
Therapeutic Targeting of DNA Replication Stress in Cancer

Abstract
:1. Introduction
2. DNA Alkylating Agents
3. Targeting Nucleotide Metabolism
4. Targeting Topoisomerase I/II
5. Targeting DNA Repair Signaling Pathways
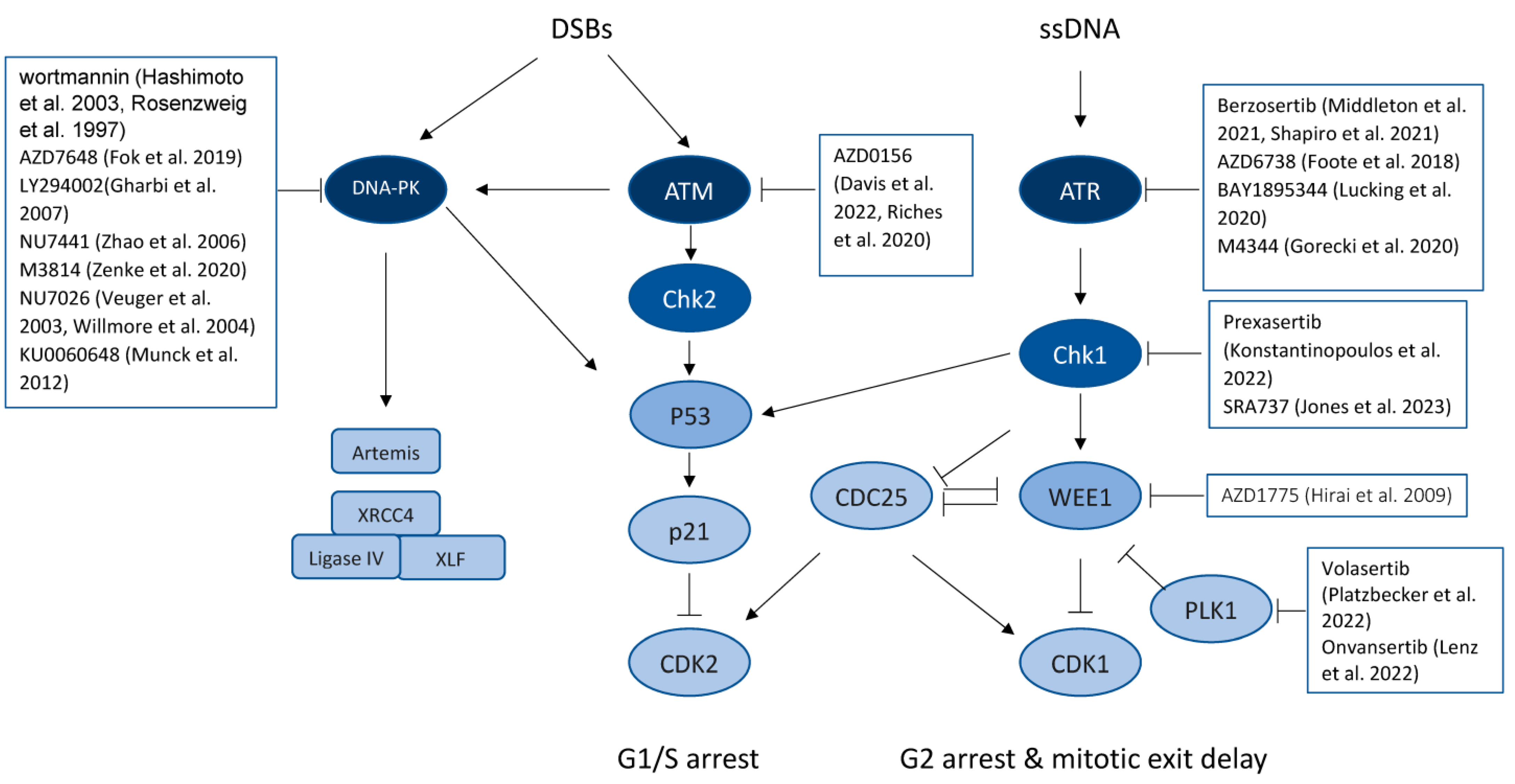
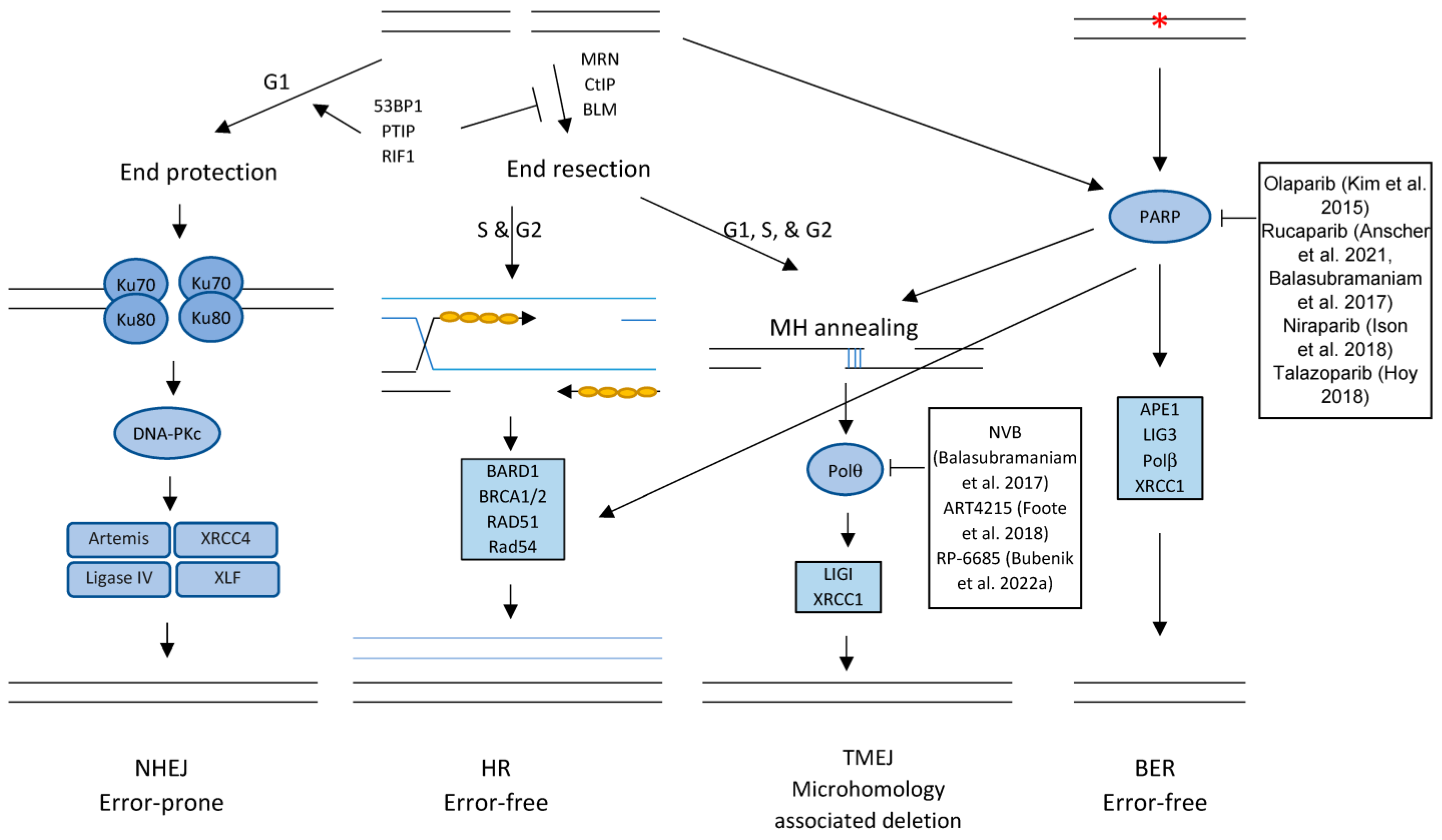
5.1. Targeting the DNA-PK Signaling Pathway
5.2. Targeting ATM/CHK2 and ATR/CHK1 Signaling
5.3. Inhibiting WEE1
6. Targeting DNA Repairing Proteins
6.1. Targeting the PARP Pathway
6.2. Inhibiting DNA Polymerase Theta
6.3. Inhibiting RECQ Helicases
7. Targeting PCNA, the “hub” Protein of DNA Replication and Repair Networks
8. Challenges and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [Green Version]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Malkas, L.H.; Herbert, B.S.; Abdel-Aziz, W.; Dobrolecki, L.E.; Liu, Y.; Agarwal, B.; Hoelz, D.; Badve, S.; Schnaper, L.; Arnold, R.J.; et al. A cancer-associated PCNA expressed in breast cancer has implications as a potential biomarker. Proc. Natl. Acad. Sci. USA 2006, 103, 19472–19477. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.; Li, M.; Lingeman, R.; Hickey, R.J.; Liu, Y.; Malkas, L.H. Mechanistic study of the superior anti-cancer properties of a first in-class small molecule targeting PCNA. Mol. Cancer Ther. 2019, 18. [Google Scholar] [CrossRef]
- Gu, L.; Smith, S.; Li, C.; Hickey, R.J.; Stark, J.M.; Fields, G.B.; Lang, W.H.; Sandoval, J.A.; Malkas, L.H. A PCNA-Derived Cell Permeable Peptide Selectively Inhibits Neuroblastoma Cell Growth. PLoS ONE 2014, 9, e94773. [Google Scholar] [CrossRef] [Green Version]
- Beranek, D.T. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat. Res. Mol. Mech. Mutagen. 1990, 231, 11–30. [Google Scholar] [CrossRef]
- Pullman, A.; Pullman, B. Molecular electrostatic potential of the nucleic acids. Q. Rev. Biophys. 1981, 14, 289–380. [Google Scholar] [CrossRef]
- Drabløs, F.; Feyzi, E.; Aas, P.A.; Vaagbø, C.B.; Kavli, B.; Bratlie, M.S.; Peña-Diaz, J.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Alkylation damage in DNA and RNA—Repair mechanisms and medical significance. DNA Repair 2004, 3, 1389–1407. [Google Scholar] [CrossRef]
- Ponti, M.; Souhami, R.L.; Fox, B.W.; Hartley, J.A. DNA interstrand crosslinking and sequence selectivity of dimethanesulphonates. Br. J. Cancer 1991, 63, 743–747. [Google Scholar] [CrossRef] [Green Version]
- Low, J.E.; Borch, R.F.; Sladek, N.E. Conversion of 4-hydroperoxycyclophosphamide and 4-hydroxycyclophosphamide to phosphoramide mustard and acrolein mediated by bifunctional catalysis. Cancer Res. 1982, 42, 830–837. [Google Scholar]
- Graham, J.E.; Marians, K.; Kowalczykowski, S. Independent and Stochastic Action of DNA Polymerases in the Replisome. Cell 2017, 169, 1201–1213.e17. [Google Scholar] [CrossRef] [Green Version]
- Marians, K.J. Lesion Bypass and the Reactivation of Stalled Replication Forks. Annu. Rev. Biochem. 2018, 87, 217–238. [Google Scholar] [CrossRef]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and cancer: Golden anniversary. Nat. Rev. Clin. Oncol. 2009, 6, 638–647. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [Green Version]
- Bush, H.; Thatcher, N.; Barnard, R. Chemotherapy in the management of invasive bladder cancer. Cancer Chemother. Pharmacol. 1979, 3, 87–96. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, Z.; You, X.-Z. Hydrolysis Theory for Cisplatin and Its Analogues Based on Density Functional Studies. J. Am. Chem. Soc. 2001, 123, 9378–9387. [Google Scholar] [CrossRef]
- Jamieson, E.R.; Lippard, S.J. Structure, Recognition, and Processing of Cisplatin-DNA Adducts. Chem. Rev. 2010, 30, 2467–2498. [Google Scholar] [CrossRef]
- Dasika, G.K.; Lin, S.-C.J.; Zhao, S.; Sung, P.; Tomkinson, A.; Lee, E.Y.-H.P. DNA damage-induced cell cycle checkpoints and DNA strand break repair in development and tumorigenesis. Oncogene 1999, 18, 7883–7899. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Hara, R.; Singh, G.; Sancar, A.; Lippard, S.J. Nucleotide Excision Repair from Site-Specifically Platinum-Modified Nucleosomes. Biochemistry 2003, 42, 6747–6753. [Google Scholar] [CrossRef]
- Sawant, A.; Kothandapani, A.; Zhitkovich, A.; Sobol, R.W.; Patrick, S.M. Role of mismatch repair proteins in the processing of cisplatin interstrand cross-links. DNA Repair 2015, 35, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, K.; Yoshioka, Y.; Hsieh, P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol. Cell 2006, 22, 501–510. [Google Scholar] [CrossRef]
- Topping, R.P.; Wilkinson, J.C.; Scarpinato, K.D. Mismatch Repair Protein Deficiency Compromises Cisplatin-induced Apoptotic Signaling. J. Biol. Chem. 2009, 284, 14029–14039. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.P.; Wang, Y.; Scherer, S.J.; Clark, A.B.; Yang, K.; Avdievich, E.; Jin, B.; Werling, U.; Parris, T.; Kurihara, N.; et al. An Msh2 Point Mutation Uncouples DNA Mismatch Repair and Apoptosis. Cancer Res 2004, 64, 517–522. [Google Scholar] [CrossRef] [Green Version]
- Prestayko, A.; D’Aoust, J.; Issell, B.; Crooke, S. Cisplatin (cis-diamminedichloroplatinum II). Cancer Treat. Rev. 1979, 6, 17–39. [Google Scholar] [CrossRef]
- Lebwohl, D.; Canetta, R. Clinical development of platinum complexes in cancer therapy: An historical perspective and an update. Eur. J. Cancer 1998, 34, 1522–1534. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Kryczka, J.; Kryczka, J.; Czarnecka-Chrebelska, K.H.; Brzeziańska-Lasota, E. Molecular Mechanisms of Chemoresistance Induced by Cisplatin in NSCLC Cancer Therapy. Int. J. Mol. Sci. 2021, 22, 8885. [Google Scholar] [CrossRef]
