, R.; Stamm, S.; Andreadis, A. Tau exon 10, whose missplicing causes frontotemporal dementia, is regulated by an intricate interplay of cis elements and trans factors. J. Neurochem. 2004, 88, 1078–1090. [Google Scholar] [CrossRef]
- Lin, J.C.; Tarn, W.Y. RBM4 down-regulates PTB and antagonizes its activity in muscle cell–specific alternative splicing. J. Cell Biol. 2011, 193, 509–520. [Google Scholar] [CrossRef]
- Drouin-Ouellet, J.; Sawiak, S.J.; Cisbani, G.; Lagacé, M.; Kuan, W.-L.; Saint-Pierre, M.; Dury, R.J.; Alata, W.; St-Amour, I.; Mason, S.L.; et al. Cerebrovascular and blood–brain barrier impairments in Huntington’s disease: Potential implications for its pathophysiology. Ann. Neurol. 2015, 78, 160–177. [Google Scholar] [CrossRef]
- Kamphaus, G.D.; Colorado, P.C.; Panka, D.J.; Hopfer, H.; Ramchandran, R.; Torre, A.; Maeshima, Y.; Mier, J.W.; Sukhatme, V.P.; Kalluri, R. Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J. Biol. Chem. 2000, 275, 1209–1215. [Google Scholar] [CrossRef]
- Bi, J.; Wang, W.; Zhang, M.; Zhang, B.; Liu, M.; Su, G.; Chen, F.; Chen, B.; Shi, T.; Zheng, Y.; et al. KLF4 inhibits early neural differentiation of ESCs by coordinating specific 3D chromatin structure. Nucleic Acids Res. 2022, 50, 12235–12250. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Liu, M.; Niu, W.; Zhang, C.L. Dysregulation of Kruppel-like factor 4 during brain development leads to hydrocephalus in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 21117–21121. [Google Scholar] [CrossRef] [PubMed]
- Semon, J.A.; Nagy, L.H.; Llamas, C.B.; Tucker, H.A.; Lee, R.H.; Prockop, D.J. Integrin expression and integrin-mediated adhesion in vitro of human multipotent stromal cells (MSCs) to endothelial cells from various blood vessels. Cell Tissue Res. 2010, 341, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Pace, J.M.; Wiese, M.; Drenguis, A.S.; Kuznetsova, N.; Leikin, S.; Schwarze, U.; Chen, D.; Mooney, S.H.; Unger, S.; Byers, P.H. Defective C-propeptides of the proα2 (I) chain of type I procollagen impede molecular assembly and result in osteogenesis imperfecta. J. Biol. Chem. 2008, 283, 16061–16067. [Google Scholar] [CrossRef]
- Greene, C.S.; Krishnan, A.; Wong, A.K.; Ricciotti, E.; Zelaya, R.A.; Himmelstein, D.S.; Zhang, R.; Hartmann, B.M.; Zaslavsky, E.; Sealfon, S.C.; et al. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 2015, 47, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Baloni, P.; Dinalankara, W.; Earls, J.C.; Knijnenburg, T.A.; Geman, D.; Marchionni, L.; Price, N.D. Identifying Personalized Metabolic Signatures in Breast Cancer. Metabolites 2021, 11, 20. [Google Scholar] [CrossRef]
