
Clinical and Genetic Characteristics of a Patient with Cystic Fibrosis with a Complex Allele [E217G;G509D] and Functional Evaluation of the CFTR Channel
 , ,
, ,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
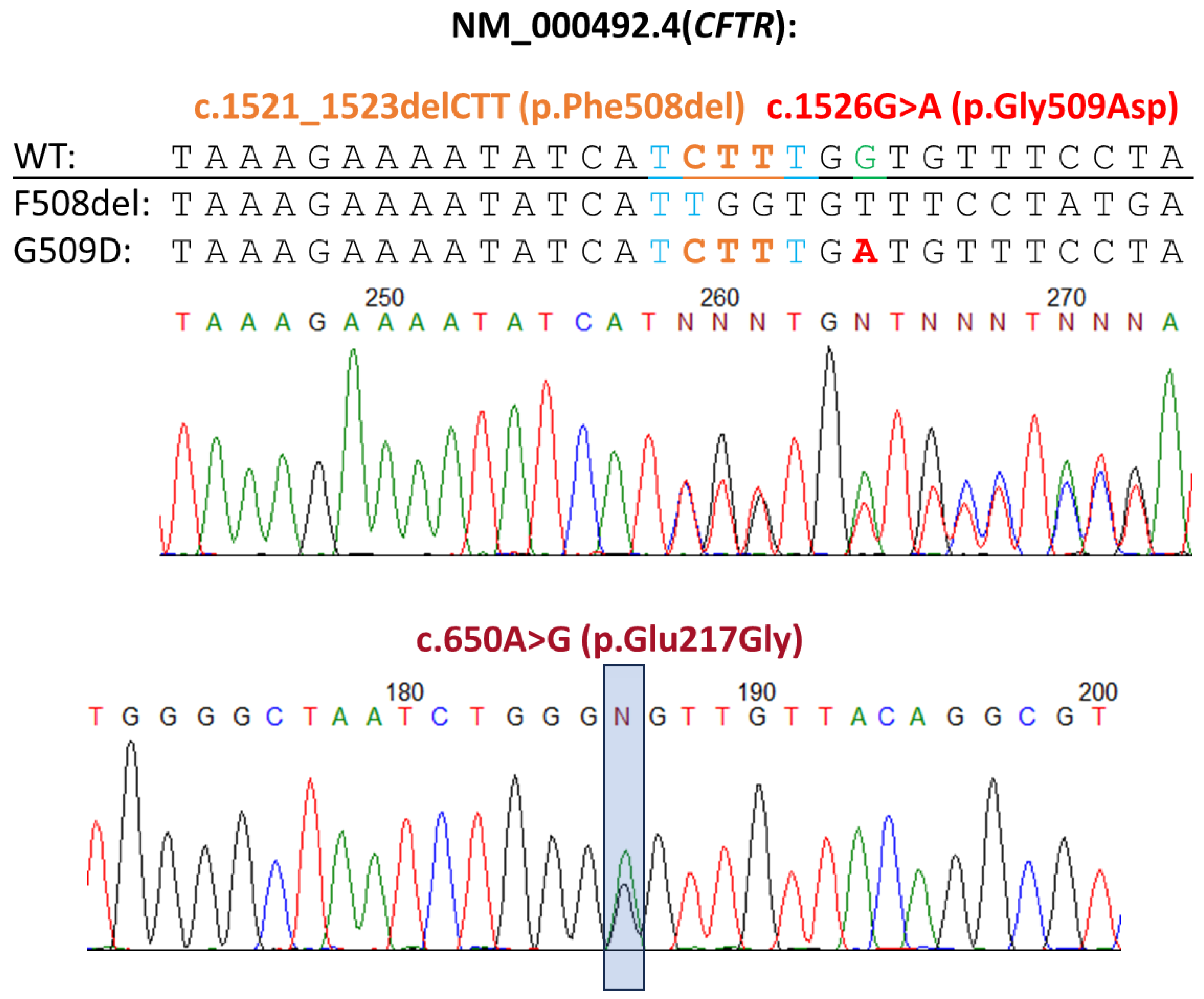
2.1. Genetic Diagnostics
2.2. Intestinal Current Measurements (ICM)
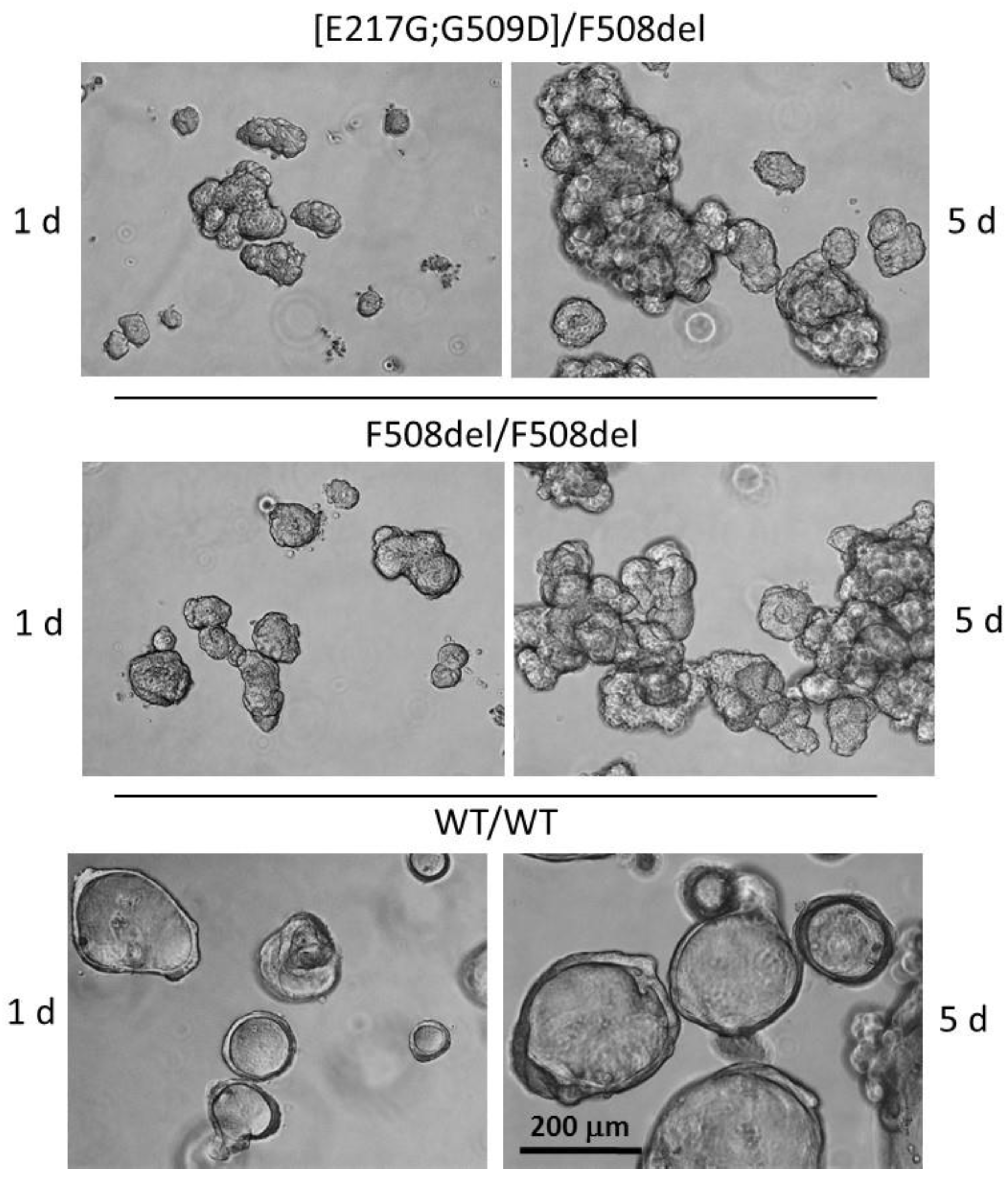
2.3. Human Intestinal Organoids Culture
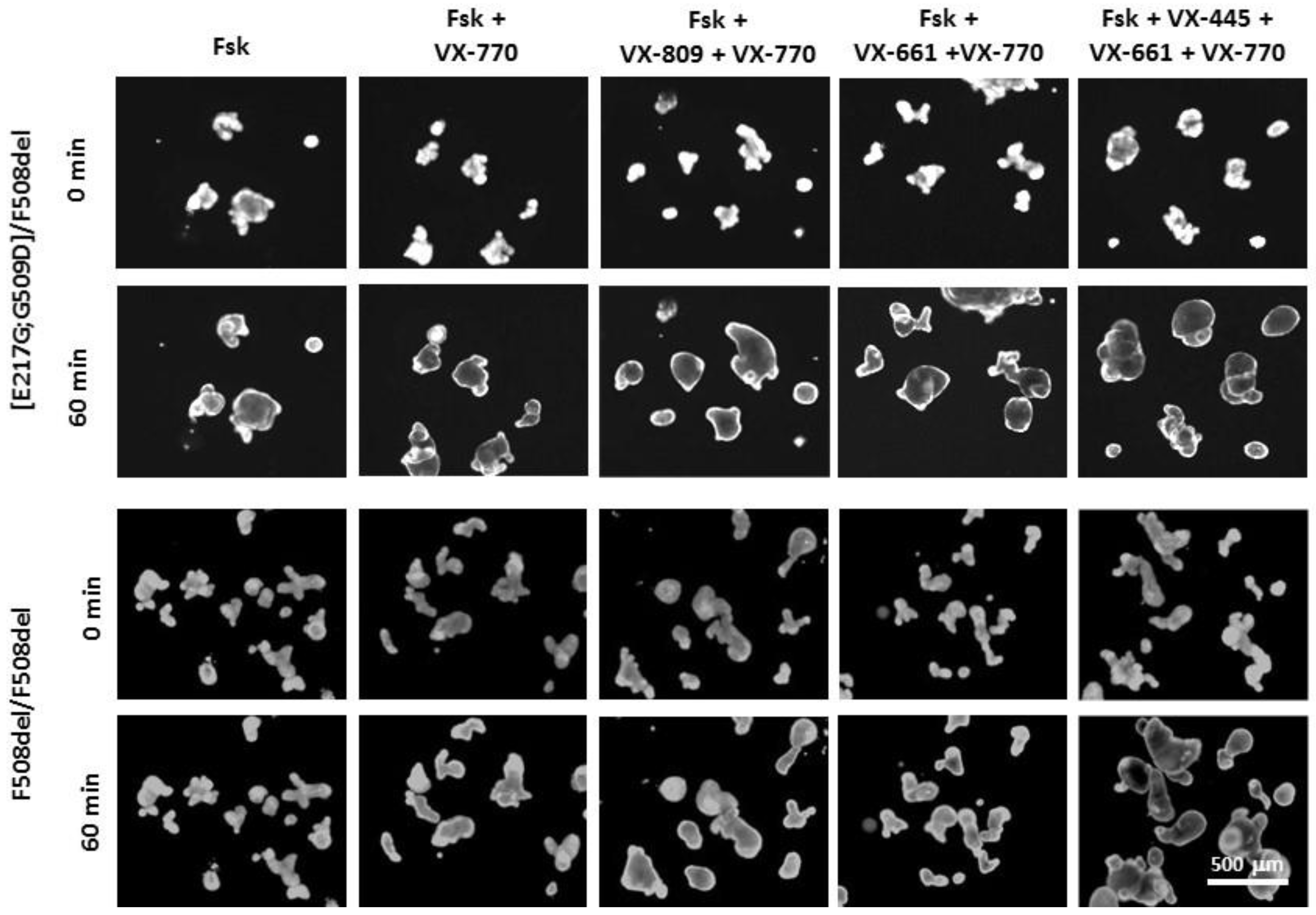
2.4. Forskolin-Induced Swelling (FIS) Assay
3. Results
