
Congenital Myopathy as a Phenotypic Expression of CACNA1S Gene Mutation: Case Report and Systematic Review of the Literature

 , , ,
, , ,
Abstract
:1. Introduction
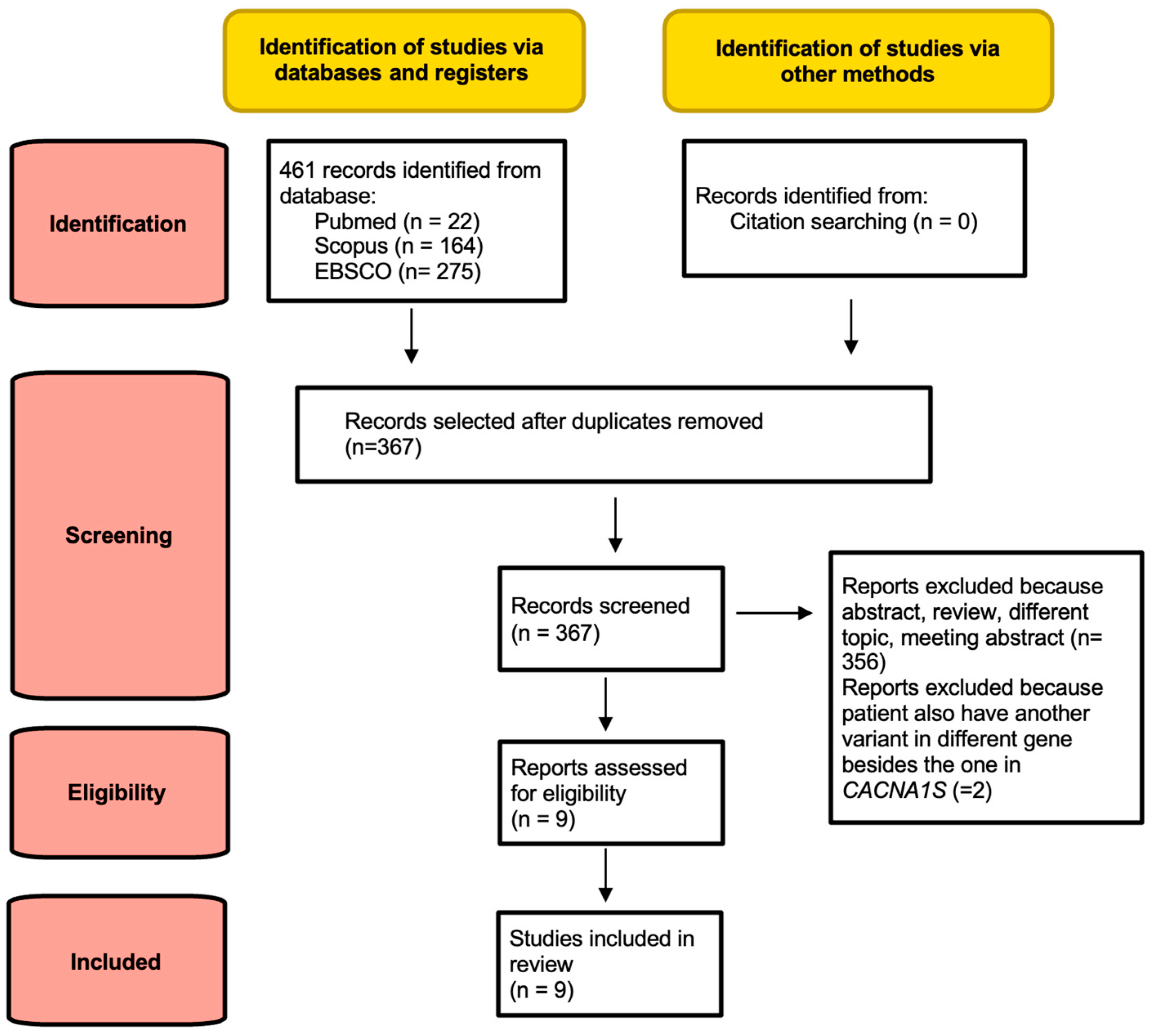
2. Materials and Methods
2.1. Search Strategy and Selection Criteria
2.2. Data Collection Process
2.3. Genetic Analysis
2.4. Clinical Grading
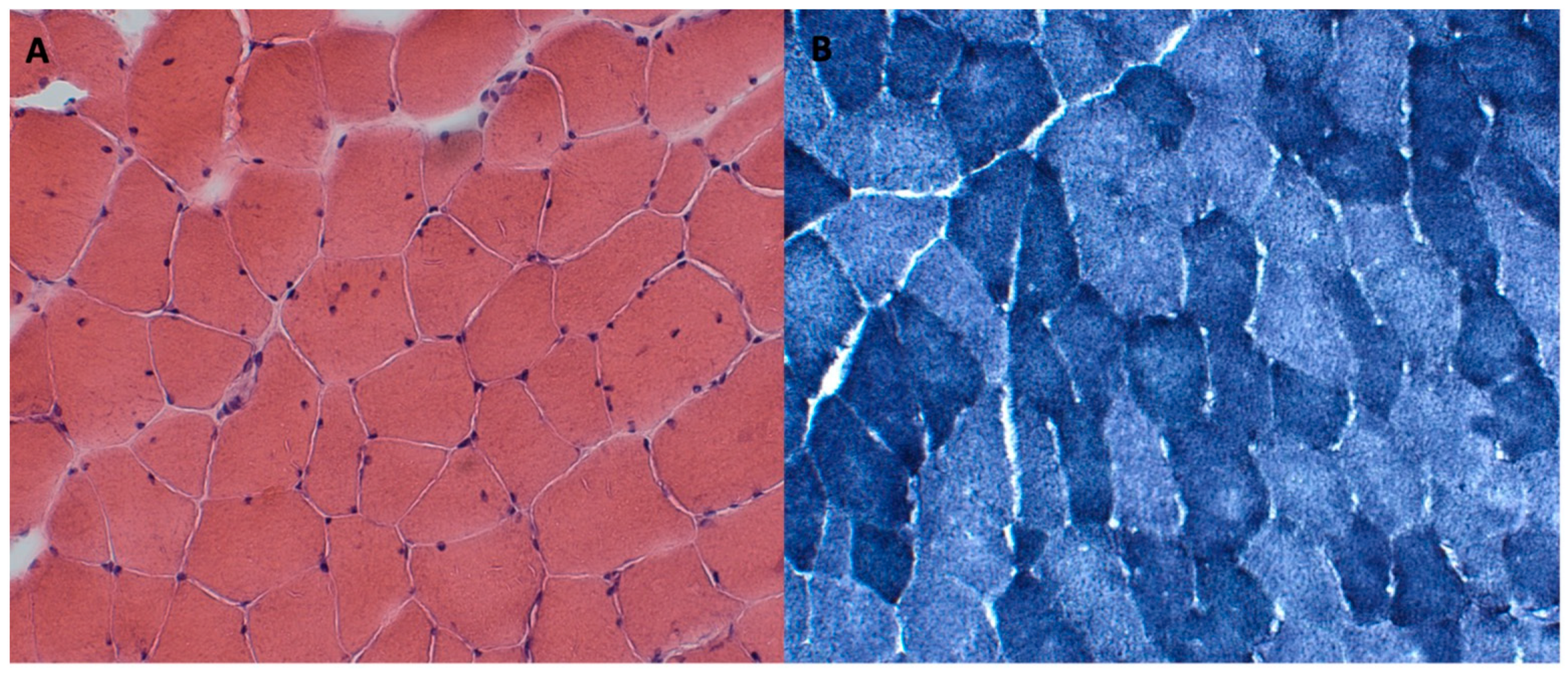
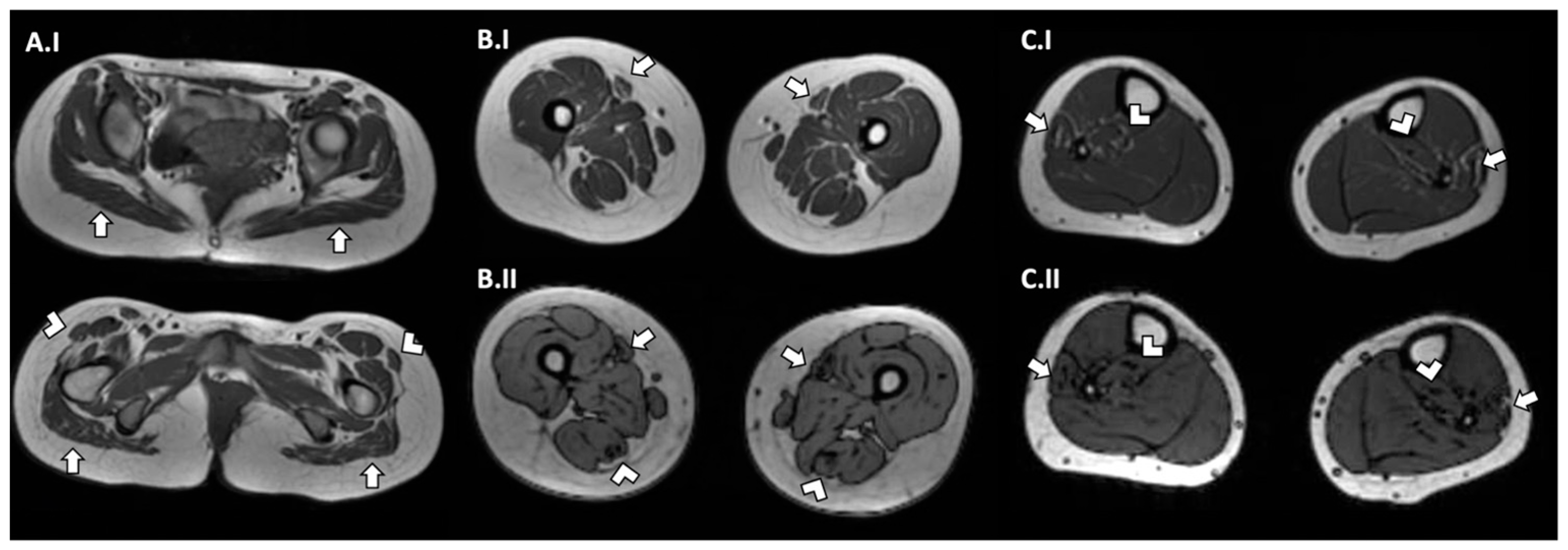
2.5. Case Report
3. Results
Descriptive Findings
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cassandrini, D.; Trovato, R.; Rubegni, A.; Lenzi, S.; Fiorillo, C.; Baldacci, J.; Minetti, C.; Astrea, G.; Bruno, C.; Santorelli, F.M. Italian Network on Congenital Myopathies. Congenital myopathies: Clinical phenotypes and new diagnostic tools. Ital. J. Pediatr. 2017, 43, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schorling, D.C.; Kirschner, J.; Bönnemann, C.G. Congenital Muscular Dystrophies and Myopathies: An Overview and Update. Neuropediatrics 2017, 48, 247–261. [Google Scholar] [CrossRef] [PubMed]
- North, K.N.; Wang, C.H.; Clarke, N.; Jungbluth, H.; Vainzof, M.; Dowling, J.J.; Amburgey, K.; Quijano-Roy, S.; Beggs, A.H.; Sewry, C.; et al. Approach to the diagnosis of congenital myopathies. Neuromuscul. Disord. 2014, 24, 97–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isackson, P.J.; Wang, J.; Zia, M.; Spurgeon, P.; Levesque, A.; Bard, J.; James, S.; Nowak, N.; Lee, T.K.; Vladutiu, G.D. RYR1 and CACNA1S genetic variants identified with statin-associated muscle symptoms. Pharmacogenomics 2018, 19, 1235–1249. [Google Scholar] [CrossRef] [PubMed]
- Yiş, U.; Hiz, S.; Güneş, S.; Diniz, G.; Baydan, F.; Töpf, A.; Sonmezler, E.; Lochmüller, H.; Horvath, R.; Oktay, Y. Dihydropyridine Receptor Congenital Myopathy In A Consangineous Turkish Family. J. Neuromuscul. Dis. 2019, 6, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Malfatti, E.; Romero, N.B. Nemaline myopathies: State of the art. Rev. Neurol. 2016, 172, 614–619. [Google Scholar] [CrossRef]
- Sangkuhl, K.; Dirksen, R.T.; Alvarellos, M.L.; Altman, R.B.; Klein, T.E. PharmGKB summary: Very important pharmacogene information for CACNA1S. Pharm. Genom. 2020, 30, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, R.T.; Karunasekara, Y.; Board, P.G.; Beard, N.A.; Casarotto, M.G.; Dulhunty, A.F. Skeletal muscle excitation-contraction coupling: Who are the dancing partners? Int. J. Biochem. Cell Biol. 2014, 48, 28–38. [Google Scholar] [CrossRef]