- Punzi, G.; Porcelli, V.; Ruggiu, M.; Hossain, F.; Menga, A.; Scarcia, P.; Castegna, A.; Gorgoglione, R.; Pierri, C.L.; Laera, L.; et al. SLC25A10 biallelic mutations in intractable epileptic encephalopathy with complex I deficiency. Hum. Mol. Genet. 2018, 27, 499–504. [Google Scholar] [CrossRef]
- Chen, S.; Wang, X.; Ye, X.; Jin, J. Gemcitabine and Doxorubicin Combination Enhance the Cytotoxic Effect to Pancreatic Cancer Cells BxPC3 and PANC1 through UMP/CMP Kinase 1. J. Biosci. Med. 2017, 5, 64–74. [Google Scholar] [CrossRef]
- Sagawa, M.; Ohguchi, H.; Harada, T.; Samur, M.K.; Tai, Y.T.; Munshi, N.C.; Anderson, K.C. Ribonucleotide Reductase Catalytic Subunit M1 (RRM1) as a Novel Therapeutic Target in Multiple MyelomaTargeting RRM1 as a Novel Treatment for Multiple Myeloma. Clin. Cancer Res. 2017, 23, 5225–5237. [Google Scholar] [CrossRef]
- Akman, H.O.; Sheiko, T.; Tay, S.K.; Finegold, M.J.; DiMauro, S.; Craigen, W.J. Generation of a novel mouse model that recapitulates early and adult onset glycogenosis type IV. Hum. Mol. Genet. 2011, 20, 4430–4439. [Google Scholar] [CrossRef]
- Wilson, L.H.; Cho, J.; Estrella, A.; Smyth, J.A.; Wu, R.; Chengsupanimit, T.; Brown, L.M.; Weinstein, D.A.; Lee, Y.M. Liver glycogen phosphorylase deficiency leads to profibrogenic phenotype in a murine model of glycogen storage disease type VI. Hepatol. Commun. 2019, 3, 1544–1555. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.; Bodhicharla, R.; Ståhlman, M.; Svensk, E.; Busayavalasa, K.; Palmgren, H.; Ruhanen, H.; Boren, J.; Pilon, M. Evolutionarily conserved long-chain Acyl-CoA synthetases regulate membrane composition and fluidity. Elife 2019, 8, e47733. [Google Scholar] [CrossRef]
- Han, J.; Deng, X.; Sun, R.; Luo, M.; Liang, M.; Gu, B.; Zhang, T.; Peng, Z.; Lu, Y.; Tian, C.; et al. GPI is a prognostic biomarker and correlates with immune infiltrates in lung adenocarcinoma. Front. Oncol. 2021, 11, 752642. [Google Scholar] [CrossRef] [PubMed]
- Salleron, L.; Magistrelli, G.; Mary, C.; Fischer, N.; Bairoch, A.; Lane, L. DERA is the human deoxyribose phosphate aldolase and is involved in stress response. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2014, 1843, 2913–2925. [Google Scholar] [CrossRef]
- Moriyama, T.; Tanaka, S.; Nakayama, Y.; Fukumoto, M.; Tsujimura, K.; Yamada, K.; Bamba, T.; Yoneda, Y.; Fukusaki, E.; Oka, M. Two isoforms of TALDO1 generated by alternative translational initiation show differential nucleocytoplasmic distribution to regulate the global metabolic network. Sci. Rep. 2016, 6, 34648. [Google Scholar] [CrossRef]
- Liu, Y.; Yuan, Z.; Song, C. Methylcrotonoyl-CoA carboxylase 2 overexpression predicts an unfavorable prognosis and promotes cell proliferation in breast cancer. Biomark. Med. 2019, 13, 427–436. [Google Scholar] [CrossRef]
