
Genetic Basis of Inflammatory Demyelinating Diseases of the Central Nervous System: Multiple Sclerosis and Neuromyelitis Optica Spectrum
 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
- Diffuse or multifocal deficit;
- Sudden or subacute onset, particularly in young adults;
- Onset within a few weeks of infection or vaccination;
- Relapsing-remitting symptoms;
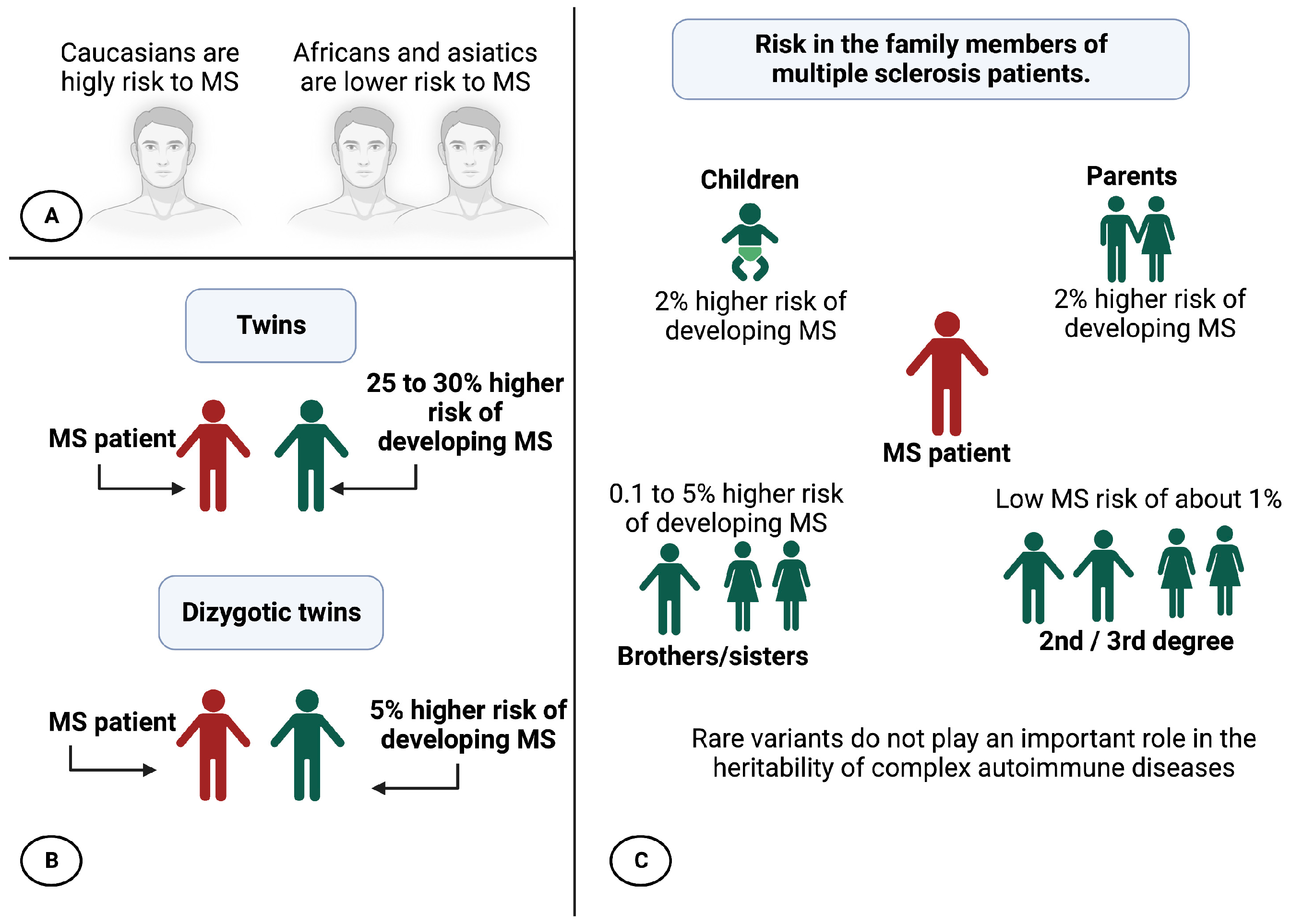
2. Epidemiology and Prevalence of MS
3. Influence of Migration in MS
4. Factors That Increase the Risk of MS
5. MS a Multifactorial Disease
5.1. Sun and Vitamin D
5.2. Viral Exposure
- The hygiene hypothesis postulates that a succession of infections by different pathogens during childhood would protect against the disease. At the same time, first contact with these same viruses in adulthood would trigger MS. This hypothesis is currently the most unifying.
- The prevalence hypothesis postulates that a more common pathogen in regions with a high prevalence of the disease is the cause of the disease. This pathogen would be globally present and cause persistent asymptomatic infection until the onset of symptoms in rare cases several years after the primary infection.
5.3. The Genetic Component of MS
- The common disease/common variants hypothesis [15,16]: the genetic predisposition to common diseases is determined by a few genetic variants, frequent in the population (frequency greater than 5%), each of which would confer only a low risk of developing the disease, with an odds ratio (OR) of between 1.1 and 1.5.
- The hypothesis of multiple rare variants or hypothesis of heterogeneity [17]: the genetic predisposition to frequent diseases is due to a combination of rare variants in the population (frequency between 0.1% and 5%) but each with a strong effect, with an OR of between 1.5 and 20.