- Nakai, J.; Dirksen, R.T.; Nguyen, H.T.; Pessah, I.N.; Beam, K.G.; Allen, P.D. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature 1996, 380, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Grabner, M.; Dirksen, R.T.; Suda, N.; Beam, K.G. The II-III loop of the skeletal muscle dihydropyridine receptor is responsible for the Bi-directional coupling with the ryanodine receptor. J. Biol. Chem. 1999, 274, 21913–21919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monnier, N.; Procaccio, V.; Stieglitz, P.; Lunardi, J. Malignant-hyperthermia susceptibility is associated with a mutation of the alpha 1-subunit of the human dihydropyridine-sensitive L-type voltage-dependent calcium-channel receptor in skeletal muscle. Am. J. Hum. Genet. 1997, 60, 1316–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, P.M.; Rüffert, H.; Snoeck, M.M.; Girard, T.; Glahn, K.P.; Ellis, F.R.; Müller, C.R.; Urwyler, A.; European Malignant Hyperthermia Group; Bandschapp, O.; et al. European Malignant Hyperthermia Group. European Malignant Hyperthermia Group guidelines for investigation of malignant hyperthermia susceptibility. Br. J. Anaesth. 2015, 115, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Jurkat-Rott, K.; Lehmann-Horn, F.; Elbaz, A.; Heine, R.; Gregg, R.G.; Hogan, K.; Powers, P.A.; Laple, P.; Vale-Santos, J.E.; Weissenbach, J.; et al. A calcium channel mutation causing hypokalemic periodic paralysis. Hum. Mol. Genet. 1994, 3, 1415–1419. [Google Scholar] [CrossRef]
- Ptáček, L.J.; Tawil, R.; Griggs, R.C.; Engel, A.G.; Layzer, R.B.; Kwieciński, H.; McManis, P.G.; Santiago, L.; Moore, M.; Fouad, G.; et al. Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell 1994, 77, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Kung, A.W.; Lau, K.S.; Fong, G.C.; Chan, V. Association of novel single nucleotide polymorphisms in the calcium channel alpha 1 subunit gene (Ca(v)1.1) and thyrotoxic periodic paralysis. J. Clin. Endocrinol. Metab. 2004, 89, 1340–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: Explanation and elaboration. J. Clin. Epidemiol. 2009, 62, e1–e34. [Google Scholar] [CrossRef] [Green Version]