- Callejo, A.; Sedó-Cabezón, L.; Juan, I.D.; Llorens, J. Cisplatin-Induced Ototoxicity: Effects, Mechanisms and Protection Strategies. Toxics 2015, 3, 268–293. [Google Scholar] [CrossRef] [Green Version]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Latcha, S.; Jaimes, E.A.; Patil, S.; Glezerman, I.G.; Mehta, S.; Flombaum, C.D. Long–Term Renal Outcomes after Cisplatin Treatment. Clin. J. Am. Soc. Nephrol. 2016, 11, 1173–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, W.B.; Cheng, Y.C. Metabolism and mechanism of action of 5-fluorouracil. Pharmacol. Ther. 1990, 48, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.; Barr, R.; Shulman, L.N.; Forte, G.B.; Magrini, N. Essential medicines for cancer: WHO recommendations and national priorities. Bull. World Health Organ. 2016, 94, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Love, W.E.; Bernhard, J.; Bordeaux, J. Topical imiquimod or fluorouracil therapy for basal and squamous cell carcinoma: A systematic review. Arch. Dermatol. 2009, 145, 1431–1438. [Google Scholar] [CrossRef]
- Gross, K.; Kircik, L.; Kricorian, G. Faculty Opinions recommendation of 5% 5-Fluorouracil cream for the treatment of small superficial Basal cell carcinoma: Efficacy, tolerability, cosmetic outcome, and patient satisfaction. Dermatol. Surg. 2007, 33, 433–439; discussion 440. [Google Scholar]
- Van Cutsem, E.; Hoff, P.M.; Harper, P.; Bukowski, R.M.; Cunningham, D.; Dufour, P.; Graeven, U.; Lokich, J.; Madajewicz, S.; Maroun, J.A.; et al. Oral capecitabine vs intravenous 5-fluorouracil and leucovorin: Integrated efficacy data and novel analyses from two large, randomised, phase III trials. Br. J. Cancer 2004, 90, 1190–1197. [Google Scholar] [CrossRef]
- Lam, S.; Guchelaar, H.; Boven, E. The role of pharmacogenetics in capecitabine efficacy and toxicity. Cancer Treat. Rev. 2016, 50, 9–22. [Google Scholar] [CrossRef]
- Wethington, S.L.; Wright, J.D.; Herzog, T.J. Key role of topoisomerase I inhibitors in the treatment of recurrent and refractory epithelial ovarian carcinoma. Expert Rev. Anticancer. Ther. 2008, 8, 819–831. [Google Scholar] [CrossRef]
- Wu, J.; Phatnani, H.P.; Hsieh, T.-S.; Greenleaf, A.L. The phosphoCTD-interacting domain of Topoisomerase I. Biochem. Biophys. Res. Commun. 2010, 397, 117–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinemann, V.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Comparison of the cellular pharmacokinetics and toxicity of 2′,2′-difluorodeoxycytidine and 1-β-D-arabinofuranosylcytosine. Cancer Res. 1988, 48, 4024–4031. [Google Scholar] [PubMed]
- Heinemann, V.; Xu, Y.Z.; Chubb, S.; Sen, A.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2′,2′-difluorodeoxycytidine. Mol. Pharmacol. 1990, 38, 567–572. [Google Scholar]
- Baker, C.H.; Banzon, J.; Bollinger, J.M.; Stubbe, J.; Samano, V.; Robins, M.J.; Lippert, B.; Jarvi, E.; Resvick, R. 2′-Deoxy-2′-methylenecytidine and 2′-deoxy-2′,2′-difluorocytidine 5′-diphosphates: Potent mechanism-based inhibitors of ribonucleotide reductase. J. Med. Chem. 1991, 34, 1879–1884. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Chubb, S.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991, 51, 6110–6117. [Google Scholar]
- Kim, E.S. Chemotherapy Resistance in Lung Cancer. Adv. Exp. Med. Biol. 2015, 893, 189–209. [Google Scholar]
- Liu, K.; Geng, Y.; Wang, L.; Xu, H.; Zou, M.; Li, Y.; Zhao, Z.; Chen, T.; Xu, F.; Sun, L.; et al. Systematic exploration of the underlying mechanism of gemcitabine resistance in pancreatic adenocarcinoma. Mol. Oncol. 2022, 16, 3034–3051. [Google Scholar] [CrossRef]
- Bush, N.G.; Evans-Roberts, K.; Maxwell, A. DNA Topoisomerases. EcoSal Plus 2015, 6. [Google Scholar] [CrossRef]
- Collin, F.; Karkare, S.; Maxwell, A. Exploiting bacterial DNA gyrase as a drug target: Current state and perspectives. Appl. Microbiol. Biotechnol. 2011, 92, 479–497. [Google Scholar] [CrossRef] [Green Version]
- Gilmour, D.S. Topoisomerase I interacts with transcribed regions in Drosophila cells. Cell 1986, 44, 401–407. [Google Scholar] [CrossRef]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [Green Version]
- Laponogov, I.; Veselkov, D.A.; Crevel, I.M.-T.; Pan, X.-S.; Fisher, L.M.; Sanderson, M.R. Structure of an ‘open’ clamp type II topoisomerase-DNA complex provides a mechanism for DNA capture and transport. Nucleic Acids Res. 2013, 41, 9911–9923. [Google Scholar] [CrossRef] [PubMed]
- Redinbo, M.R.; Stewart, L.; Kuhn, P.; Champoux, J.J.; Hol, W.G.J. Crystal Structures of Human Topoisomerase I in Covalent and Noncovalent Complexes with DNA. Science 1998, 279, 1504–1513. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.L.; Hsieh, C.-M.; Chan, N.-L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef]
- Nicum, S.J.; O’brien, M.E. Topotecan for the treatment of small-cell lung cancer. Expert Rev. Anticancer. Ther. 2007, 7, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Brave, M.; Dagher, R.; Farrell, A.; Abraham, S.; Ramchandani, R.; Gobburu, J.; Booth, B.; Jiang, X.; Sridhara, R.; Justice, R.; et al. Topotecan in combination with cisplatin for the treatment of stage IVB, recurrent, or persistent cervical cancer. Oncology 2006, 20, 1401–1404, 1410; discussion 1410–1411, 1415–1416. [Google Scholar]
- Mathijssen, R.H.; Van Alphen, R.J.; Verweij, J.; Loos, W.J.; Nooter, K.; Stoter, G.; Sparreboom, A. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin. Cancer Res. 2001, 7, 2182–2194. [Google Scholar]
- Wang-Gillam, A.; Li, C.-P.; Bodoky, G.; Dean, A.; Shan, Y.-S.; Jameson, G.; Macarulla, T.; Lee, K.-H.; Cunningham, D.; Blanc, J.F.; et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): A global, randomised, open-label, phase 3 trial. Lancet 2015, 387, 545–557. [Google Scholar] [CrossRef]