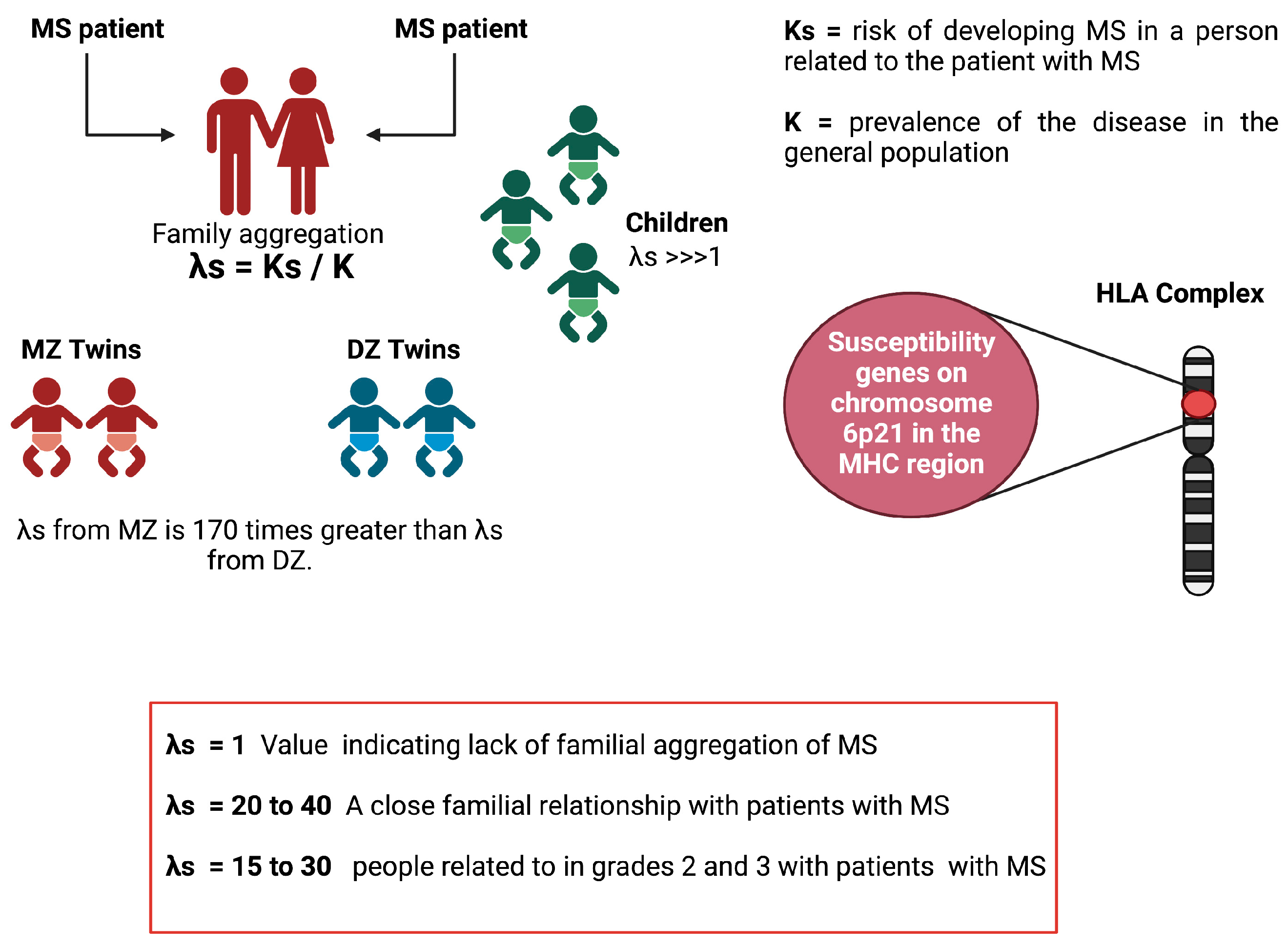
6. The Locus of the MHC and MS
6.1. Linkage Studies
6.2. Association Studies
- to the confirmation of the association of 23 of the 26 suspicious chromosomal regions, highlighted by the studies published between 2007 and 2010;
- to the identification of 29 new regions;
- to the highlighting of five potentials. Each of the identified variants confers only a low risk of developing the disease, 1.1 to 1.3 times higher than that of a non-carrier individual.
6.3. Heritability Calculation
- Unidentified gene interactions (also called epistasis phenomena);
- The importance of gene interactions.