- Macias, A.; Fichna, J.P.; Topolewska, M.; Rȩdowicz, M.J.; Kaminska, A.M.; Kostera-Pruszczyk, A. Targeted Next-Generation Sequencing Reveals Mutations in Non-coding Regions and Potential Regulatory Sequences of Calpain-3 Gene in Polish Limb-Girdle Muscular Dystrophy Patients. Front. Neurosci. 2021, 15, 692482. [Google Scholar] [CrossRef] [PubMed]
- Schartner, V.; Romero, N.B.; Donkervoort, S.; Treves, S.; Munot, P.; Pierson, T.M.; Dabaj, I.; Malfatti, E.; Zaharieva, I.T.; Zorzato, F.; et al. Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy. Acta Neuropathol. 2017, 133, 517–533. [Google Scholar] [CrossRef]
- Juntas Morales, R.; Perrin, A.; Solé, G.; Lacourt, D.; Pegeot, H.; Walther-Louvier, U.; Cintas, P.; Cances, C.; Espil, C.; Theze, C.; et al. An Integrated Clinical-Biological Approach to Identify Interindividual Variability and Atypical Phenotype-Genotype Correlations in Myopathies: Experience on A Cohort of 156 Families. Genes 2021, 12, 1199. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.M.; Ahearn, M.E.; Balak, C.D.; Liang, W.S.; Kurdoglu, A.; Corneveaux, J.J.; Russell, M.; Huentelman, M.J.; Craig, D.W.; Carpten, J.; et al. Novel pathogenic variants and genes for myopathies identified by whole exome sequencing. Mol. Genet. Genom. Med. 2015, 3, 283–301. [Google Scholar] [CrossRef] [PubMed]
- François-Heude, M.C.; Walther-Louvier, U.; Espil-Taris, C.; Beze-Beyrie, P.; Rivier, F.; Baudou, E.; Uro-Coste, E.; Rigau, V.; Negrier, M.L.; Rendu, J.; et al. Evaluating next-generation sequencing in neuromuscular diseases with neonatal respiratory distress. Eur. J. Paediatr. Neurol. 2021, 31, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Mauri, E.; Piga, D.; Pagliarani, S.; Magri, F.; Manini, A.; Sciacco, M.; Ripolone, M.; Napoli, L.; Borellini, L.; Cinnante, C.; et al. CACNA1S mutation associated with a case of juvenile-onset congenital myopathy. J. Neurol. Sci. 2021, 431, 120047. [Google Scholar] [CrossRef] [PubMed]
- Ravenscroft, G.; Clayton, J.S.; Faiz, F.; Sivadorai, P.; Milnes, D.; Cincotta, R.; Moon, P.; Kamien, B.; Edwards, M.; Delatycki, M.; et al. Neurogenetic fetal akinesia and arthrogryposis: Genetics, expanding genotype-phenotypes and functional genomics. J. Med. Genet. 2021, 58, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Grunseich, C.; Sarkar, N.; Lu, J.; Owen, M.; Schindler, A.; Calabresi, P.A.; Sumner, C.J.; Roda, R.H.; Chaudhry, V.; Lloyd, T.E.; et al. Improving the efficacy of exome sequencing at a quaternary care referral centre: Novel mutations, clinical presentations and diagnostic challenges in rare neurogenetic diseases. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1186–1196. [Google Scholar] [CrossRef] [PubMed]