3.1. Description of the Clinical Picture
3.2. Evaluation of the Functional Activity of the CFTR Channel by the ICM Method
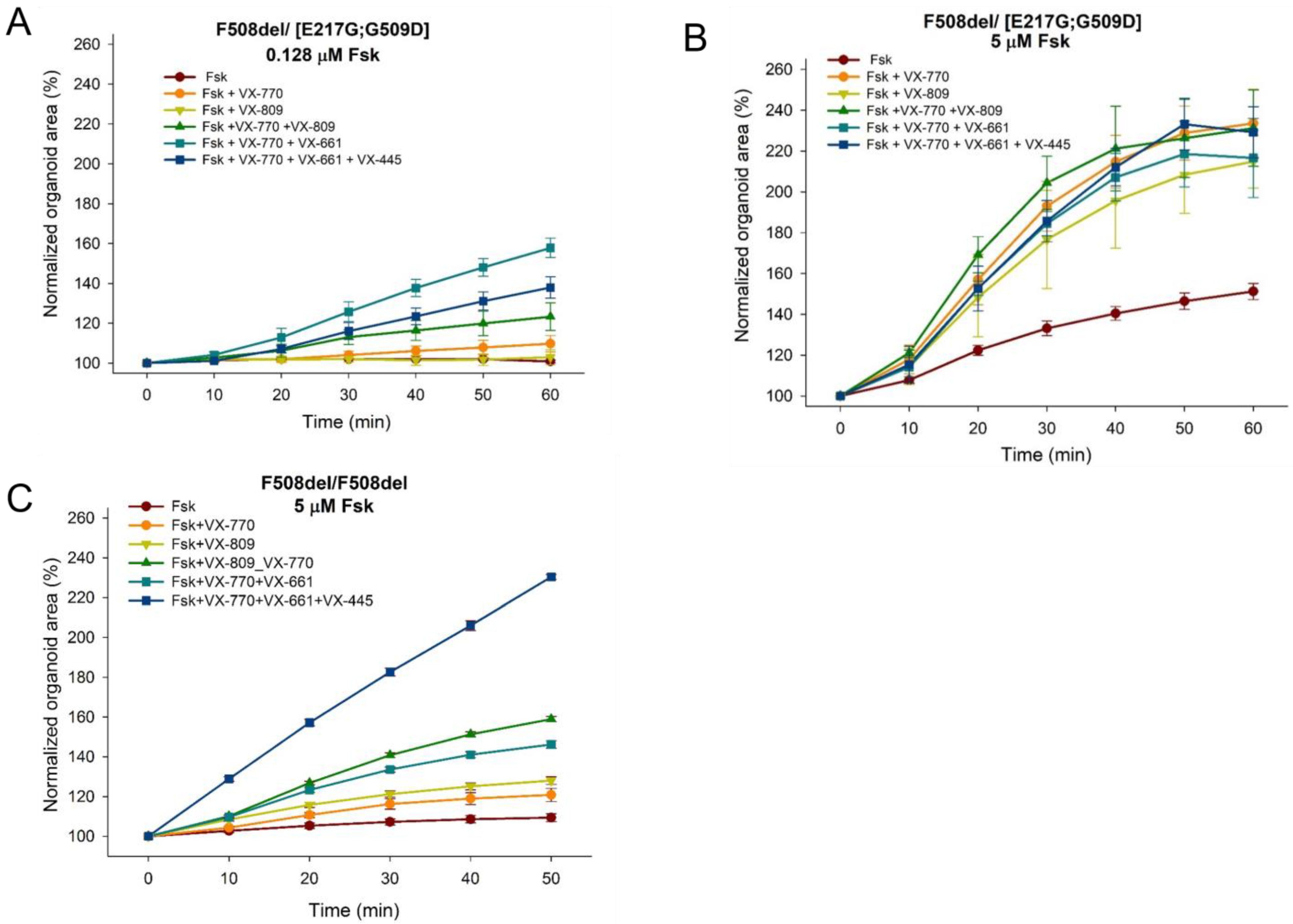
3.3. Evaluation of the Effect of CFTR Modulators on the Restoration of the CFTR Channel Function on the Model of Intestinal Organoids
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bareil, C.; Bergougnoux, A. CFTR gene variants, epidemiology and molecular pathology. Arch. Pediatr. 