6.4. Epistasis
6.5. Gene-Environment Interactions: GxE
6.6. Lambda-S Parameter
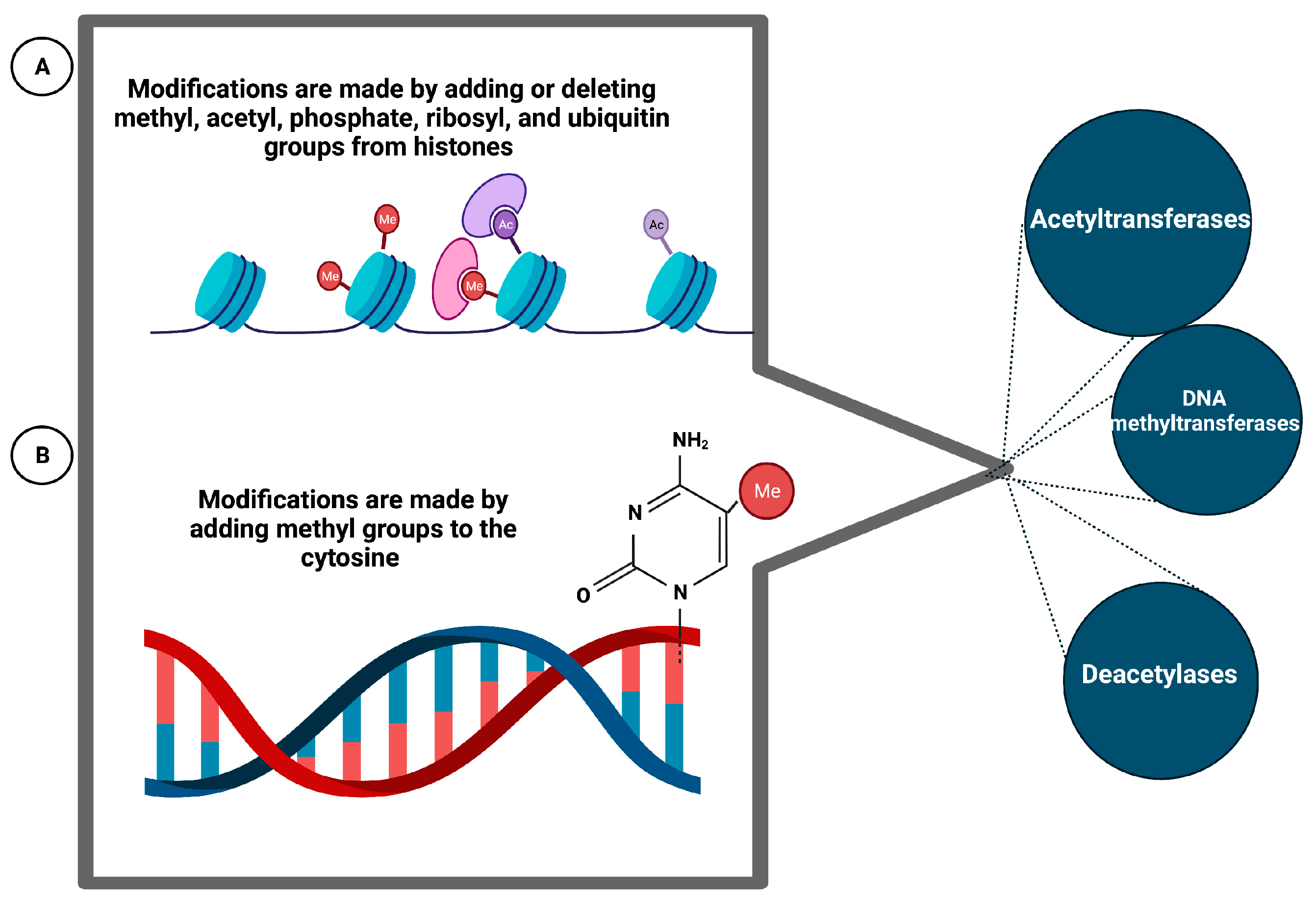
6.7. Epigenetic Phenomena
7. Genetic Changes in MS, Connections and Mechanism of Action
8. Genetic Factors of the NMOSD
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; van der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS; 3rd ed. Mult. Scler. J. 2020, 26, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.P.; Fraser, M.; Deans, J.; Clayton, D.; Walker, N.; Compston, D.A. Age-adjusted recurrence risks for relatives of patients with multiple sclerosis. Brain 1996, 119 Pt 2, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Tourbah, A. La Sclérose en Plaques—Aujourd’hui et Demain; Eurotext, J.L., Ed.; John Libbey Eurotext: Paris, France, 2003. [Google Scholar]
- Yang, J.; Hamade, M.; Wu, Q.; Wang, Q.; Axtell, R.; Giri, S.; Mao-Draayer, Y. Current and Future Biomarkers in Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 5877. [Google Scholar] [CrossRef]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk-Sowa, M.; Gebka-Kepinska, B.; Kepinski, M. Multiple Sclerosis—Risk Factors. Wiadomości Lekarskie 2020, 73, 2677–2682. [Google Scholar] [CrossRef]
- Handel, A.E.; Giovannoni, G.; Ebers, G.C.; Ramagopalan, S.V. Environmental factors and their timing in adult-onset multiple sclerosis. Nat. Rev. Neurol. 2010, 6, 156–166. [Google Scholar] [CrossRef]
- Kampman, M.T.; Wilsgaard, T.; Mellgren, S.I. Outdoor activities and diet in childhood and adolescence relate to MS risk above the Arctic Circle. J. Neurol. 2007, 254, 471–477. [Google Scholar] [CrossRef]
- Marrie, R.A. Environmental risk factors in multiple sclerosis aetiology. Lancet Neurol. 2004, 3, 709–718. [Google Scholar] [CrossRef]
- Sloka, S.; Silva, C.; Pryse-Phillips, W.; Patten, S.; Metz, L.; Yong, V.W. A quantitative analysis of suspected environmental causes of MS. Can. J. Neurol. Sci. 2011, 38, 98–105. [Google Scholar] [CrossRef] [Green Version]
- van der Mei, I.A.; Ponsonby, A.L.; Blizzard, L.; Dwyer, T. Regional variation in multiple sclerosis prevalence in Australia and its association with ambient ultraviolet radiation. Neuroepidemiology 2001, 20, 168–174. [Google Scholar] [CrossRef]
- Ascherio, A.; Munger, K.L. Environmental risk factors for multiple sclerosis. Part II: Noninfectious factors. Ann. Neurol. 2007, 61, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Munger, K.L. Environmental risk factors for multiple sclerosis. Part I: The role of infection. Ann. Neurol. 2007, 61, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Milo, R.; Kahana, E. Multiple sclerosis: Geoepidemiology, genetics and the environment. Autoimmun. Rev. 2010, 9, A387–A394. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Cox, N.J. The allelic architecture of human disease genes: Common disease-common variant...or not? Hum. Mol. Genet. 2002, 11, 2417–2423. [Google Scholar] [CrossRef] [Green Version]
- Reich, D.E.; Lander, E.S. On the allelic spectrum of human disease. Trends Genet. 2001, 17, 502–510. [Google Scholar] [CrossRef]
- Smith, D.J.; Lusis, A.J. The allelic structure of common disease. Hum. Mol. Genet. 2002, 11, 2455–2461. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.Y.; Barratt, B.J.; Clayton, D.G.; Todd, J.A. Genome-wide association studies: Theoretical and practical concerns. Nat. Rev. Genet. 2005, 6, 109–118. [Google Scholar] [CrossRef]
- Sahraian, M.A.; Radue, E.W.; Minagar, A. Neuromyelitis optica: Clinical manifestations and neuroimaging features. Neurol. Clin. 2013, 31, 139–152. [Google Scholar] [CrossRef]
- Lennon, V.A.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet 2004, 364, 2106–2112. [Google Scholar] [CrossRef]
- Clifford Allbutt, T. On the ophthalmoscopic signs of spinal disease. Lancet 1870, 95, 76–78. [Google Scholar] [CrossRef] [Green Version]
- Dreschfeld, J. Acute myélitis associated with optica neuritis. Lancet 1882, 119, 8. [Google Scholar] [CrossRef]
- Devic, E. Myélite subaiguë compliquée de névrite optique. Bull. Med. 1894, 8, 1033–1034. [Google Scholar]
- Gault, F. De la Neuromyélite Optique Aiguë; Rey, A., Ed.; Imprimeur de la Faculté de Médecine: Paris, France, 1894. [Google Scholar]