- Kausthubham, N.; Shukla, A.; Gupta, N.; Bhavani, G.S.; Kulshrestha, S.; Das Bhowmik, A.; Moirangthem, A.; Bijarnia-Mahay, S.; Kabra, M.; Puri, R.D.; et al. A data set of variants derived from 1455 clinical and research exomes is efficient in variant prioritization for early-onset monogenic disorders in Indians. Hum. Mutat. 2021, 42, e15–e61. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [Green Version]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledge- base and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Marinella, G.; Astrea, G.; Buchignani, B.; Cassandrini, D.; Doccini, S.; Filosto, M.; Galatolo, D.; Gallone, S.; Giannini, F.; Lopergolo, D.; et al. A Schematic Approach to Defining the Prevalence of COL VI Variants in Five Years of Next-Generation Sequencing. Int. J. Mol. Sci. 2022, 23, 14567. [Google Scholar] [CrossRef]
- Nance, J.R.; Dowling, J.J.; Gibbs, E.M.; Bönnemann, C.G. Congenital myopathies: An update. Curr. Neurol. Neurosci. Rep. 2012, 12, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Romero, N.B.; Clarke, N.F. Congenital myopathies. Handb. Clin. Neurol. 2013, 113, 1321–1336. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N. A single nucleotide deletion in the skeletal muscle-specific calcium channel transcript of muscular dysgenesis (mdg) mice. J. Biol. Chem. 1992, 267, 25636–25639. [Google Scholar] [CrossRef]
- Eberl, D.F.; Ren, D.; Feng, G.; Lorenz, L.J.; Van Vactor, D.; Hall, L.M. Genetic and developmental characterization of Dmca1D, a calcium channel alpha1 subunit gene in Drosophila melanogaster. Genetics 1998, 148, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Knudson, C.M.; Chaudhari, N.; Sharp, A.H.; Powell, J.A.; Beam, K.G.; Campbell, K.P. Specific absence of the alpha 1 subunit of the dihydropyridine receptor in mice with muscular dysgenesis. J. Biol. Chem. 1989, 264, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Herasse, M.; Bitoun, M.; Barragán-Campos, H.M.; Chiras, J.; Laforêt, P.; Fardeau, M.; Eymard, B.; Guicheney, P.; Romero, N.B. Characterization of the muscle involvement in dynamin 2-related centronuclear myopathy. Brain 2006, 129 Pt 6, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Susman, R.D.; Quijano-Roy, S.; Yang, N.; Webster, R.; Clarke, N.F.; Dowling, J.; Kennerson, M.; Nicholson, G.; Biancalana, V.; Ilkovski, B.; et al. Expanding the clinical, pathological and MRI phenotype of DNM2-related centronuclear myopathy. Neuromuscul. Disord. 