2020, 27, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Kashirskaya, N.Y.; Kapranov, N.I.; Kondratieva, E.I. Monograph Cystic Fibrosis: 2nd Edition Revised and Supplemented; Medpraktika: Moscow, Russia, 2021; p. 680. [Google Scholar]
- Castellani, C.; Duff, A.J.A.; Bell, S.C.; Heijerman, H.G.M.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; Southern, K.W.; Barben, J.; Flume, P.A.; et al. ECFS best practice guidelines: The 2018 revision. J. Cyst. Fibros. 2018, 17, 153–178. [Google Scholar] [CrossRef] [PubMed]
- Kondratyeva, E.I.; Krasovsky, S.A.; Starinova, M.A.; Voronkova, A.Y.; Amelina, E.L.; Kashirskaya, N.Y.; Avdeeva, S.N.; Kutsev, S.I. Russian Federation Cystic Fibrosis Patient Registry; Medpraktika-M: Moscow, Russia, 2020. [Google Scholar]
- Krasovsky, S.A.; Starinova, M.A.; Voronkova, A.Y.; Amelina, E.L.; Kashirskaya, N.Y.; Kondratyeva, E.I.; Avdeev, S.N.; Kutsev, S.I. Russian Federation Cystic Fibrosis Patient Registry; MEDPRACTICA-M: Moscow, Russia, 2021; p. 95. [Google Scholar]
- Kondratyeva, E.I.; Melyanovskaya, Y.L.; Efremova, A.S.; Bulatenko, N.V.; Bukharova, T.B.; Goldstein, D.V.; Zodbinova, A.E.; Nikonova, V.S.; Zhekaite, E.K.; Kashirskaya, N.Y.; et al. Experience in the application of methods for assessing the functionality of the CFTR anion channel in patients with an established and suspected diagnosis of cystic fibrosis. Sib. Med. Rev. 2019, 2, 60–69. [Google Scholar] [CrossRef]
- Clinical Guidelines for Cystic Fibrosis 2021. Available online: https://cr.minzdrav.gov.ru/recomend/372_2 (accessed on 25 July 2023).