- Jarius, S.; Paul, F.; Weinshenker, B.G.; Levy, M.; Kim, H.J.; Wildemann, B. Neuromyelitis optica. Nat. Rev. Dis. Prim. 2020, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Mireles-Ramirez, M.A.; Cortes-Enriquez, F.; Valdivia-Tangarife, E.R.; Sanchez-Rosales, N.A.; Hernandez-Preciado, M.R.; Gonzalez-Rodriguez, C.H.; Garcia-Rivera, J.J.; Macias-Islas, M.A. Neuromyelitis optica spectrum disorder in Western Mexico. Mult. Scler. Relat. Disord. 2022, 61, 103733. [Google Scholar] [CrossRef]
- Zarei, S.; Eggert, J.; Franqui-Dominguez, L.; Carl, Y.; Boria, F.; Stukova, M.; Avila, A.; Rubi, C.; Chinea, A. Comprehensive review of neuromyelitis optica and clinical characteristics of neuromyelitis optica patients in Puerto Rico. Surg. Neurol. Int. 2018, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shlomo, Y. Multiple Sclerosis: Warren S., Warren K.G. Geneva: World Health Organization, 2001, pp. 123, SFr 35.00. ISBN: 92-4-156203-X. Int. J. Epidemiol. 2003, 32, 477. [Google Scholar] [CrossRef] [Green Version]
- Warren, S.; Warren, K.G.; World Health Organization. Multiple Sclerosis; Warren, S., Warren, K.G., Eds.; World Health Organization: Geneva, Switzerland, 2001. [Google Scholar]
- Oliva Ramirez, A.; Keenan, A.; Kalau, O.; Worthington, E.; Cohen, L.; Singh, S. Prevalence and burden of multiple sclerosis-related fatigue: A systematic literature review. BMC Neurol. 2021, 21, 468. [Google Scholar] [CrossRef]
- Nylander, A.; Hafler, D.A. Multiple sclerosis. J. Clin. Investig. 2012, 122, 1180–1188. [Google Scholar] [CrossRef] [Green Version]
- Benedikz, J.; Magnusson, H.; Guthmundsson, G. Multiple sclerosis in Iceland, with observations on the alleged epidemic in the Faroe Islands. Ann. Neurol. 1994, 36 (Suppl. 2), S175–S179. [Google Scholar] [CrossRef]
- Alonso, A.; Hernan, M.A. Temporal trends in the incidence of multiple sclerosis: A systematic review. Neurology 2008, 71, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Gale, C.R.; Martyn, C.N. Migrant studies in multiple sclerosis. Prog. Neurobiol. 1995, 47, 425–448. [Google Scholar] [CrossRef]
- Bhigjee, A.I.; Moodley, K.; Ramkissoon, K. Multiple sclerosis in KwaZulu Natal, South Africa: An epidemiological and clinical study. Mult. Scler. 2007, 13, 1095–1099. [Google Scholar] [CrossRef]
- Lavasani, S.; Dzhambazov, B.; Nouri, M.; Fak, F.; Buske, S.; Molin, G.; Thorlacius, H.; Alenfall, J.; Jeppsson, B.; Westrom, B. A novel probiotic mixture exerts a therapeutic effect on experimental autoimmune encephalomyelitis mediated by IL-10 producing regulatory T cells. PLoS ONE 2010, 5, e9009. [Google Scholar] [CrossRef] [Green Version]
- Whitacre, C.C. Sex differences in autoimmune disease. Nat. Immunol. 2001, 2, 777–780. [Google Scholar] [CrossRef]
- Orton, S.M.; Herrera, B.M.; Yee, I.M.; Valdar, W.; Ramagopalan, S.V.; Sadovnick, A.D.; Ebers, G.C.; Canadian Collaborative Study, G. Sex ratio of multiple sclerosis in Canada: A longitudinal study. Lancet Neurol. 2006, 5, 932–936. [Google Scholar] [CrossRef]
- Holick, M.F. Sunlight and vitamin D for bone health and prevention of autoimmune diseases, cancers and cardiovascular disease. Am. J. Clin. Nutr. 2004, 80, 1678S–1688S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes de Abreu, D.A.; Babron, M.C.; Rebeix, I.; Fontenille, C.; Yaouanq, J.; Brassat, D.; Fontaine, B.; Clerget-Darpoux, F.; Jehan, F.; Feron, F. Season of birth and not vitamin D receptor promoter polymorphisms is a risk factor for multiple sclerosis. Mult. Scler. 2009, 15, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortum; Wellcome Trust Case Control Consortum; Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Khoury, M.J. Evolving methods in genetic epidemiology. III. Gene-environment interaction in epidemiologic research. Epidemiol. Rev. 1997, 19, 33–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, K.A.; Mistry, V.; Bockett, N.A.; Ahmad, T.; Ban, M.; Barker, J.N.; Barrett, J.C.; Blackburn, H.; Brand, O.; Burren, O.; et al. Negligible impact of rare autoimmune-locus coding-region variants on missing heritability. Nature 2013, 498, 232–235. [Google Scholar] [CrossRef] [Green Version]
- Gourraud, P.A.; Harbo, H.F.; Hauser, S.L.; Baranzini, S.E. The genetics of multiple sclerosis: An up-to-date review. Immunol. Rev. 2012, 248, 87–103. [Google Scholar] [CrossRef] [Green Version]
- Naito, S.; Namerow, N.; Mickey, M.R.; Terasaki, P.I. Multiple Sclerosis: Association with HL-A3. Tissue Antigens 1972, 2, 1–4. [Google Scholar] [CrossRef]
- Jersild, C.; Svejgaard, A.; Fog, T. HL-A antigens and multiple sclerosis. Lancet 1972, 1, 1240–1241. [Google Scholar] [CrossRef] [PubMed]