2010, 20, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Cacciari, E.; Milani, S.; Balsamo, A.; Spada, E.; Bona, G.; Cavallo, L.; Cerutti, F.; Gargantini, L.; Greggio, N.; Tonini, G.; et al. Italian cross-sectional growth charts for height, weight and BMI (2 to 20 yr). J. Endocrinol. Investig. 2006, 29, 581–593. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
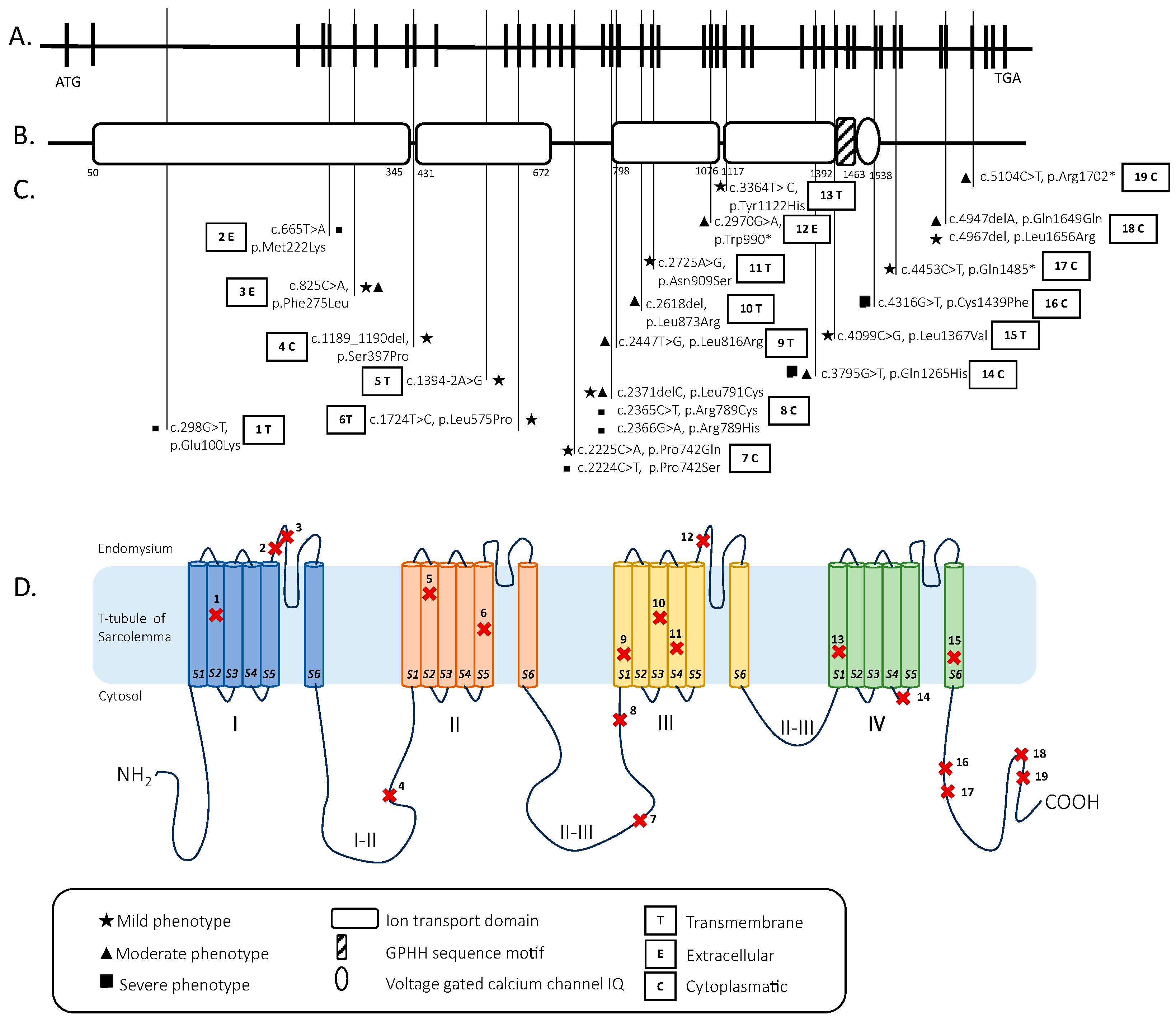
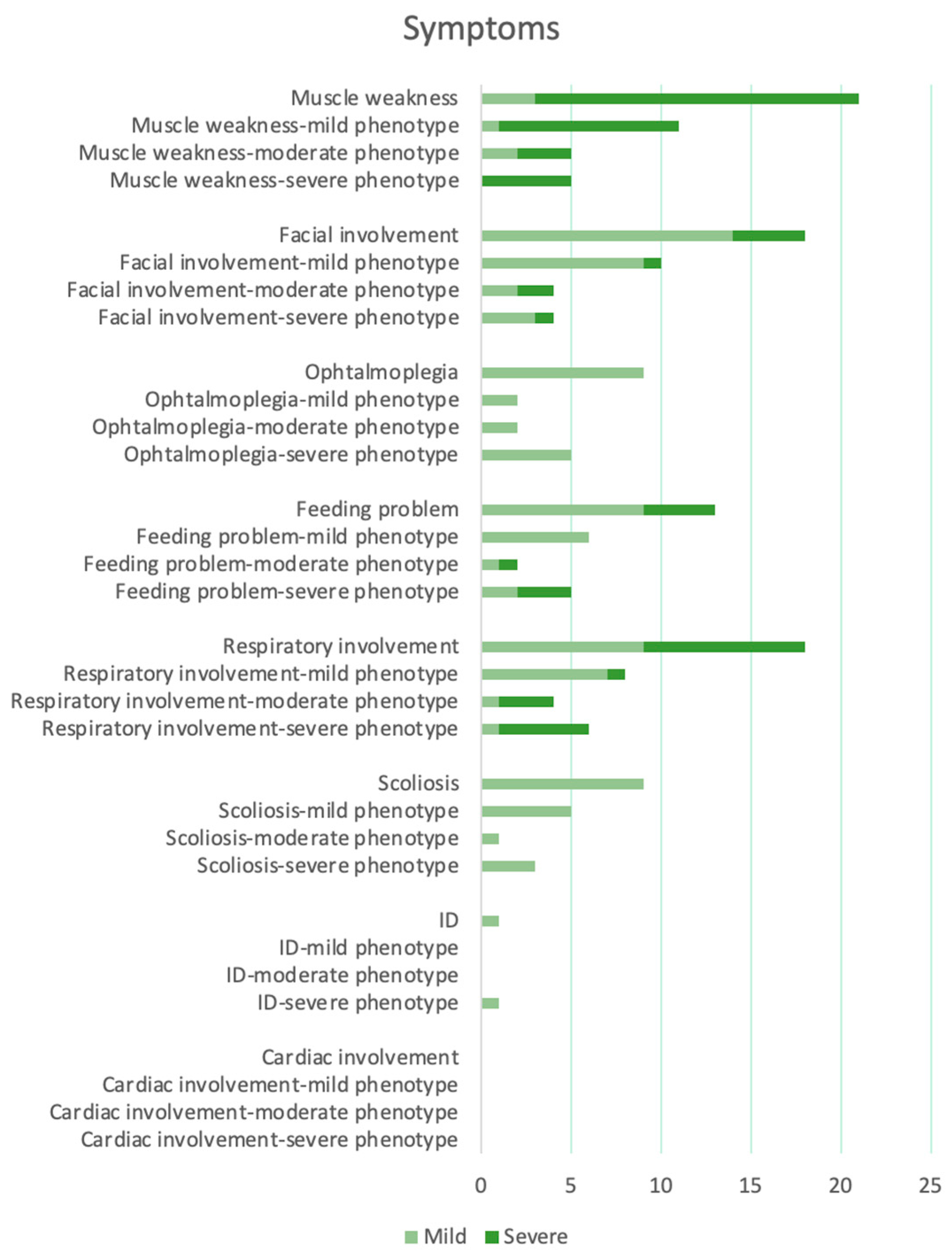
| Number and Sex | Mutations | Onset | Motor Devel. | Muscle Weakness | Facial Involvement | Ophtalmo- Plegia | Feeding | Respiratory Inv. | Scoliosis | Deep Tendon Reflexes | ID | Cardiac Inv. | CK Level | Muscle MRI | Muscle Biopsy | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Our case | 1 F | c.1394-2A>G; c.1724T>C, p.L575P | Neonatal | Delayed | Generalized (A and P > D) | Moderate | No | Slight swallowing problem | Mild | Yes | Reduced | No | No | Normal | Hypotrophy of pelvis and thigh muscles (> medio-posterior). Adipose infiltration of gluteus, tensor fasciae latae semitendinosus, sartorius, peroneus, and postero tibialis muscles | Fiber size variability, occasional internal nuclei, and disorganization of intermyofibrillar network |
| Schartner et al. [18] | 2 M | c.1189_1190del, p.Ser397Pro; c.4967del, p.Leu1656Arg | Antenatal/ neonatal | Delayed | Generalized (A and P > D) | Mild with high arched palate | Yes | Slight swallowing problem | Normal | No | NA | NA | No | Normal | Muscle atrophy | Centralized nuclei; focal disorganization; fiber size variability; alveolar aspect of the intermyofibrillar network; and uniformity of type I fibers |
| 3 M | c.4453C>T, p.Gln1485*; c.4967del, p.Leu1656Arg | Antenatal/ neonatal | Delayed | Proximal and axial | Mild with high arched palate | Yes | Slight swallowing problem | Mild | No | NA | NA | No | Normal | Fatty replacement and anterior muscle involvement in lower limb | Centralized nuclei; minicore; alveolar aspect of the inter-myofibrillar network; and uniformity of type I fibers | |
| 4 F | c.825C>A, p.Phe275Leu; c.2371delC, p.Leu791Cysfs*37 | Neonatal | Delayed | Generalized (P > D) | Mild with high arched palate | No | Normal | Normal | Yes | Normal | NA | No | Normal | NA | NA | |