- Batsios, G.; Taglang, C.; Cao, P.; Gillespie, A.M.; Najac, C.; Subramani, E.; Wilson, D.M.; Flavell, R.R.; Larson, P.E.Z.; Ronen, S.M.; et al. Imaging 6-Phosphogluconolactonase Activity in Brain Tumors In Vivo Using Hyperpolarized δ-[1-13C] gluconolactone. Front. Oncol. 2021, 11, 589570. [Google Scholar] [CrossRef]
- Chen, W.; Guo, X.; Jin, Z.; Li, R.; Shen, L.; Li, W.; Cai, W.; Zhang, G. Transcriptional alterations of genes related to fertility decline in male rats induced by chronic sleep restriction. Syst. Biol. Reprod. Med. 2020, 66, 99–111. [Google Scholar] [CrossRef]
- Kishk, A.; Pacheco, M.P.; Sauter, T. DCcov: Repositioning of drugs and drug combinations for SARS-CoV-2 infected lung through constraint-based modeling. Iscience 2021, 24, 103331. [Google Scholar] [CrossRef]
- Khan, N.; Shah, P.P.; Ban, D.; Trigo-Mouriño, P.; Carneiro, M.G.; DeLeeuw, L.; Dean, W.L.; Trent, J.O.; Beverly, L.J.; Konrad, M.; et al. Solution structure and functional investigation of human guanylate kinase reveals allosteric networking and a crucial role for the enzyme in cancer. J. Biol. Chem. 2019, 294, 11920–11933. [Google Scholar] [CrossRef]
- Li, H.; Ericsson, M.; Rabasha, B.; Budnik, B.; Chan, S.H.; Freinkman, E.; Lewis, C.A.; Doench, J.G.; Wagner, B.K.; Garraway, L.A.; et al. 6-Phosphogluconate dehydrogenase links cytosolic carbohydrate metabolism to protein secretion via modulation of glutathione levels. Cell Chem. Biol. 2019, 26, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Yu, X.; Liang, Y.; Zhu, Y.; He, Y.; Liao, L.; Wang, D.; Yang, Y.; Yin, X.; Li, A.; et al. Targeting PFKL with penfluridol inhibits glycolysis and suppresses esophageal cancer tumorigenesis in an AMPK/FOXO3a/BIM-dependent manner. Acta Pharm. Sin. B 2022, 12, 1271–1287. [Google Scholar] [CrossRef] [PubMed]
- Budry, L.; Bouyakdan, K.; Tobin, S.; Rodaros, D.; Marcher, A.-B.; Mandrup, S.; Fulton, S.; Alquier, T. DBI/ACBP loss-of-function does not affect anxiety-like behaviour but reduces anxiolytic responses to diazepam in mice. Behav. Brain Res. 2016, 313, 201–207. [Google Scholar] [CrossRef] [PubMed]
- De Giorgi, M.; Jarrett, K.E.; Burton, J.C.; Doerfler, A.M.; Hurley, A.; Li, A.; Hsu, R.H.; Furgurson, M.; Patel, K.R.; Han, J.; et al. Depletion of essential isoprenoids and ER stress induction following acute liver-specific deletion of HMG-CoA reductase. J. Lipid Res. 2020, 61, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Nong, X.; Zhang, C.; Wang, J.; Ding, P.; Ji, G.; Wu, T. The mechanism of branched-chain amino acid transferases in different diseases: Research progress and future prospects. Front. Oncol. 2022, 12, 988290. [Google Scholar] [CrossRef]
- Schulz, J.G.; Laranjeira, A.; Van Huffel, L.; Gärtner, A.; Vilain, S.; Bastianen, J.; Van Veldhoven, P.P.; Dotti, C.G. Glial β-oxidation regulates Drosophila energy metabolism. Sci. Rep. 2015, 5, 7805. [Google Scholar] [CrossRef]