- Bertrams, J.; Kuwert, E.K. HL-A Antigen Frequencies in Multiple Sclerosis. Eur. Neurol. 1972, 7, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Jersild, C.; Svejgaard, A.; Fog, T.; Ammitzboll, T. HL-A antigens and diseases. I. Multiple sclerosis. Tissue Antigens 1973, 3, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Barcellos, L.F.; Kamdar, B.B.; Ramsay, P.P.; DeLoa, C.; Lincoln, R.R.; Caillier, S.; Schmidt, S.; Haines, J.L.; Pericak-Vance, M.A.; Oksenberg, J.R.; et al. Clustering of autoimmune diseases in families with a high-risk for multiple sclerosis: A descriptive study. Lancet Neurol. 2006, 5, 924–931. [Google Scholar] [CrossRef]
- Yaouanq, J.; Semana, G.; Eichenbaum, S.; Quelvennec, E.; Roth, M.P.; Clanet, M.; Edan, G.; Clerget-Darpoux, F. Evidence for linkage disequilibrium between HLA-DRB1 gene and multiple sclerosis. The French Research Group on Genetic Susceptibility to MS. Science 1997, 276, 664–665. [Google Scholar] [CrossRef] [PubMed]
- Miretti, M.M.; Walsh, E.C.; Ke, X.; Delgado, M.; Griffiths, M.; Hunt, S.; Morrison, J.; Whittaker, P.; Lander, E.S.; Cardon, L.R.; et al. A high-resolution linkage-disequilibrium map of the human major histocompatibility complex and first generation of tag single-nucleotide polymorphisms. Am. J. Hum. Genet. 2005, 76, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Oksenberg, J.R.; Baranzini, S.E.; Sawcer, S.; Hauser, S.L. The genetics of multiple sclerosis: SNPs to pathways to pathogenesis. Nat. Rev. Genet. 2008, 9, 516–526. [Google Scholar] [CrossRef]
- Ramagopalan, S.V.; Ebers, G.C. Multiple sclerosis: Major histocompatibility complexity and antigen presentation. Genome Med. 2009, 1, 105. [Google Scholar] [CrossRef]
- Beecham, A.H.; McCauley, J.L. Fine-Mapping Array Design for Multi-Ethnic Studies of Multiple Sclerosis. Genes 2019, 10, 903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Multiple Sclerosis Genetics Consortium; Hafler, D.A.; Compston, A.; Sawcer, S.; Lander, E.S.; Daly, M.J.; De Jager, P.L.; de Bakker, P.I.; Gabriel, S.B.; Mirel, D.B.; et al. Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med. 2007, 357, 851–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Multiple Sclerosis Genetics Consortium; ANZgene; IIBDGC; WTCCC2. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaid, D.J.; Chen, W.; Larson, N.B. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 2018, 19, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.G.; Schmidt, S.; Seth, P.; Oksenberg, J.R.; Hart, J.; Prokop, A.; Caillier, S.J.; Ban, M.; Goris, A.; Barcellos, L.F.; et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat. Genet. 2007, 39, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Lundmark, F.; Duvefelt, K.; Iacobaeus, E.; Kockum, I.; Wallstrom, E.; Khademi, M.; Oturai, A.; Ryder, L.P.; Saarela, J.; Harbo, H.F.; et al. Variation in interleukin 7 receptor alpha chain (IL7R) influences risk of multiple sclerosis. Nat. Genet. 2007, 39, 1108–1113. [Google Scholar] [CrossRef]
- Baranzini, S.E.; Galwey, N.W.; Wang, J.; Khankhanian, P.; Lindberg, R.; Pelletier, D.; Wu, W.; Uitdehaag, B.M.; Kappos, L.; GeneMSA Consortium. Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Human. Mol. Gen. 2009, 18, 2078–2090. [Google Scholar] [CrossRef] [Green Version]
- Mitrovič, M.; Patsopoulos, N.A.; Beecham, A.H.; Dankowski, T.; Goris, A.; Dubois, B.; D’hooghe, M.B.; Lemmens, R.; Van Damme, P.; Søndergaard, H.B.; et al. Low-Frequency and Rare-Coding Variation Contributes to Multiple Sclerosis Risk. Cell 2018, 175, 1679–1687.e1677. [Google Scholar] [CrossRef] [Green Version]
- Steri, M.; Orrù, V.; Idda, M.L.; Pitzalis, M.; Pala, M.; Zara, I.; Sidore, C.; Faà, V.; Floris, M.; Deiana, M.; et al. Overexpression of the Cytokine BAFF and Autoimmunity Risk. N. Engl. J. Med. 2017, 376, 1615–1626. [Google Scholar] [CrossRef]
- De Jager, P.L.; Jia, X.; Wang, J.; de Bakker, P.I.W.; Ottoboni, L.; Aggarwal, N.T.; Piccio, L.; Raychaudhuri, S.; Tran, D.; Aubin, C.; et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat. Genet. 2009, 41, 776–782. [Google Scholar] [CrossRef] [Green Version]
- Sawcer, S. The complex genetics of multiple sclerosis: Pitfalls and prospects. Brain 2008, 131, 3118–3131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirota, M.; Schaub, M.A.; Batzoglou, S.; Robinson, W.H.; Butte, A.J. Autoimmune Disease Classification by Inverse Association with SNP Alleles. PLoS Genet. 2009, 5, e1000792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manolio, T.A.; Collins, F.S. The HapMap and genome-wide association studies in diagnosis and therapy. Annu. Rev. Med. 2009, 60, 443–456. [Google Scholar] [CrossRef] [Green Version]