| 5 F | c.825C>A, p.Phe275Leu; c.2371delC, p.Leu791Cysfs*37 | Neonatal | Delayed | Generalized (P > D) | Mild with high arched palate | No | Swallowing issues (G-tube) | Normal | Yes | Normal | NA | No | Normal | Mild involvement of the anterior thigh, predominantly in the vastus lateralis | NA | |
| 6 M | c.298G>T, p.Glu100Lys; c.3795G>T, p.Gln1265Hisfs*57 | Neonatal | Delayed | Generalized (A and P > D) | Mild with high arched palate and ptosis | Yes | Occasional swallowing difficulties | Severe | Yes | NA | NA | No | Normal | Marked atrophy involving all muscle groups of the upper leg bilaterally. Fatty infiltration, particularly of the extensor groups. | Fiber size variability; endomysial connective tissue around most fibers. Predominance of type I fibers | |
| 7 F | c.2225C>A, p.Pro742Gln | Early childhood | Normal | Generalized | Mild with high arched palate | No | Normal | Mild | No | NA | NA | No | Normal | NA | Fiber size variability and alveolar aspect of the intermyofibrillar network | |
| 8 M | c.2225C>A, p.Pro742Gln | Early childhood | Delayed | Generalized | Mild with high arched palate | No | Normal | Mild | yes | NA | NA | No | Normal | Muscle wasting of the anterior thigh and no fatty infiltration | Fiber size variability and alveolar aspect of the intermyofibrillar network | |
| 9 M | c.2225C>A, p.Pro742Gln | Early childhood | Normal | Generalized | Mild with high arched palate | No | Normal | Mild | No | NA | NA | No | Normal | Muscle wasting of the anterior thigh and no fatty infiltration | Fiber size variability and alveolar aspect of the intermyofibrillar network | |
| 10 M | c.2224C>T, p.Pro742Ser | Antenatal/neonatal | Delayed | Generalized | Mild with high arched palate | Yes | Occasional swallowing difficulties | Severe | Yes | NA | NA | No | Normal | Diffuse atrophy and more severe fatty infiltration in anterior compartment of the thigh and soleus and peroneal muscles in the leg | Rare internalized nuclei; fiber size variability; alveolar aspect of the inter-myofibrillar network; and uniformity of type I fibers | |
| 11 M | c.4099C>G, p.Leu1367Val | Early onset (6–7 months) | Delayed | Generalized (more severe at upper member and P > D at lower members) | Mild with high arched palate | No | Occasional swallowing difficulties | Mild | Yes | NA | NA | No | Elevated (1000–2000) | Severe changes in thigh (anterior > posterior) and relative sparing of anterior compartment in the leg | Internalized nuclei; core-like structures; fiber size variability; and endomysial fibrosis | |