- Dona, M.; Waaijers, S.; Richter, S.; Eisenhofer, G.; Korving, J.; Kamel, S.M.; Bakkers, J.; Rapizzi, E.; Rodenburg, R.J.; Zethof, J.; et al. Loss of sdhb in zebrafish larvae recapitulates human paraganglioma characteristics. Endocr. Relat. Cancer 2021, 28, 65–77. [Google Scholar] [CrossRef]
- Jiang, W.; Liu, C.; Deng, M.; Wang, F.; Ren, X.; Fan, Y.; Du, J.; Wang, Y. H2S promotes developmental brain angiogenesis via the NOS/NO pathway in zebrafish. Stroke Vasc. Neurol. 2021, 6, 244–251. [Google Scholar] [CrossRef]
- Nyman, L.R.; Cox, K.B.; Hoppel, C.L.; Kerner, J.; Barnoski, B.L.; Hamm, D.A.; Tian, L.; Schoeb, T.R.; Wood, P.A. Homozygous carnitine palmitoyltransferase 1a (liver isoform) deficiency is lethal in the mouse. Mol. Genet. Metab. 2005, 86, 179–187. [Google Scholar] [CrossRef]
- Moore, B.A.; Leonard, B.C.; Sebbag, L.; Edwards, S.G.; Cooper, A.; Imai, D.M.; Straiton, E.; Santos, L.; Reilly, C.; Griffey, S.M.; et al. Identification of genes required for eye development by high-throughput screening of mouse knockouts. Commun. Biol. 2018, 1, 236. [Google Scholar] [CrossRef]
- Hallmann, K.; Kudin, A.P.; Zsurka, G.; Kornblum, C.; Reimann, J.; Stüve, B.; Waltz, S.; Hattingen, E.; Thiele, H.; Nürnberg, P.; et al. Loss of the smallest subunit of cytochrome c oxidase, COX8A, causes Leigh-like syndrome and epilepsy. Brain 2016, 139, 338–345. [Google Scholar] [CrossRef]
- Yalgin, C.; Rovenko, B.; Andjelković, A.; Neefjes, M.; Oymak, B.; Dufour, E.; Hietakangas, V.; Jacobs, H.T. Effects on Dopaminergic Neurons Are Secondary in COX-Deficient Locomotor Dysfunction in Drosophila. Iscience 2020, 23, 101362. [Google Scholar] [CrossRef] [PubMed]
- Kriebel, M.; Ebel, J.; Battke, F.; Griesbach, S.; Volkmer, H. Interference with complex IV as a model of age-related decline in synaptic connectivity. Front. Mol. Neurosci. 2020, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Indrieri, A.; van Rahden, V.A.; Tiranti, V.; Morleo, M.; Iaconis, D.; Tammaro, R.; D’amato, I.; Conte, I.; Maystadt, I.; Demuth, S.; et al. Mutations in COX7B cause microphthalmia with linear skin lesions, an unconventional mitochondrial disease. Am. J. Hum. Genet. 2012, 91, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Kemppainen, K.K.; Rinne, J.; Sriram, A.; Lakanmaa, M.; Zeb, A.; Tuomela, T.; Popplestone, A.; Singh, S.; Sanz, A.; Rustin, P.; et al. Expression of alternative oxidase in Drosophila ameliorates diverse phenotypes due to cytochrome oxidase deficiency. Hum. Mol. Genet. 2014, 23, 2078–2093. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.H.; Oh, Y.; Lee, H.; Lee, W.; Chang, J.W.; Pyo, H.K.; Jung, Y.K. Identification of glucose-6-phosphate transporter as a key regulator functioning at the autophagy initiation step. FEBS Lett. 2015, 589, 2100–2109. [Google Scholar] [CrossRef]