- Hemminki, K.; Li, X.; Sundquist, K.; Sundquist, J. Familial risks for type 2 diabetes in Sweden. Diabetes Care 2010, 33, 293–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, B. Personal genomes: The case of the missing heritability. Nature 2008, 456, 18–21. [Google Scholar] [CrossRef] [Green Version]
- Marian, A.J. Elements of ‘missing heritability’. Curr. Opin. Cardiol. 2012, 27, 197–201. [Google Scholar] [CrossRef]
- Dempfle, A.; Scherag, A.; Hein, R.; Beckmann, L.; Chang-Claude, J.; Schäfer, H. Gene–environment interactions for complex traits: Definitions, methodological requirements and challenges. Eur. J. Hum. Genet. 2008, 16, 1164–1172. [Google Scholar] [CrossRef]
- Oksenberg, J.R.; Baranzini, S.E.; Barcellos, L.F.; Hauser, S.L. Multiple sclerosis: Genomic rewards. J. Neuroimmunol. 2001, 113, 171–184. [Google Scholar] [CrossRef]
- Dyment, D.A.; Ebers, G.C.; Sadovnick, A.D. Genetics of multiple sclerosis. Lancet Neurol. 2004, 3, 104–110. [Google Scholar] [CrossRef] [Green Version]
- Ebers, G.C.; Sadovnick, A.D.; Risch, N.J.; Canadian Collaborative Study Group. A genetic basis for familial aggregation in multiple sclerosis. Nature 1995, 377, 150–151. [Google Scholar] [CrossRef]
- Sadovnick, A.D.; Ebers, G.C.; Dyment, D.A.; Risch, N.J.; The Canadian Collaborative Study Group. Evidence for genetic basis of multiple sclerosis. Lancet 1996, 347, 1728–1730. [Google Scholar] [CrossRef]
- Robertson, N.P.; O’Riordan, J.I.; Chataway, J.; Kingsley, D.P.; Miller, D.H.; Clayton, D.; Compston, D.A. Offspring recurrence rates and clinical characteristics of conjugal multiple sclerosis. Lancet 1997, 349, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Dyment, D.A.; Risch, N.J.; Sadovnick, A.D.; Ebers, G.C.; Canadian Collaborative Study Group. Twin concordance and sibling recurrence rates in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 12877–12882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtuncu, M.; Tuzun, E. Multiple sclerosis: Could it be an epigenetic disease? Med. Hypotheses 2008, 71, 945–947. [Google Scholar] [CrossRef] [PubMed]
- Baranzini, S.E.; Mudge, J.; van Velkinburgh, J.C.; Khankhanian, P.; Khrebtukova, I.; Miller, N.A.; Zhang, L.; Farmer, A.D.; Bell, C.J.; Kim, R.W.; et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature 2010, 464, 1351–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagodic, M.; Colacios, C.; Nohra, R.; Dejean, A.S.; Beyeen, A.D.; Khademi, M.; Casemayou, A.; Lamouroux, L.; Duthoit, C.; Papapietro, O.; et al. A role for VAV1 in experimental autoimmune encephalomyelitis and multiple sclerosis. Sci. Transl. Med. 2009, 1, 10ra21. [Google Scholar] [CrossRef]
- Galarza-Munoz, G.; Briggs, F.B.S.; Evsyukova, I.; Schott-Lerner, G.; Kennedy, E.M.; Nyanhete, T.; Wang, L.; Bergamaschi, L.; Widen, S.G.; Tomaras, G.D.; et al. Human Epistatic Interaction Controls IL7R Splicing and Increases Multiple Sclerosis Risk. Cell 2017, 169, 72–84.e13. [Google Scholar] [CrossRef] [Green Version]
- Maier, L.M.; Lowe, C.E.; Cooper, J.; Downes, K.; Anderson, D.E.; Severson, C.; Clark, P.M.; Healy, B.; Walker, N.; Aubin, C.; et al. IL2RA genetic heterogeneity in multiple sclerosis and type 1 diabetes susceptibility and soluble interleukin-2 receptor production. PLoS Genet. 2009, 5, e1000322. [Google Scholar] [CrossRef]
- Gregory, A.P.; Dendrou, C.A.; Attfield, K.E.; Haghikia, A.; Xifara, D.K.; Butter, F.; Poschmann, G.; Kaur, G.; Lambert, L.; Leach, O.A.; et al. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature 2012, 488, 508–511. [Google Scholar] [CrossRef] [Green Version]
- Didonna, A.; Isobe, N.; Caillier, S.J.; Li, K.H.; Burlingame, A.L.; Hauser, S.L.; Baranzini, S.E.; Patsopoulos, N.A.; Oksenberg, J.R. A non-synonymous single-nucleotide polymorphism associated with multiple sclerosis risk affects the EVI5 interactome. Hum. Mol. Genet. 2015, 24, 7151–7158. [Google Scholar] [CrossRef] [Green Version]