| 12 F | c.4099C>G, p.Leu1367Val | Neonatal | Delayed | Generalized (more severe at upper member and P > D at lower members) | Mild with high arched palate | No | Occasional swallowing difficulties | Mild | Yes | NA | NA | No | Elevated (1000–2000) | Severe changes in thigh (anterior > posterior) and relative sparing of anterior compartment in the leg | NA | |
| yis et al. [5] | 13 M (died 3 m) | c.2366G>A, p.R789H *homo | Neonatal | NA | Generalized | NA | Yes | Absent suck | Severe | NA | NA | NA | No | NA | NA | Mild dystrophic changes such as contraction, regeneration, degeneration, nuclear internalization, and fibrosis were visible |
| 14 F | c.2366G>A, p.R789H *homo | Neonatal | Delayed | Generalized | Mild with high arched palate | Yes | Swallowing issues (Gastrostomy) | Severe | Yes | Absent | Yes | No | NA | NA | Marked variation in fiber size and shape; increased nuclear internalization; and grouping fascicules of large and small myofibers | |
| 15 F (died 3 m) | c.2366G>A, p.R789H *homo | Neonatal | NA | Generalized | NA | Yes | Absent suck | Severe | NA | NA | NA | No | NA | NA | NA | |
| Morales et al. [19] | 16 F | c.2970G>A, p.Trp990*; c.5104C>T, p.Arg1702* | Neonatal | NA | Proximal and axial (mild) | NA | NA | Slight swallowing problem | Severe | NA | NA | NA | NA | Normal | Upper and lower limbs atrophy. No fat tissue replacement | Non-specific myopathic pattern with type 1 fiber predominance and mild myofibrillar disorganization |
| 17 M | c.2447T>G, p.(Leu816Arg) | Neonatal | NA | Proximal lower members | Mild with high arched palate | NA | Slight swallowing problem | Severe | NA | NA | NA | NA | Normal | Involvement of gluteus maximum | Non-specific myopathic pattern with type 1 fiber predominance | |
| Hunter et al. [20] | 18 M | c.4947delA, p.Gln1649Glnfs *72; c.3795G>T, p.Gln1265His | Neonatal | NA | Generalized (A and P > D) | Moderate with high arched palate | Yes | Swallowing issues (Gastrostomy) | Mild | NA | Absent | NA | NA | Normal | NA | Considerable myofiber size variation, polygonal small and large fibers, and occasional internal nuclei; and moderate architectural alterations in the form of coarse whorled fibers |