- Keenan, B.T.; Galante, R.J.; Lian, J.; Zhang, L.; Guo, X.; Veatch, O.J.; Chesler, E.J.; O’brien, W.T.; Svenson, K.L.; Churchill, G.A.; et al. The dihydropyrimidine dehydrogenase gene contributes to heritable differences in sleep in mice. Curr. Biol. 2021, 31, 5238–5248. [Google Scholar] [CrossRef]
- Arikketh, D.; Nelson, R.; Vance, J.E. Defining the importance of phosphatidylserine synthase-1 (PSS1): Unexpected viability of PSS1-deficient mice. J. Biol. Chem. 2008, 283, 12888–12897. [Google Scholar] [CrossRef]
- Jiang, P.; Gan, M.; Lin, W.L.; Yen, S.H.C. Nutrient deprivation induces α-synuclein aggregation through endoplasmic reticulum stress response and SREBP2 pathway. Front. Aging Neurosci. 2014, 6, 268. [Google Scholar] [CrossRef]
- Wan, F.; Anderson, D.E.; Barnitz, R.A.; Snow, A.; Bidere, N.; Zheng, L.; Hegde, V.; Lam, L.T.; Staudt, L.M.; Levens, D.; et al. Ribosomal protein S3: A KH domain subunit in NF-κB complexes that mediates selective gene regulation. Cell 2007, 131, 927–939. [Google Scholar] [CrossRef]
- Lamb, L.E.; Zarif, J.C.; Miranti, C.K. The androgen receptor induces integrin α6β1 to promote prostate tumor cell survival via NF-κB and Bcl-xL Independently of PI3K signaling. Cancer Res. 2011, 71, 2739–2749. [Google Scholar] [CrossRef]
- Martín-Villar, E.; Fernández-Muñoz, B.; Parsons, M.; Yurrita, M.M.; Megías, D.; Pérez-Gómez, E.; Jones, G.E.; Quintanilla, M. Podoplanin associates with CD44 to promote directional cell migration. Mol. Biol. Cell 2010, 21, 4387–4399. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Shi, Y.; Mulligan, P.; Gay, F.; Landry, J.; Liu, H.; Lu, J.; Qi, H.H.; Wang, W.; Nickoloff, J.A.; et al. A YY1–INO80 complex regulates genomic stability through homologous recombination–based repair. Nat. Struct. Mol. Biol. 2007, 14, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Short, J.D.; Pfarr, C.M. Translational regulation of the JunD messenger RNA. J. Biol. Chem. 2002, 277, 32697–32705. [Google Scholar] [CrossRef]
- Albagli, O.; Lantoine, D.; Quief, S.; Quignon, F.; Englert, C.; Kerckaert, J.-P.; Montarras, D.; Pinset, C.; Lindon, C. Overexpressed BCL6 (LAZ3) oncoprotein triggers apoptosis, delays S phase progression and associates with replication foci. Oncogene 1999, 18, 5063–5075. [Google Scholar] [CrossRef] [PubMed]
- Mittal, V.; Hernandez, N. Role for the amino-terminal region of human TBP in U6 snRNA transcription. Science 1997, 275, 1136–1140. [Google Scholar] [CrossRef]