- International Multiple Sclerosis Genetics Consortum. Network-based multiple sclerosis pathway analysis with GWAS data from 15,000 cases and 30,000 controls. Am. J. Hum. Genet. 2013, 92, 854–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussman, J.P.; Beecham, A.H.; Schmidt, M.; Martin, E.R.; McCauley, J.L.; Vance, J.M.; Haines, J.L.; Pericak-Vance, M.A. GWAS analysis implicates NF-kappaB-mediated induction of inflammatory T cells in multiple sclerosis. Genes. Immun. 2016, 17, 305–312. [Google Scholar] [CrossRef] [Green Version]
- International Multiple Sclerosis Genetics Consortum; Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masterman, T.; Ligers, A.; Olsson, T.; Andersson, M.; Olerup, O.; Hillert, J. HLA-DR15 is associated with lower age at onset in multiple sclerosis. Ann. Neurol. 2000, 48, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Fusco, C.; Andreone, V.; Coppola, G.; Luongo, V.; Guerini, F.; Pace, E.; Florio, C.; Pirozzi, G.; Lanzillo, R.; Ferrante, P.; et al. HLA-DRB1*1501 and response to copolymer-1 therapy in relapsing-remitting multiple sclerosis. Neurology 2001, 57, 1976–1979. [Google Scholar] [CrossRef]
- Tur, C.; Ramagopalan, S.; Altmann, D.R.; Bodini, B.; Cercignani, M.; Khaleeli, Z.; Miller, D.H.; Thompson, A.J.; Ciccarelli, O. HLA-DRB1*15 influences the development of brain tissue damage in early PPMS. Neurology 2014, 83, 1712–1718. [Google Scholar] [CrossRef] [Green Version]
- Healy, B.C.; Liguori, M.; Tran, D.; Chitnis, T.; Glanz, B.; Wolfish, C.; Gauthier, S.; Buckle, G.; Houtchens, M.; Stazzone, L.; et al. HLA B*44: Protective effects in MS susceptibility and MRI outcome measures. Neurology 2010, 75, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Isobe, N.; Keshavan, A.; Gourraud, P.A.; Zhu, A.H.; Datta, E.; Schlaeger, R.; Caillier, S.J.; Santaniello, A.; Lizee, A.; Himmelstein, D.S.; et al. Association of HLA Genetic Risk Burden With Disease Phenotypes in Multiple Sclerosis. JAMA Neurol. 2016, 73, 795–802. [Google Scholar] [CrossRef]
- Ogasawara, M.; Meguro, A.; Sakai, T.; Mizuki, N.; Takahashi, T.; Fujihara, K.; Tsuneoka, H.; Shikishima, K. Genetic analysis of the aquaporin-4 gene for anti-AQP4 antibody-positive neuromyelitis optica in a Japanese population. Jpn. J. Ophthalmol. 2016, 60, 198–205. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Azimi, T.; Taheri, M. A Comprehensive Review on the Role of Genetic Factors in Neuromyelitis Optica Spectrum Disorder. Front. Immunol. 2021, 12, 737673. [Google Scholar] [CrossRef]
- Zhou, L.; He, Z.; Zhu, L.; Zhu, J.J.; Zhu, J.H.; Pan, J. Association Analysis between HLA-DQA1 Loci and Neuromyelitis Optica Spectrum Disorder in a Han Chinese Population. Neurologist 2022, 27, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Romero-Hidalgo, S.; Flores-Rivera, J.; Rivas-Alonso, V.; Barquera, R.; Villarreal-Molina, M.T.; Antuna-Puente, B.; Macias-Kauffer, L.R.; Villalobos-Comparán, M.; Ortiz-Maldonado, J.; Yu, N.; et al. Native American ancestry significantly contributes to neuromyelitis optica susceptibility in the admixed Mexican population. Sci. Rep. 2020, 10, 13706. [Google Scholar] [CrossRef] [PubMed]
- Estrada, K.; Whelan, C.W.; Zhao, F.; Bronson, P.; Handsaker, R.E.; Sun, C.; Carulli, J.P.; Harris, T.; Ransohoff, R.M.; McCarroll, S.A.; et al. A whole-genome sequence study identifies genetic risk factors for neuromyelitis optica. Nat. Commun. 2018, 9, 1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Li, H.; Li, Y.; Dong, S.A.; Yi, M.; Zhang, Q.X.; Feng, B.; Yang, L.; Shi, F.D.; Yang, C.S. Multi-Level Analyses of Genome-Wide Association Study to Reveal Significant Risk Genes and Pathways in Neuromyelitis Optica Spectrum Disorder. Front. Genet. 2021, 12, 690537. [Google Scholar] [CrossRef]
- Mo, Y.; Wang, S.; Chang, Y.; Sun, X.; Liu, Z.; Sun, P.; Xu, Y.; Zhong, X.; Peng, L. A novel rare variant of CNPY3 from familial NMOSD impairs the TLR-mediated immune response. J. Neuroimmunol. 2023, 377, 578065. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Zhou, L.; Zhong, X.; Shi, Z.; Sun, X.; Wang, Y.; Li, R.; Long, Y.; Zhou, H.; Quan, C.; et al. Clinical and genetic analysis of familial neuromyelitis optica spectrum disorder in Chinese: Associated with ubiquitin-specific peptidase USP18 gene variants. J. Neurol. Neurosurg. Psychiatry 2022, 93, 1269–1275. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Associated Factors of Multiple Sclerosis | ||
|---|---|---|
| Environmental factors | Sunlight [4,6,7,8,9,10,11] | Inverse correlation between the prevalence of metabolic syndrome (MetS) and the annual rate of ultraviolet skin cancers related to exposure to sunlight were significantly less common in patients with MS than in matched controls, implying that greater exposure was protective against MS. |