- Milano, G.; Innocenti, F.; Minami, H. Liposomal irinotecan (Onivyde): Exemplifying the benefits of nanotherapeutic drugs. Cancer Sci. 2022, 113, 2224–2231. [Google Scholar] [CrossRef]
- Chen, A.Y.; Liu, L.F. DNA topoisomerases: Essential enzymes and lethal targets. Annu. Rev. Pharmacol. Toxicol. 1994, 34, 191–218. [Google Scholar] [CrossRef]
- Kathiravan, M.K.; Khilare, M.M.; Nikoomanesh, K.; Chothe, A.S.; Jain, K.S. Topoisomerase as target for antibacterial and anticancer drug discovery. J. Enzym. Inhib. Med. Chem. 2012, 28, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Froelich-Ammon, S.J.; Osheroff, N. Topoisomerase Poisons: Harnessing the Dark Side of Enzyme Mechanism. J. Biol. Chem. 1995, 270, 21429–21432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evison, B.J.; Sleebs, B.E.; Watson, K.G.; Phillips, D.R.; Cutts, S.M. Mitoxantrone, More than Just Another Topoisomerase II Poison. Med. Res. Rev. 2015, 36, 248–299. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The Good, the Bad and the Ugly Effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [Green Version]
- Kowalczykowski, S.C. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a016410. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [Green Version]
- Kantidze, O.L.; Velichko, A.K.; Luzhin, A.V.; Petrova, N.V.; Razin, S.V. Synthetically Lethal Interactions of ATM, ATR, and DNA-PKcs. Trends Cancer 2018, 4, 755–768. [Google Scholar] [CrossRef]
- Hartley, K.O.; Gell, D.; Smith, G.C.; Zhang, H.; Divecha, N.; A Connelly, M.; Admon, A.; Lees-Miller, S.P.; Anderson, C.W.; Jackson, S.P. DNA-dependent protein kinase catalytic subunit: A relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell 1995, 82, 849–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, T.M.; Jackson, S.P. The DNA-dependent protein kinase: Requirement for DNA ends and association with Ku antigen. Cell 1993, 72, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Dvir, A.; Peterson, S.R.; Knuth, M.W.; Lu, H.; Dynan, W.S. Ku autoantigen is the regulatory component of a template-associated protein kinase that phosphorylates RNA polymerase II. Proc. Natl. Acad. Sci. USA 1992, 89, 11920–11924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimori, T.; Akizuki, M.; Yamagata, H.; Inada, S.; Yoshida, S.; Homma, M. Characterization of a high molecular weight acidic nuclear protein recognized by autoantibodies in sera from patients with polymyositis-scleroderma overlap. J. Clin. Investig. 1981, 68, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Featherstone, C.; Jackson, S.P. Ku, a DNA repair protein with multiple cellular functions? Mutat. Res. 1999, 434, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; MacDonald, J.J.; Lees-Miller, S.; Tjian, R. GC box binding induces phosphorylation of Sp1 by a DNA-dependent protein kinase. Cell 1990, 63, 155–165. [Google Scholar] [CrossRef]
- Jiang, W.; Crowe, J.L.; Liu, X.; Nakajima, S.; Wang, Y.; Li, C.; Lee, B.J.; Dubois, R.L.; Liu, C.; Yu, X.; et al. Differential Phosphorylation of DNA-PKcs Regulates the Interplay between End-Processing and End-Ligation during Nonhomologous End-Joining. Mol. Cell 2015, 58, 172–185. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Reddy, Y.V.R.; Wang, W.; Woods, T.; Douglas, P.; Ramsden, D.A.; Lees-Miller, S.P.; Meek, K. Autophosphorylation of the Catalytic Subunit of the DNA-Dependent Protein Kinase Is Required for Efficient End Processing during DNA Double-Strand Break Repair. Mol. Cell. Biol. 2003, 23, 5836–5848. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, R.A.; Myler, L.R.; Soniat, M.M.; Makharashvili, N.; Lee, L.; Lees-Miller, S.P.; Finkelstein, I.J.; Paull, T.T. DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP. Sci. Adv. 2020, 6, eaay0922. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef]
- Neal, J.A.; Dang, V.; Douglas, P.; Wold, M.S.; Lees-Miller, S.P.; Meek, K. Inhibition of Homologous Recombination by DNA-Dependent Protein Kinase Requires Kinase Activity, Is Titratable, and Is Modulated by Autophosphorylation. Mol. Cell. Biol. 2011, 31, 1719–1733. [Google Scholar] [CrossRef] [Green Version]
- Rosenzweig, K.E.; Youmell, M.B.; Palayoor, S.T.; Price, B.D. Radiosensitization of human tumor cells by the phosphatidylinositol3-kinase inhibitors wortmannin and LY294002 correlates with inhibition of DNA-dependent protein kinase and prolonged G2-M delay. Clin. Cancer Res. 1997, 3, 1149–1156. [Google Scholar] [PubMed]
- Hashimoto, M.; Rao, S.; Tokuno, O.; Yamamoto, K.-I.; Takata, M.; Takeda, S.; Utsumi, H. DNA-PK: The Major Target for Wortmannin-mediated Radiosensitization by the Inhibition of DSB Repair via NHEJ Pathway. J. Radiat. Res. 2003, 44, 151–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenke, F.T.; Zimmermann, A.; Sirrenberg, C.; Dahmen, H.; Kirkin, V.; Pehl, U.; Grombacher, T.; Wilm, C.; Fuchss, T.; Amendt, C.; et al. Pharmacologic Inhibitor of DNA-PK, M3814, Potentiates Radiotherapy and Regresses Human Tumors in Mouse Models. Mol. Cancer Ther. 2020, 19, 1091–1101. [Google Scholar] [CrossRef] [Green Version]
- Smithson, M.; Irwin, R.K.; Williams, G.; McLeod, M.C.; Choi, E.K.; Ganguly, A.; Pepple, A.; Cho, C.S.; Willey, C.D.; Leopold, J.; et al. Inhibition of DNA-PK may improve response to neoadjuvant chemoradiotherapy in rectal cancer. Neoplasia 2022, 25, 53–61. [Google Scholar] [CrossRef]
- Wang, M.; Chen, S.; Wei, Y.; Wei, X. DNA-PK inhibition by M3814 enhances chemosensitivity in non-small cell lung cancer. Acta Pharm. Sin. B 2021, 11, 3935–3949. [Google Scholar] [CrossRef]