- Nakata, Y.; Brignier, A.C.; Jin, S.; Shen, Y.; Rudnick, S.I.; Sugita, M.; Gewirtz, A.M. c-Myb, Menin, GATA-3, and MLL form a dynamic transcription complex that plays a pivotal role in human T helper type 2 cell development. Blood J. Am. Soc. Hematol. 2010, 116, 1280–1290. [Google Scholar] [CrossRef]
- Chang, C.C.; Zhang, Q.Y.; Liu, Z.; Clynes, R.A.; Suciu-Foca, N.; Vlad, G. Downregulation of inflammatory microRNAs by Ig-like transcript 3 is essential for the differentiation of human CD8+ T suppressor cells. J. Immunol. 2012, 188, 3042–3052. [Google Scholar] [CrossRef]
- Dirkx, E.; Gladka, M.M.; Philippen, L.E.; Armand, A.S.; Kinet, V.; Leptidis, S.; El Azzouzi, H.; Salic, K.; Bourajjaj, M.; da Silva, G.J.; et al. Nfat and miR-25 cooperate to reactivate the transcription factor Hand2 in heart failure. Nat. Cell Biol. 2013, 15, 1282–1293. [Google Scholar] [CrossRef]
- Armstrong, R.J.; Watts, C.; Svendsen, C.N.; Dunnett, S.B.; Rosser, A.E. Survival, neuronal differentiation, and fiber outgrowth of propagated human neural precursor grafts in an animal model of Huntington’s disease. Cell Transplant. 2000, 9, 55–64. [Google Scholar] [CrossRef]
- Francelle, L.; Lotz, C.; Outeiro, T.; Brouillet, E.; Merienne, K. Contribution of neuroepigenetics to Huntington’s disease. Front. Hum. Neurosci. 2017, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Dowie, M.J.; Scotter, E.L.; Molinari, E.; Glass, M. The therapeutic potential of G-protein coupled receptors in Huntington’s disease. Pharmacol. Ther. 2010, 128, 305–323. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.A. Vesicular dysfunction and pathways to neurodegeneration. Essays Biochem. 2021, 65, 941–948. [Google Scholar] [PubMed]
- Breza, M.; Emmanouilidou, E.; Leandrou, E.; Kartanou, C.; Bougea, A.; Panas, M.; Stefanis, L.; Karadima, G.; Vekrellis, K.; Koutsis, G. Elevated serum α-synuclein levels in huntington’s disease patients. Neuroscience 2020, 431, 34–39. [Google Scholar] [CrossRef]
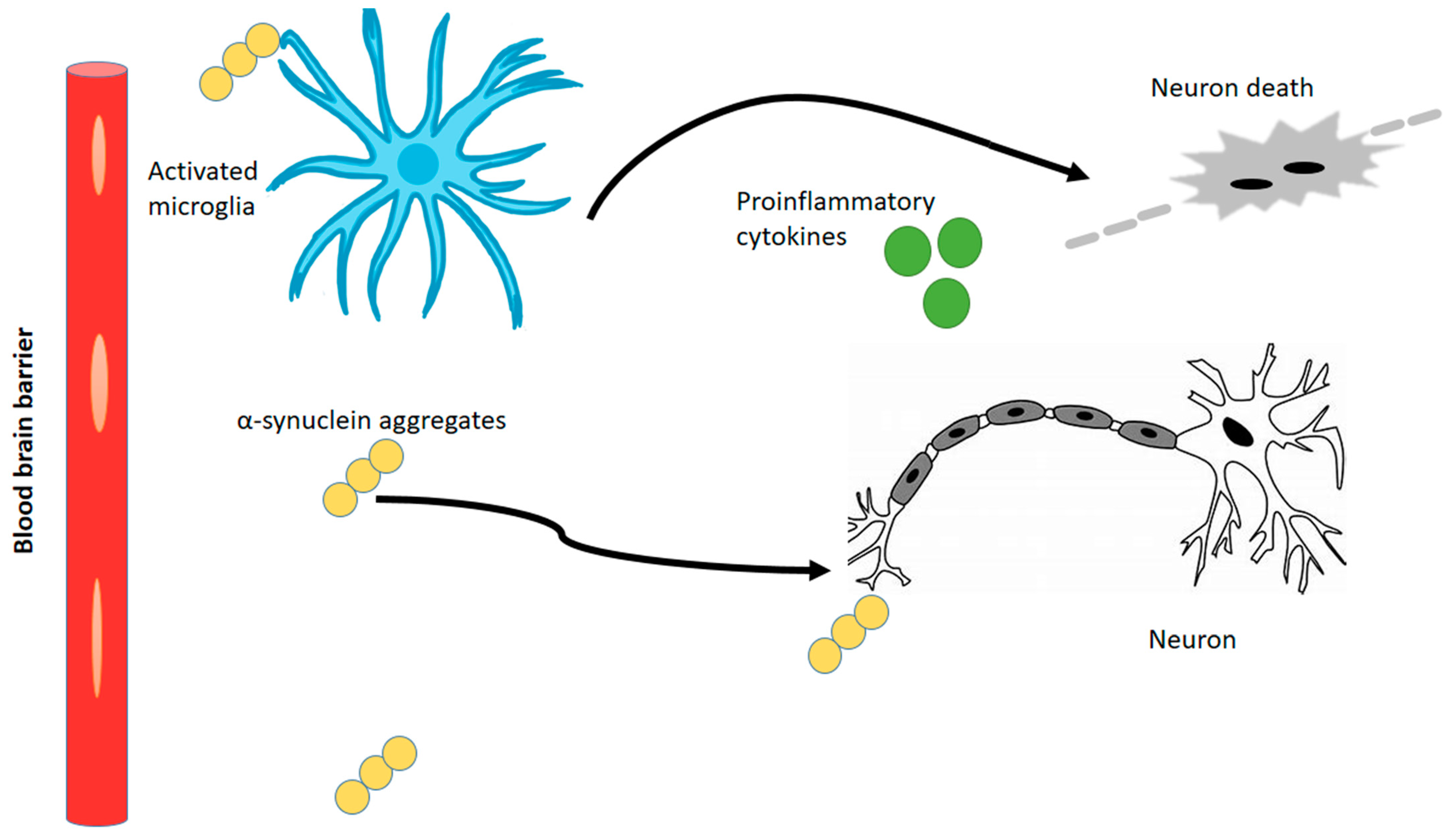
- Jia, Q.; Li, S.; Li, X.J.; Yin, P. Neuroinflammation in Huntington’s disease: From animal models to clinical therapeutics. Front. Immunol. 2022, 13, 1088124. [Google Scholar] [CrossRef]
- Lois, C.; González, I.; Izquierdo-García, D.; Zürcher, N.R.; Wilkens, P.; Loggia, M.L.; Hooker, J.M.; Rosas, H.D. Neuroinflammation in Huntington’s disease: New insights with 11C-PBR28 PET/MRI. ACS Chem. Neurosci. 2018, 9, 2563–2571. [Google Scholar] [CrossRef]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. The role of microglia and astrocytes in Huntington’s disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease—A double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Procaccini, C.; Santopaolo, M.; Faicchia, D.; Colamatteo, A.; Formisano, L.; de Candia, P.; Galgani, M.; De Rosa, V.; Matarese, G. Role of metabolism in neurodegenerative disorders. Metabolism 2016, 65, 1376–1390. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr | REF/ALT | Gene (Expression) | Variant | Transcripts | Variant Novel/Reported | QTL Category | Transcription Factor Affected | Transcription Factor Expression |