| Francois-Heude et al. [21] | 19 M | c.2618del, p.(Leu873Argfs*21); c.5104C>T, p.(Arg1702*) | Neonatal | NA | Generalized | Moderate | Yes | Swallowing issues (Gastrostomy) | NA | NA | NA | No | NA | Normal | Normal | Centronuclear myopathy |
| Mauri et al. [22] | 20 F | c.3364 T>C, p.Tyr1122His | Adult | NA | Generalized (P > D) | Mild with ptosis | NA | Slight swallowing problem | Moderate | NA | NA | NA | No | Normal | Diffuse hypotrophy, more prominent in the right deltoid muscle | Rare nuclear clumps, fiber size variability and few type II angulated and grouped hypotrophic fibers; and focal zones of myofibrillar disorganization |
| Ravenscroft et al. [23] | 21 M (died 10d) | c.665T>A, p.Met222Lys; c.2365C>T, p.Arg789Cys | Antenatal/neonatal | NA | NA | NA | NA | NA | Severe | NA | NA | NA | NA | NA | NA | NA |
| 22 M (died 26wg) | c.665T>A, p.Met222Lys; c.2365C>T, p.Arg789Cys | Antenatal | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Atrophy or myofibre disorganization | |
| Grunseich C. et al. [24] | 23 F | c.2725A>G, p.Asn909Ser | Neonatal | Delayed | Generalized (> A) | NA | NA | No | No | NA | NA | NA | No | NA | NA | Non-specific myopathy |
| Kausthubham et al. [25] | 24 (died early) | c.4316G>T, p.Cys1439Phe | Neonatal | NA | NA | Cleft palate | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marinella, G.; Orsini, A.; Scacciati, M.; Costa, E.; Santangelo, A.; Astrea, G.; Frosini, S.; Pasquariello, R.; Rubegni, A.; Sgherri, G.; et al. Congenital Myopathy as a Phenotypic Expression of CACNA1S Gene Mutation: Case Report and Systematic Review of the Literature. Genes 2023, 14, 1363. https://doi.org/10.3390/genes14071363
Marinella G, Orsini A, Scacciati M, Costa E, Santangelo A, Astrea G, Frosini S, Pasquariello R, Rubegni A, Sgherri G, et al. Congenital Myopathy as a Phenotypic Expression of CACNA1S Gene Mutation: Case Report and Systematic Review of the Literature. Genes. 2023; 14(7):1363. https://doi.org/10.3390/genes14071363
Chicago/Turabian StyleMarinella, Gemma, Alessandro Orsini, Massimo Scacciati, Elisa Costa, Andrea Santangelo, Guja Astrea, Silvia Frosini, Rosa Pasquariello, Anna Rubegni, Giada Sgherri, and et al. 2023. "Congenital Myopathy as a Phenotypic Expression of CACNA1S Gene Mutation: Case Report and Systematic Review of the Literature" Genes 14, no. 7: 1363. https://doi.org/10.3390/genes14071363