- Gordhandas, S.B.; Manning-Geist, B.; Henson, C.; Iyer, G.; Gardner, G.J.; Sonoda, Y.; Moore, K.N.; Aghajanian, C.; Chui, M.H.; Grisham, R.N. Pre-clinical activity of the oral DNA-PK inhibitor, peposertib (M3814), combined with radiation in xenograft models of cervical cancer. Sci. Rep. 2022, 12, 974. [Google Scholar] [CrossRef]
- Wise, H.C.; Iyer, G.V.; Moore, K.; Temkin, S.M.; Gordon, S.; Aghajanian, C.; Grisham, R.N. Activity of M3814, an Oral DNA-PK Inhibitor, In Combination with Topoisomerase II Inhibitors in Ovarian Cancer Models. Sci. Rep. 2019, 9, 18882. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y. Development and Evolution of DNA-Dependent Protein Kinase Inhibitors toward Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 4264. [Google Scholar] [CrossRef]
- Bubenik, M.; Mader, P.; Mochirian, P.; Vallée, F.; Clark, J.; Truchon, J.-F.; Perryman, A.L.; Pau, A.; Kurinov, I.; Zahn, K.E.; et al. Identification of RP-6685, an Orally Bioavailable Compound that Inhibits the DNA Polymerase Activity of Poltheta. J. Med. Chem. 2022, 65, 13198–13215. [Google Scholar] [CrossRef]
- Saleh, N.; Copur, M.S. Overview: FDA Approval of Olaparib Maintenance for BRCA-Mutated Ovarian Cancer. Oncology 2019, 33, 293–294. [Google Scholar]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keisner, S.V. Rucaparib and olaparib for the treatment of prostate cancer: A clinician’s guide to choice of therapy. J. Oncol. Pharm. Pract. 2022, 28, 1624–1633. [Google Scholar] [CrossRef]
- Wolford, J.E.; Bai, J.; Moore, K.N.; Kristeleit, R.; Monk, B.J.; Tewari, K.S. Cost-effectiveness of niraparib, rucaparib, and olaparib for treatment of platinum-resistant, recurrent ovarian carcinoma. Gynecol. Oncol. 2020, 157, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.; Goble, S.; Isaacson, J.; Maloney, L.; Cameron, T.; Bedel, J. Comment on: “Cost-Effectiveness of Niraparib Versus Routine Surveillance, Olaparib and Rucaparib for the Maintenance Treatment of Patients with Ovarian Cancer in the United States”. Pharmacoeconomics 2019, 37, 1065–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ison, G.; Howie, L.J.; Amiri-Kordestani, L.; Zhang, L.; Tang, S.; Sridhara, R.; Pierre, V.; Charlab, R.; Ramamoorthy, A.; Song, P.; et al. FDA Approval Summary: Niraparib for the Maintenance Treatment of Patients with Recurrent Ovarian Cancer in Response to Platinum-Based Chemotherapy. Clin. Cancer. Res. 2018, 24, 4066–4071. [Google Scholar] [CrossRef] [Green Version]
- Hoy, S.M. Talazoparib: First Global Approval. Drugs 2018, 78, 1939–1946. [Google Scholar] [CrossRef]
- Nam, A.-R.; Jin, M.H.; Park, J.E.; Bang, J.-H.; Oh, D.-Y.; Bang, Y.-J. Therapeutic Targeting of the DNA Damage Response Using an ATR Inhibitor in Biliary Tract Cancer. Cancer Res. Treat. 2019, 51, 1167–1179. [Google Scholar] [CrossRef] [Green Version]
- Lecona, E.; Fernandez-Capetillo, O. Targeting ATR in cancer. Nat. Rev. Cancer 2018, 18, 586–595. [Google Scholar] [CrossRef]
- Lee, J.H.; Paull, T.T. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 2004, 304, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.V.; Graham, M.E.; Peng, C.; Chen, P.; Robinson, P.J.; Lavin, M.F. Involvement of novel autophosphorylation sites in ATM activation. EMBO J. 2006, 25, 3504–3514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Räschle, M.; Knipscheer, P.; Enoiu, M.; Angelov, T.; Sun, J.; Griffith, J.D.; Ellenberger, T.E.; Schärer, O.D.; Walter, J.C. Mechanism of Replication-Coupled DNA Interstrand Crosslink Repair. Cell 2008, 134, 969–980. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Cortez, D.; Elledge, S.J. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 2002, 16, 198–208. [Google Scholar] [CrossRef] [Green Version]
- Bartek, J.; Lukas, C.; Lukas, J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004, 5, 792–804. [Google Scholar] [CrossRef]
- O’Connell, M.J.; Raleigh, J.M.; Verkade, H.; Nurse, P. Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J. 1997, 16, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Matheson, C.J.; Backos, D.S.; Reigan, P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol. Sci. 2016, 37, 872–881. [Google Scholar] [CrossRef]
- Geenen, J.J.; Schellens, J.H. Molecular Pathways: Targeting the Protein Kinase Wee1 in Cancer. Clin. Cancer Res. 2017, 23, 4540–4544. [Google Scholar] [CrossRef] [Green Version]
- Moiseeva, T.N.; Qian, C.; Sugitani, N.; Osmanbeyoglu, H.U.; Bakkenist, C.J. WEE1 kinase inhibitor AZD1775 induces CDK1 kinase-dependent origin firing in unperturbed G1- and S-phase cells. Proc. Natl. Acad. Sci. USA 2019, 116, 23891–23893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez-Kelly, R.; Martín, Y.; Koundrioukoff, S.; Tanenbaum, M.E.; Smits, V.A.; Medema, R.; Debatisse, M.; Freire, R. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J. Cell Biol. 2011, 194, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.; Nähse-Kumpf, V.; Larsen, M.S.Y.; O’Hanlon, K.A.; Patzke, S.; Holmberg, C.; Mejlvang, J.; Groth, A.; Nielsen, O.; Syljuåsen, R.G.; et al. Cyclin-Dependent Kinase Suppression by WEE1 Kinase Protects the Genome through Control of Replication Initiation and Nucleotide Consumption. Mol. Cell. Biol. 2012, 32, 4226–4236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Zhuang, L.; Wang, Y.; Jiang, Y.; Tu, Z.; Dong, C.; Chen, Y.; Zhu, Y. Inhibitors of cell cycle checkpoint target Wee1 kinase—A patent review (2003–2022). Expert Opin. Ther. Pat. 2022, 32, 1217–1244. [Google Scholar] [CrossRef] [PubMed]
- Kong, A.; Mehanna, H. WEE1 Inhibitor: Clinical Development. Curr. Oncol. Rep. 2021, 23, 107. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Hirai, H.; Iwasawa, Y.; Okada, M.; Arai, T.; Nishibata, T.; Kobayashi, M.; Kimura, T.; Kaneko, N.; Ohtani, J.; Yamanaka, K.; et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 2009, 8, 2992–3000. [Google Scholar] [CrossRef] [Green Version]