- Kondratyeva, E.I.; Kashirskaya, N.Y.; Kapranov, N.I. National Consensus “Cystic Fibrosis: Definition, Diagnostic Criteria, Therapy”; LLC BORGHES Company: Moscow, Russia, 2016; 205p. [Google Scholar]
- Derichs, N.; Sanz, J.; Von Kanel, T.; Stolpe, C.; Zapf, A.; Tümmler, B.; Gallati, S.; Ballmann, M. Intestinal current measurement for diagnostic classification of patients with questionable cystic fibrosis: Validation and reference data. Thorax 2010, 65, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Melyanovskaya, Y.L.; Kondratyeva, E.I.; Kuzev, S.I. Determination of reference values for the method of intestinal current measurement in the Russian Federation. Med. News North Cauc. 2020, 15, 162–166. [Google Scholar] [CrossRef]
- Dekkers, J.F.; van der Ent, C.K.; Beekman, J.M. Novel opportunities for CFTR-targeting drug development using organoids. Rare Dis. 2013, 1, e27112. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Berkers, G.; Kruisselbrink, E.; Vonk, A.; De Jonge, H.R.; Janssens, H.M.; Bronsveld, I.; Van De Graaf, E.A.; Nieuwenhuis, E.E.S.; Houwen, R.H.J.; et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 2016, 8, 344. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; Vonk, A.M.; Statia, M.; Su, J.; Vries, R.R.G.; Beekman, J.M.; and Clevers, H. Forskolin-induced swelling in intestinal organoids: An in vitro assay for assessing drug response in cystic fibrosis patients. J. Vis. Exp. 2017, 120, e55159. [Google Scholar]
- Vonk, A.M.; van Mourik, P.; Ramalho, A.S.; Silva, I.A.; Statia, M.; Kruisselbrink, E.; Suen, S.W.; Dekkers, J.F.; Vleggaar, F.P.; Houwen, R.H.; et al. Protocol for Application, Standardization and Validation of the Forskolin-Induced Swelling Assay in Cystic Fibrosis Human Colon Organoids. STAR Protoc. 2020, 1, 100019. [Google Scholar] [CrossRef]
- Kondratyeva, E.; Bulatenko, N.; Melyanovskaya, Y.; Efremova, A.; Zhekaite, E.; Sherman, V.; Voronkova, A.; Asherova, I.; Polyakov, A.; Adyan, T.; et al. Personalized selection of a CFTR modulator for a patient with a complex allele [L467F;F508del]. Curr. Issues Mol. Biol. 2022, 44, 5126–5138. [Google Scholar] [CrossRef]
- Kondratyeva, E.; Melyanovskaya, Y.; Bulatenko, N.; Davydenko, K.; Filatova, A.; Efremova, A.; Skoblov, M.; Bukharova, T.; Sherman, V.; Voronkova, A.; et al. Clinical and Functional Characteristics of the E92K CFTR Gene Variant in the Russian and Turkish Population of People with Cystic Fibrosis. Int. J. Mol. Sci. 2023, 24, 6351. [Google Scholar] [CrossRef]
- Kondratyeva, E.; Bukharova, T.; Efremova, A.; Melyanovskaya, Y.; Bulatenko, N.; Davydenko, K.; Filatova, A.; Skoblov, M.; Krasovsky, S.; Petrova, N.; et al. Health Characteristics of Patients with Cystic Fibrosis Whose Genotype Includes a Variant of the Nucleotide Sequence c.3140-16T > A and Functional Analysis of This Variant. Genes 2021, 12, 837. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, P.; Wilschanski, M.; Muallem, S.; Lukacs, G.L.; Sahin-Tóth, M.; Uc, A.; Gray, M.A.; Rakonczay, Z.; Maléth, J., Jr. CFTR: A New Horizon in the Pathomechanism and Treatment of Pancreatitis. Rev. Physiol. Biochem. Pharmacol. 2016, 170, 37–66. [Google Scholar]
- Baldwin, C.; Zerofsky, M.; Sathe, M.; Troendle, D.M.; Perito, E.R. Acute Recurrent and Chronic Pancreatitis as Initial Manifestations of Cystic Fibrosis and Cystic Fibrosis Transmembrane Conductance Regulator-Related Disorders. Pancreas 2019, 48, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Dörk, T.; Richter, T.; Neumann, T.; Wolfes, H.; Wulf, B.; Dörk, T.; Maass, G.; Tümmler, B. Cystic fibrosis with three mutations in the cystic fibrosis transmembrane conductance regulator gene. Hum. Genet. 1991, 87, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Claustres, M.; Altiéri, J.P.; Guittard, C.; Templin, C.; Chevalier-Porst, F.; Georges, M.D. Are p.1148T, p.R74W and p.D1270N cystic fibrosis causing mutations? BMC Med. Genet. 2004, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://cftr.iurc.montp.inserm.fr/cgi-bin/affiche.cgi?variant=c.650A%3EG (accessed on 25 July 2023).
- Available online: https://www.ncbi.nlm.nih.gov/clinvar/RCV000007660 (accessed on 25 July 2023).