|---|---|---|---|---|---|---|---|---|
| Chr7 | G/A | COX19 (UP) | rs10282027 | ENST00000457254.5 | Novel | eQTL | ADAP1 | DOWN |
| Chr3 | T/C | UBA3 (UP) | rs3853156 | ENST00000415609.6 | AD, PD, AMD | eQTL | EOGT | DOWN |
| Chr1 | T/C | CDK11A (DOWN) | rs1137005 | ENST00000356200.7 | NOVEL | eQTL, sQTL | GNB1 | DOWN |
| Chr6 | T/A | PAQR8 (DOWN) | rs78305768 | ENST00000512121.1 | AD, AMD | eQTL | IRF1 | DOWN |
| Chr16 | C/T | C16orf95 (UP) | rs12148919 | ENST00000562840.1 | AD, AMD | eQTL | ZCCHC14 | DOWN |
| Chr1 | C/T | CDK11A (DOWN) | rs61777471 | ENST00000356200.7 | Novel | eQTL, sQTL | PAX5 | DOWN |
| Chr1 | T/C | CDK11A (DOWN) | rs74378830 | ENST00000356200.7 | Novel | mQTL | ZFX | DOWN |
| Chr11 | T/G | STARD10 (DOWN) | rs72964856 | ENST00000537351.5 | AD, AMD | eQTL, mQTL, sQTL | ARAP1 | DOWN |
| Chr10 | C/T | KIAA1217 (UP) | rs41279872 | ENST00000376462.5 | AMD, AD | eQTL | SOX5 | UP |
| Chr18 | C/A | CABLES1 (UP) | rs748717 | ENST00000400473.6 | PD, AD, AMD | eQTL | TCF7L2 | DOWN |
| Variant Gene/DEG (Expression) | miRNA | miRNA Expression | Targets | Target Expression | The Function of Target Genes |
|---|---|---|---|---|---|
| HOXA10 (DOWN), HOXD9 (DOWN) | Mir-29-3p | UP | COL4A2, KLF4, ITGB1, COL1A2 | Down | Vasculature development |
| KLC1 (DOWN) | miR-34a-5p | UP | ERBB2, SRC, GAS1, PDGFR | Down | Response to growth factor |
| SEMA6A (DOWN) | miR-124 | UP | ITGB1, PTBP1, MTDH, GNAI3, KLF6 | DOWN | Kinase signaling, angiogenesis |
| HOXA9 (DOWN), HOXA7 (DOWN) | miR-196a-5p | UP | RANBP9, NOTCH2, SRRT, TRAP1 | DOWN | ERK1/2 cascade, regulation of gene expression |
| PRKCI (UP), DLC1 (UP), TRIM37 (UP) | miR-146a-5p | DOWN | SOX2, ERBB4, BCLAF1, RAC1 | UP | Positive regulation of MAPK cascade, cellular component biogenesis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sneha, N.P.; Dharshini, S.A.P.; Taguchi, Y.-h.; Gromiha, M.M. Investigating Neuron Degeneration in Huntington’s Disease Using RNA-Seq Based Transcriptome Study. Genes 2023, 14, 1801. https://doi.org/10.3390/genes14091801
Sneha NP, Dharshini SAP, Taguchi Y-h, Gromiha MM. Investigating Neuron Degeneration in Huntington’s Disease Using RNA-Seq Based Transcriptome Study. Genes. 2023; 14(9):1801. https://doi.org/10.3390/genes14091801
Chicago/Turabian StyleSneha, Nela Pragathi, S. Akila Parvathy Dharshini, Y.-h. Taguchi, and M. Michael Gromiha. 2023. "Investigating Neuron Degeneration in Huntington’s Disease Using RNA-Seq Based Transcriptome Study" Genes 14, no. 9: 1801. https://doi.org/10.3390/genes14091801