- Leijen, S.; van Geel, R.M.; Pavlick, A.C.; Tibes, R.; Rosen, L.; Razak, A.R.A.; Lam, R.; Demuth, T.; Rose, S.; Lee, M.A.; et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination with Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4371–4380. [Google Scholar] [CrossRef]
- Li, Z.; Pinch, B.J.; Olson, C.M.; Donovan, K.A.; Nowak, R.P.; Mills, C.E.; Scott, D.A.; Doctor, Z.M.; Eleuteri, N.A.; Chung, M.; et al. Development and Characterization of a Wee1 Kinase Degrader. Cell Chem. Biol. 2019, 27, 57–65.e9. [Google Scholar] [CrossRef]
- Manasaryan, G.; Suplatov, D.; Pushkarev, S.; Drobot, V.; Kuimov, A.; Švedas, V.; Nilov, D. Bioinformatic Analysis of the Nicotinamide Binding Site in Poly(ADP-Ribose) Polymerase Family Proteins. Cancers 2021, 13, 1201. [Google Scholar] [CrossRef]
- Vyas, S.; Chang, P. New PARP targets for cancer therapy. Nat. Rev. Cancer 2014, 14, 502–509. [Google Scholar] [CrossRef]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5. [Google Scholar] [CrossRef]
- Isabelle, M.; Moreel, X.; Gagné, J.-P.; Rouleau, M.; Ethier, C.; Gagné, P.; Hendzel, M.J.; Poirier, G.G. Investigation of PARP-1, PARP-2, and PARG interactomes by affinity-purification mass spectrometry. Proteome Sci. 2010, 8, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.-S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegan, D.C.; Lu, Y.; Stachelek, G.C.; Crosby, M.E.; Bindra, R.S.; Glazer, P.M. Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc. Natl. Acad. Sci. USA 2010, 107, 2201–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Petit, S.A.; Ficarro, S.B.; Toomire, K.J.; Xie, A.; Lim, E.; Cao, S.A.; Park, E.; Eck, M.J.; Scully, R.; et al. PARP1-Driven Poly-ADP-Ribosylation Regulates BRCA1 Function in Homologous Recombination–Mediated DNA Repair. Cancer Discov. 2014, 4, 1430–1447. [Google Scholar] [CrossRef] [Green Version]
- Anscher, M.S.; Chang, E.; Gao, X.; Gong, Y.; Weinstock, C.; Bloomquist, E.; Adeniyi, O.; Charlab, R.; Zimmerman, S.; Serlemitsos-Day, M.; et al. FDA Approval Summary: Rucaparib for the Treatment of Patients with Deleterious BRCA-Mutated Metastatic Castrate-Resistant Prostate Cancer. Oncol. 2020, 26, 139–146. [Google Scholar] [CrossRef]
- Balasubramaniam, S.; Beaver, J.A.; Horton, S.; Fernandes, L.L.; Tang, S.; Horne, H.N.; Liu, J.; Liu, C.; Schrieber, S.J.; Yu, J.; et al. FDA Approval Summary: Rucaparib for the Treatment of Patients with Deleterious BRCA Mutation–Associated Advanced Ovarian Cancer. Clin. Cancer Res. 2017, 23, 7165–7170. [Google Scholar] [CrossRef] [Green Version]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; Van Der Burg, E.; Nygren, A.O.H.; Zander, S.A.L.; Derksen, P.W.B.; De Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, C.R.; Almassy, R.; Barton, S.; Batey, M.A.; Calvert, A.H.; Canan-Koch, S.; Durkacz, B.W.; Hostomsky, Z.; Kumpf, R.A.; Kyle, S.; et al. Anticancer Chemosensitization and Radiosensitization by the Novel Poly(ADP-ribose) Polymerase-1 Inhibitor AG14361. Gynecol. Oncol. 2004, 96, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Daniel, R.A.; Rozanska, A.L.; Thomas, H.D.; Mulligan, E.A.; Drew, Y.; Castelbuono, D.J.; Hostomsky, Z.; Plummer, E.R.; Boddy, A.V.; Tweddle, D.A.; et al. Inhibition of Poly(ADP-Ribose) Polymerase-1 Enhances Temozolomide and Topotecan Activity against Childhood Neuroblastoma. Clin. Cancer Res. 2009, 15, 1241–1249. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Zhou, F.; Jiang, F.; Lu, H.; Wang, J.; Cheng, C. PARP inhibitor reduces proliferation and increases apoptosis in breast cancer cells. Chin. J. Cancer Res. 2014, 26, 142–147. [Google Scholar]
- Sachdev, E.; Tabatabai, R.; Roy, V.; Rimel, B.J.; Mita, M.M. PARP Inhibition in Cancer: An Update on Clinical Development. Target. Oncol. 2019, 14, 657–679. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Wyatt, D.W.; Takata, K.-I.; Mu, Y.; Hensley, S.C.; Tomida, J.; Bylund, G.O.; Doublié, S.; Johansson, E.; Ramsden, D.A.; et al. Mechanism of Suppression of Chromosomal Instability by DNA Polymerase POLQ. PLOS Genet. 2014, 10, e1004654. [Google Scholar] [CrossRef] [Green Version]
- Roerink, S.F.; van Schendel, R.; Tijsterman, M. Polymerase theta-mediated end joining of replication-associated DNA breaks in C. elegans. Genome Res. 2014, 24, 954–962. [Google Scholar] [CrossRef] [Green Version]
- Schimmel, J.; van Schendel, R.; Dunnen, J.T.D.; Tijsterman, M. Templated Insertions: A Smoking Gun for Polymerase Theta-Mediated End Joining. Trends Genet. 2019, 35, 632–644. [Google Scholar] [CrossRef]
- Higgins, G.S.; Prevo, R.; Lee, Y.-F.; Helleday, T.; Muschel, R.J.; Taylor, S.; Yoshimura, M.; Hickson, I.D.; Bernhard, E.J.; McKenna, W.G. A Small Interfering RNA Screen of Genes Involved in DNA Repair Identifies Tumor-Specific Radiosensitization by POLQ Knockdown. Cancer Res. 2010, 70, 2984–2993. [Google Scholar] [CrossRef] [Green Version]
- Lemée, F.; Bergoglio, V.; Fernandez-Vidal, A.; Machado-Silva, A.; Pillaire, M.-J.; Bieth, A.; Gentil, C.; Baker, L.; Martin, A.-L.; Leduc, C.; et al. DNA polymerase theta up-regulation is associated with poor survival in breast cancer, perturbs DNA replication, and promotes genetic instability. Proc. Natl. Acad. Sci. USA 2010, 107, 13390–13395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyatt, D.W.; Feng, W.; Conlin, M.P.; Yousefzadeh, M.J.; Roberts, S.A.; Mieczkowski, P.; Wood, R.D.; Gupta, G.P.; Ramsden, D.A. Essential Roles for Polymerase theta-Mediated End Joining in the Repair of Chromosome Breaks. Mol. Cell 2016, 63, 662–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yücel, H.; Davis, R.E.; Färkkilä, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. A first-in-class polymerase theta inhibitor selectively targets homologous-recombination-deficient tumors. Nat. Cancer 2021, 2, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Zatreanu, D.; Robinson, H.M.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Perryman, A.L.; Pau, V.; Kurinov, I.; Zahn, K.E.; et al. Poltheta inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat. Commun. 2021, 12, 3636. [Google Scholar] [CrossRef]