- Terlizzi, V.; Centrone, C.; Ferrari, B.; Castellani, C.; Gunawardena, T.N.A.; Taccetti, G.; Laselva, O. Modulator Therapy in Cystic Fibrosis Patients with cis Variants in F508del Complex Allele: A Short-Term Observational Case Series. J. Pers. Med. 2022, 12, 1421. [Google Scholar] [CrossRef] [PubMed]
- Romey, M.C.; Guittard, C.; Chazalette, J.P.; Frossard, P.; Dawson, K.P.; Patton, M.A.; Casals, T.; Bazarbachi, T.; Girodon, E.; Rault, G.; et al. Complex allele [−102T > A+S549R(T > G)] is associated with milder forms of cystic fibrosis than allele S549R(T > G) alone. Hum. Genet. 1999, 105, 145–150. [Google Scholar] [PubMed]
- Baatallah, N.; Bitam, S.; Martin, N.; Servel, N.; Costes, B.; Mekki, C.; Chevalier, B.; Pranke, I.; Simonin, J.; Girodon, E.; et al. Cis variants identified in F508del complex alleles modulate CFTR channel rescue by small molecules. Hum. Mutat. 2018, 39, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Kondratyeva, E.; Efremova, A.; Melyanovskaya, Y.; Voronkova, A.; Polyakov, A.; Bulatenko, N.; Adyan, T.; Sherman, V.; Kovalskaia, V.; Petrova, N.; et al. Evaluation of the Complex p.[Leu467Phe;Phe508del] CFTR Allele in the Intestinal Organoids Model: Implications for Therapy. Int. J. Mol. Sci. 2022, 23, 10377. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΔISC, µA/cm2 | Amiloride | Forskolin | Genistein | Carbachol | DIDS | Histamine |
|---|---|---|---|---|---|---|
| M ± m patient | −13.67 ± 4.56 | 20.5 ± 3.37 | 0 | 22 ± 1.97 | 0 | 15.67 ± 2.41 |
| F508del/F508del [*, **] | −18.39 ± 5.62 | 3.06 ± 0.89 | 1.83 ± 0.35 | - | 1.83 ± 0.35 | 21.5 ± 5.46 |
| Control group [*, **] | −8.98 ± 3.42 | 25.78 ± 4.41 | 2 ± 0.29 | 117.44 ± 4.32 | 1.8 ± 0.26 | 101.68 ± 10.99 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kondratyeva, E.; Melyanovskaya, Y.; Efremova, A.; Krasnova, M.; Mokrousova, D.; Bulatenko, N.; Petrova, N.; Polyakov, A.; Adyan, T.; Kovalskaia, V.; et al. Clinical and Genetic Characteristics of a Patient with Cystic Fibrosis with a Complex Allele [E217G;G509D] and Functional Evaluation of the CFTR Channel. Genes 2023, 14, 1705. https://doi.org/10.3390/genes14091705
Kondratyeva E, Melyanovskaya Y, Efremova A, Krasnova M, Mokrousova D, Bulatenko N, Petrova N, Polyakov A, Adyan T, Kovalskaia V, et al. Clinical and Genetic Characteristics of a Patient with Cystic Fibrosis with a Complex Allele [E217G;G509D] and Functional Evaluation of the CFTR Channel. Genes. 2023; 14(9):1705. https://doi.org/10.3390/genes14091705
Chicago/Turabian StyleKondratyeva, Elena, Yuliya Melyanovskaya, Anna Efremova, Mariya Krasnova, Diana Mokrousova, Nataliya Bulatenko, Nika Petrova, Alexander Polyakov, Tagui Adyan, Valeriia Kovalskaia, and et al. 2023. "Clinical and Genetic Characteristics of a Patient with Cystic Fibrosis with a Complex Allele [E217G;G509D] and Functional Evaluation of the CFTR Channel" Genes 14, no. 9: 1705. https://doi.org/10.3390/genes14091705