| Vitamin D [4,6,12,13] |
| |
| Hygiene hypothesis [4,13] | The succession of infections by different pathogens during childhood would protect against the disease while a first contact with these same viruses in adulthood would trigger MS. | |
| Other factors [6,12,14] | Tobacco, fats (diet), chemical compounds | |
| Infective agents [15,16] | Epstein–Barr, Acinetobacter and Pseudomonas were significantly elevated in MS. | |
| Genetic factors | The common variants hypothesis [15,16] | The genetic predisposition is determined by few genetic variants, frequent in the population (greater than 5%) but each confers a low risk of developing the disease (OR 1.1 to 1.5). |
| The heterogeneity hypothesis [17,18] | The genetic predisposition is due to a combination of very rare variants in population (between 0.1% and 5%) but each confers a strong effect (OR 1.5 to 20). | |
| Major Histocompatibility Complex Locus Associated with MS | ||
|---|---|---|
| HLA-class I | Allele A3 Allele B7 | A3 allele has shown to be secondary to allele B7. It has been probable secondary association with HLA-DR2 and DQw6. |
| HLA-class II | HLA-DR2 DQw6 | HLA-DQB1*0602 is present in most populations with MS, it has not been possible to discern has any independent role in MS. HLA-DQA1*0102 and HLA-DRB1*1501 are the known association with the HLA class II DR2 haplotype. |
| HLA-DRB1 | HLA-DRB1 would act on the shape and charge of the antigen-binding site, and therefore could affect the efficiency of presentation of these antigens to cells. HLA-DRB1*15: The highest risk is attributed to HLA-DRB1*15 homozygotes, three to four times higher risk of developing MS. HLA-DRB1*08 allele only modestly increases the risk of developing MS. | |
| HLA-class III | NOTCH4 | A Japanese population genotyped for 3534 SNPs in the MHC region showed independent associations to both an HLA class III marker in the NOTCH4 gene. |
| Association Studies | ||
|---|---|---|
| Biological Pathway | Encodes | Characteristics |
| IL7RA | Interleukin 7 receptor subunit and 73 genes with putative relations. | Genes that had significantly associated single-nucleotide polymorphisms in an independent case-control dataset.
|
| IL2RA | Interleukin 2RA | Encoding the alpha chain of the interleukin-2 receptor. It is not a specific marker of regulatory T cells. The effect of IL2RA might be better described by several SNPs rather than by a single one. |
| Genetic Factors NMOSD | |
|---|---|
| Ethnicity HLA [93,94] | DRB116:02 in southern Han Chinese, Japanese, and southern Brazilian patients. DQB104:02 in a cohort of European descent. DRB104:05 in southern Brazilians. |
| Non-HLA [20,92,93] | PD-1, IL-17, IL-7R, CD6, and CD58. |
| Familial cases [96,97] | Including siblings, parent–child, and aunt–niece pairs, with more than 80% of them being female. HLA-A*31, B*61, *51, DRB1*0802, and DPB1*0501. More than 75% of cases had AQP4-IgG. |
| Genomic studies [93,96,97] | HLA-DQB1*05:02-DRB1*15:01” haplotype has been higher in the NMO group compared with controls. The SNP rs1964995 in the MHC region as a risk locus. Additionally, genotyped eight SNPs in AQP4 in a group of AQP4-IgG-positive. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz, G.G.; Torres-Mendoza, B.M.G.; Ramírez-Jirano, J.; Marquez-Pedroza, J.; Hernández-Cruz, J.J.; Mireles-Ramirez, M.A.; Torres-Sánchez, E.D. Genetic Basis of Inflammatory Demyelinating Diseases of the Central Nervous System: Multiple Sclerosis and Neuromyelitis Optica Spectrum. Genes 2023, 14, 1319. https://doi.org/10.3390/genes14071319
Ortiz GG, Torres-Mendoza BMG, Ramírez-Jirano J, Marquez-Pedroza J, Hernández-Cruz JJ, Mireles-Ramirez MA, Torres-Sánchez ED. Genetic Basis of Inflammatory Demyelinating Diseases of the Central Nervous System: Multiple Sclerosis and Neuromyelitis Optica Spectrum. Genes. 2023; 14(7):1319. https://doi.org/10.3390/genes14071319
Chicago/Turabian StyleOrtiz, Genaro Gabriel, Blanca M. G. Torres-Mendoza, Javier Ramírez-Jirano, Jazmin Marquez-Pedroza, José J. Hernández-Cruz, Mario A. Mireles-Ramirez, and Erandis D. Torres-Sánchez. 2023. "Genetic Basis of Inflammatory Demyelinating Diseases of the Central Nervous System: Multiple Sclerosis and Neuromyelitis Optica Spectrum" Genes 14, no. 7: 1319. https://doi.org/10.3390/genes14071319