- Cobb, J.A.; Bjergbaek, L. RecQ helicases: Lessons from model organisms. Nucleic Acids Res. 2006, 34, 4106–4114. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Davis, A.J. Human RecQ Helicases in DNA Double-Strand Break Repair. Front. Cell Dev. Biol. 2021, 9, 640755. [Google Scholar] [CrossRef]
- Hanada, K.; Hickson, I.D. Molecular genetics of RecQ helicase disorders. Cell. Mol. Life Sci. 2007, 64, 2306–2322. [Google Scholar] [CrossRef]
- He, Y.-J.; Qiao, Z.-Y.; Gao, B.; Zhang, X.-H.; Wen, Y.-Y. Association between RECQL5 genetic polymorphisms and susceptibility to breast cancer. Tumor Biol. 2014, 35, 12201–12204. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Kundu, S.; Laskar, S.; Choudhury, Y.; Ghosh, S.K. Assessment of DNA repair susceptibility genes identified by whole exome sequencing in head and neck cancer. DNA Repair 2018, 66–67, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Fewings, E.; Larionov, A.; Redman, J.; Goldgraben, M.A.; Scarth, J.; Richardson, S.; Brewer, C.; Davidson, R.; Ellis, I.; Evans, D.G.; et al. Germline pathogenic variants in PALB2 and other cancer-predisposing genes in families with hereditary diffuse gastric cancer without CDH1 mutation: A whole-exome sequencing study. Lancet Gastroenterol. Hepatol. 2018, 3, 489–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Shamanna, R.A.; Keijzers, G.; Anand, R.; Rasmussen, L.J.; Cejka, P.; Croteau, D.L.; Bohr, V.A. RECQL4 Promotes DNA End Resection in Repair of DNA Double-Strand Breaks. Cell Rep. 2016, 16, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Shamanna, R.A.; de Freitas, J.K.; Okur, M.; Khadka, P.; Kulikowicz, T.; Holland, P.P.; Tian, J.; Croteau, D.L.; Davis, A.J.; et al. Cell cycle-dependent phosphorylation regulates RECQL4 pathway choice and ubiquitination in DNA double-strand break repair. Nat. Commun. 2017, 8, 2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamanna, R.A.; Singh, D.K.; Lu, H.; Mirey, G.; Keijzers, G.; Salles, B.; Croteau, D.L.; Bohr, V.A. RECQ helicase RECQL4 participates in non-homologous end joining and interacts with the Ku complex. Carcinogenesis 2014, 35, 2415–2424. [Google Scholar] [CrossRef] [Green Version]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ Is a Mechanistically Distinct Pathway of Mammalian Chromosome Break Repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, M.; Banerjee, T.; Sommers, J.A.; Iannascoli, C.; Pichierri, P.; Shoemaker, R.H.; Brosh, R.M., Jr. Werner Syndrome Helicase Has a Critical Role in DNA Damage Responses in the Absence of a Functional Fanconi Anemia Pathway. Cancer Res. 2013, 73, 5497–5507. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, M.; Sommers, J.A.; Shoemaker, R.H.; Brosh, R.M., Jr. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc. Natl. Acad. Sci. USA 2011, 108, 1525–1530. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Peng, X.; Daley, J.; Yang, L.; Shen, J.; Nguyen, N.; Bae, G.; Niu, H.; Peng, Y.; Hsieh, H.-J.; et al. Inhibition of DNA2 nuclease as a therapeutic strategy targeting replication stress in cancer cells. Oncogenesis 2017, 6, e319. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Zhou, M.; Li, Z.; Li, H.; Polaczek, P.; Dai, H.; Wu, Q.; Liu, C.; Karanja, K.K.; Popuri, V.; et al. A Selective Small Molecule DNA2 Inhibitor for Sensitization of Human Cancer Cells to Chemotherapy. Ebiomedicine 2016, 6, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Cheung-Ong, K.; Giaever, G.; Nislow, C. DNA-Damaging Agents in Cancer Chemotherapy: Serendipity and Chemical Biology. Chem. Biol. 2013, 20, 648–659. [Google Scholar] [CrossRef] [Green Version]
- Burris, H.A. Dual Kinase Inhibition in the Treatment of Breast Cancer: Initial Experience with the EGFR/ErbB-2 Inhibitor Lapatinib. Oncologist 2004, 9, 10–15. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [Green Version]
- Santoro, A.; Rimassa, L.; Borbath, I.; Daniele, B.; Salvagni, S.; Van Laethem, J.L.; Van Vlierberghe, H.; Trojan, J.; Kolligs, F.T.; Weiss, A.; et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: A randomised, placebo-controlled phase 2 study. Lancet Oncol. 2012, 14, 55–63. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 719–726. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Yauch, R.L.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; et al. Inhibition of the Hedgehog Pathway in Advanced Basal-Cell Carcinoma. N. Engl. J. Med. 2009, 361, 1164–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konkel, M.J.; Packiarajan, M.; Chen, H.; Topiwala, U.P.; Jimenez, H.; Talisman, I.J.; Coate, H.; Walker, M.W. Amino substituted analogs of 1-phenyl-3-phenylimino-2-indolones with potent galanin Gal3 receptor binding affinity and improved solubility. Bioorg. Med. Chem. Lett. 2006, 16, 3950–3954. [Google Scholar] [CrossRef] [PubMed]
- Izbicka, E.; Izbicki, T. Therapeutic strategies for the treatment of neuroblastoma. Curr. Opin. Investig. Drugs (Lond. Engl. 2000) 2005, 6, 1200–1214. [Google Scholar]
- García-Vallejo, J.J.; Ambrosini, M.; Overbeek, A.; van Riel, W.E.; Bloem, K.; Unger, W.W.; Chiodo, F.; Bolscher, J.G.; Nazmi, K.; Kalay, H.; et al. Multivalent glycopeptide dendrimers for the targeted delivery of antigens to dendritic cells. Mol. Immunol. 2013, 53, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Krishna, T.S.; Kong, X.-P.; Gary, S.; Burgers, P.M.; Kuriyan, J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell 1994, 79, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Schurtenberger, P.; Egelhaaf, S.; Hindges, R.; Maga, G.; Jónsson, Z.O.; May, R.P.; Glatter, O.; Hübscher, U. The solution structure of functionally active human proliferating cell nuclear antigen determined by small-angle neutron scattering. J. Mol. Biol. 1998, 275, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Warbrick, E. The puzzle of PCNA’s many partners. Bioessays 2000, 22, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Waga, S.; Hannon, G.J.; Beach, D.; Stillman, B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature 1994, 369, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Ducoux, M.; Urbach, S.; Baldacci, G.; Hübscher, U.; Koundrioukoff, S.; Christensen, J.; Hughes, P. Mediation of proliferating cell nuclear antigen (PCNA)-dependent DNA replication through a conserved p21(Cip1)-like PCNA-binding motif present in the third subunit of human DNA polymerase delta. J. Biol. Chem. 2001, 276, 49258–49266. [Google Scholar] [CrossRef] [Green Version]
- Warbrick, E.; Lane, D.P.; Glover, D.M.; Cox, L.S. Homologous regions of Fen1 and p21Cip1 compete for binding to the same site on PCNA: A potential mechanism to co-ordinate DNA replication and repair. Oncogene 1997, 14, 2313–2321. [Google Scholar] [CrossRef] [Green Version]
- Chuang, L.S.-H.; Ian, H.-I.; Koh, T.-W.; Ng, H.-H.; Xu, G.; Li, B.F.L. Human DNA-(Cytosine-5) Methyltransferase-PCNA Complex as a Target for p21WAF1. Science 1997, 277, 1996–2000. [Google Scholar] [CrossRef]
- Levin, D.S.; McKenna, A.E.; Motycka, T.A.; Matsumoto, Y.; Tomkinson, A.E. Interaction between PCNA and DNA ligase I is critical for joining of Okazaki fragments and long-patch base-excision repair. Curr. Biol. 2000, 10, 919–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jónsson, Z.O.; Hindges, R.; Hübscher, U. Regulation of DNA replication and repair proteins through interaction with the front side of proliferating cell nuclear antigen. EMBO J. 1998, 17, 2412–2425. [Google Scholar] [CrossRef] [Green Version]
- Strzalka, W.; Ziemienowicz, A. Proliferating cell nuclear antigen (PCNA): A key factor in DNA replication and cell cycle regulation. Ann. Bot. 2010, 107, 1127–1140. [Google Scholar] [CrossRef] [Green Version]
- Punchihewa, C.; Inoue, A.; Hishiki, A.; Fujikawa, Y.; Connelly, M.; Evison, B.; Shao, Y.; Heath, R.; Kuraoka, I.; Rodrigues, P.; et al. Identification of Small Molecule Proliferating Cell Nuclear Antigen (PCNA) Inhibitor That Disrupts Interactions with PIP-box Proteins and Inhibits DNA Replication. J. Biol. Chem. 2012, 287, 14289–14300. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Wortman, M.; Dillehay, K.L.; Seibel, W.L.; Evelyn, C.R.; Smith, S.J.; Malkas, L.H.; Zheng, Y.; Lu, S.; Dong, Z. Small-Molecule Targeting of Proliferating Cell Nuclear Antigen Chromatin Association Inhibits Tumor Cell Growth. Mol. Pharmacol. 2012, 81, 811–819. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Lo, Y.-H.; Ma, L.; Waltz, S.E.; Gray, J.K.; Hung, M.-C.; Wang, S.-C. Targeting Tyrosine Phosphorylation of PCNA Inhibits Prostate Cancer Growth. Mol. Cancer Ther. 2011, 10, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Hoelz, D.J.; Arnold, R.J.; Dobrolecki, L.E.; Abdel-Aziz, W.; Loehrer, A.P.; Novotny, M.V.; Schnaper, L.; Hickey, R.J.; Malkas, L.H. The discovery of labile methyl esters on proliferating cell nuclear antigen by MS/MS. Proteomics 2006, 6, 4808–4816. [Google Scholar] [CrossRef] [PubMed]
- Lingeman, R.G.; Hickey, R.J.; Malkas, L.H. Expression of a novel peptide derived from PCNA damages DNA and reverses cisplatin resistance. Cancer Chemother. Pharmacol. 2014, 74, 981–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Chu, P.; Lingeman, R.; McDaniel, H.; Kechichian, S.; Hickey, R.J.; Liu, Z.; Yuan, Y.-C.; Sandoval, J.A.; Fields, G.B.; et al. The Mechanism by Which MYCN Amplification Confers an Enhanced Sensitivity to a PCNA-Derived Cell Permeable Peptide in Neuroblastoma Cells. Ebiomedicine 2015, 2, 1923–1931. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.J.; Hickey, R.J.; Malkas, L.H. Validating the disruption of proliferating cell nuclear antigen interactions in the development of targeted cancer therapeutics. Cancer Biol. Ther. 2016, 17, 310–319. [Google Scholar] [CrossRef] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Ashwell, S.; Zabludoff, S. DNA Damage Detection and Repair Pathways—Recent Advances with Inhibitors of Checkpoint Kinases in Cancer Therapy. Clin. Cancer Res. 2008, 14, 4032–4037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roccaro, A.; Hideshima, T.; Richardson, P.; Russo, D.; Ribatti, D.; Vacca, A.; Dammacco, F.; Anderson, K. Bortezomib as an Antitumor Agent. Curr. Pharm. Biotechnol. 2006, 7, 441–448. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]

 RPA and
RPA and  DNA damage. MRN: Mre11/Rad50/Nbs1 complex; 53BP1: p53-binding protein 1; PTIP: Pax transactivation domain-interacting protein; RIF1: Replication Timing Regulatory Factor 1; CtIP: CtBP (carboxy-terminal binding protein) interacting protein; LIGI: DNA ligase 1; LIG3: DNA ligase 3; XRCC4: X-ray Repair Cross Complementing 4; XLF: XRCC4-like factor; APE1: DNA (apurinic/apyrimidinic site) endonuclease 1; Polβ: DNA polymerase β; BARD1: BRCA1 associated RING domain 1; MH: microhomology.
RPA and DNA damage. MRN: Mre11/Rad50/Nbs1 complex; 53BP1: p53-binding protein 1; PTIP: Pax transactivation domain-interacting protein; RIF1: Replication Timing Regulatory Factor 1; CtIP: CtBP (carboxy-terminal binding protein) interacting protein; LIGI: DNA ligase 1; LIG3: DNA ligase 3; XRCC4: X-ray Repair Cross Complementing 4; XLF: XRCC4-like factor; APE1: DNA (apurinic/apyrimidinic site) endonuclease 1; Polβ: DNA polymerase β; BARD1: BRCA1 associated RING domain 1; MH: microhomology.
DNA damage. MRN: Mre11/Rad50/Nbs1 complex; 53BP1: p53-binding protein 1; PTIP: Pax transactivation domain-interacting protein; RIF1: Replication Timing Regulatory Factor 1; CtIP: CtBP (carboxy-terminal binding protein) interacting protein; LIGI: DNA ligase 1; LIG3: DNA ligase 3; XRCC4: X-ray Repair Cross Complementing 4; XLF: XRCC4-like factor; APE1: DNA (apurinic/apyrimidinic site) endonuclease 1; Polβ: DNA polymerase β; BARD1: BRCA1 associated RING domain 1; MH: microhomology.
RPA and DNA damage. MRN: Mre11/Rad50/Nbs1 complex; 53BP1: p53-binding protein 1; PTIP: Pax transactivation domain-interacting protein; RIF1: Replication Timing Regulatory Factor 1; CtIP: CtBP (carboxy-terminal binding protein) interacting protein; LIGI: DNA ligase 1; LIG3: DNA ligase 3; XRCC4: X-ray Repair Cross Complementing 4; XLF: XRCC4-like factor; APE1: DNA (apurinic/apyrimidinic site) endonuclease 1; Polβ: DNA polymerase β; BARD1: BRCA1 associated RING domain 1; MH: microhomology.

{kind=link}
{kind=link}
| Target | Agent | Cancer Type | Phase |
|---|---|---|---|
| ATR | Berzosertib | Lung Cancer | Phase II (Sources: clinicaltrials.gov) Access date: 30 May 2023 |
| AZD6738 | Bile duct cancer Clear cell renal cell carcinoma Breast cancer | Phase II (Sources: clinicaltrials.gov) Access date: 30 May 2023 | |
| BAY1895344 | Advanced solid tumor Non-Hodgkin’s lymphoma Mantle cell lymphoma | Phase I (Sources: clinicaltrials.gov) Access date: 30 May 2023 | |
| M4344 | Recurrent ovarian cancer | Phase I (Sources: clinicaltrials.gov) Access date: 30 May 2023 | |
| Chk1 | Prexasertib | Ovarian cancer Triple-negative breast cancer Small cell lung cancer | Phase II (Sources: clinicaltrials.gov) Access date: 30 May 2023 |
| SRA737 | Advanced solid tumors Non-Hodgkin’s lymphoma | Phase 1/II (Sources: clinicaltrials.gov) Access date: 30 May 2023 | |
| WEE1 | AZD1775 | Advanced solid tumor Refractory solid tumor Triple-negative breast cancer Ovarian cancer Pancreatic cancer | Phase II (Sources: clinicaltrials.gov) Access date: 30 May 2023 |
| PLK1 | Volasertib | Myeloid acute leukemia | Phase III (Sources: clinicaltrials.gov) Access date: 30 May 2023 |
| Onvansertib | Colorectal cancer Breast cancer Pancreatic cancer Small cell lung cancer | Phase II (Sources: clinicaltrials.gov) Access date: 30 May 2023 | |
| DNA-PK | AZD7648 | Advanced malignancies | Phase I (completed) (Sources: clinicaltrials.gov) Access date: 30 May 2023 |
| M3814 | Pancreatic cancer Prostate cancer Locally Advanced Rectal Cancer | Phase II (Sources: clinicaltrials.gov) Access date: 30 May 2023 | |
| DNA polymerase theta | NVB | Tumors That Have Alterations in DNA Repair Genes | Phase I (Sources: clinicaltrials.gov) Access date: 30 May 2023 |
| ART4215 | Advanced or Metastatic Solid Tumors | Phase I/II (Sources: clinicaltrials.gov) Access date: 30 May 2023 | |
| RP-6685 | BRCA-mutant breast and ovarian cancers | Preclinical development [91] | |
| PARP | Olaparib | BRCA-mutant breast cancer Ovarian cancer Prostate cancer | Approved drug [92,93] |
| Rucaparib | BRCA-mutant prostate cancer Recurrent Ovarian Cancer BRCA-mutant Ovarian cancer | Approved drug [94,95,96] | |
| Niraparib | Epithelial ovarian, Fallopian tube, or primary peritoneal cancer | Approved drug [97] | |
| Talazoparib | BRCA-mutant HER2-negative breast cancer | Approved drug [98] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, L.; Hickey, R.J.; Malkas, L.H. Therapeutic Targeting of DNA Replication Stress in Cancer. Genes 2023, 14, 1346. https://doi.org/10.3390/genes14071346
Gu L, Hickey RJ, Malkas LH. Therapeutic Targeting of DNA Replication Stress in Cancer. Genes. 2023; 14(7):1346. https://doi.org/10.3390/genes14071346
Chicago/Turabian StyleGu, Long, Robert J. Hickey, and Linda H. Malkas. 2023. "Therapeutic Targeting of DNA Replication Stress in Cancer" Genes 14, no. 7: 1346. https://doi.org/10.3390/genes14071346