
Lipid-Associated GWAS Loci Predict Antiatherogenic Effects of Rosuvastatin in Patients with Coronary Artery Disease

 , ,
, ,  ,
,
Abstract
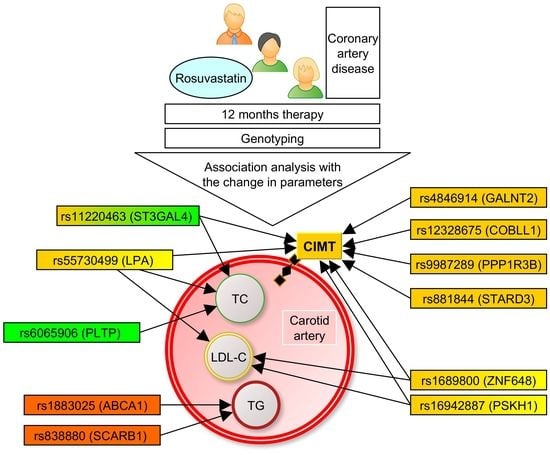
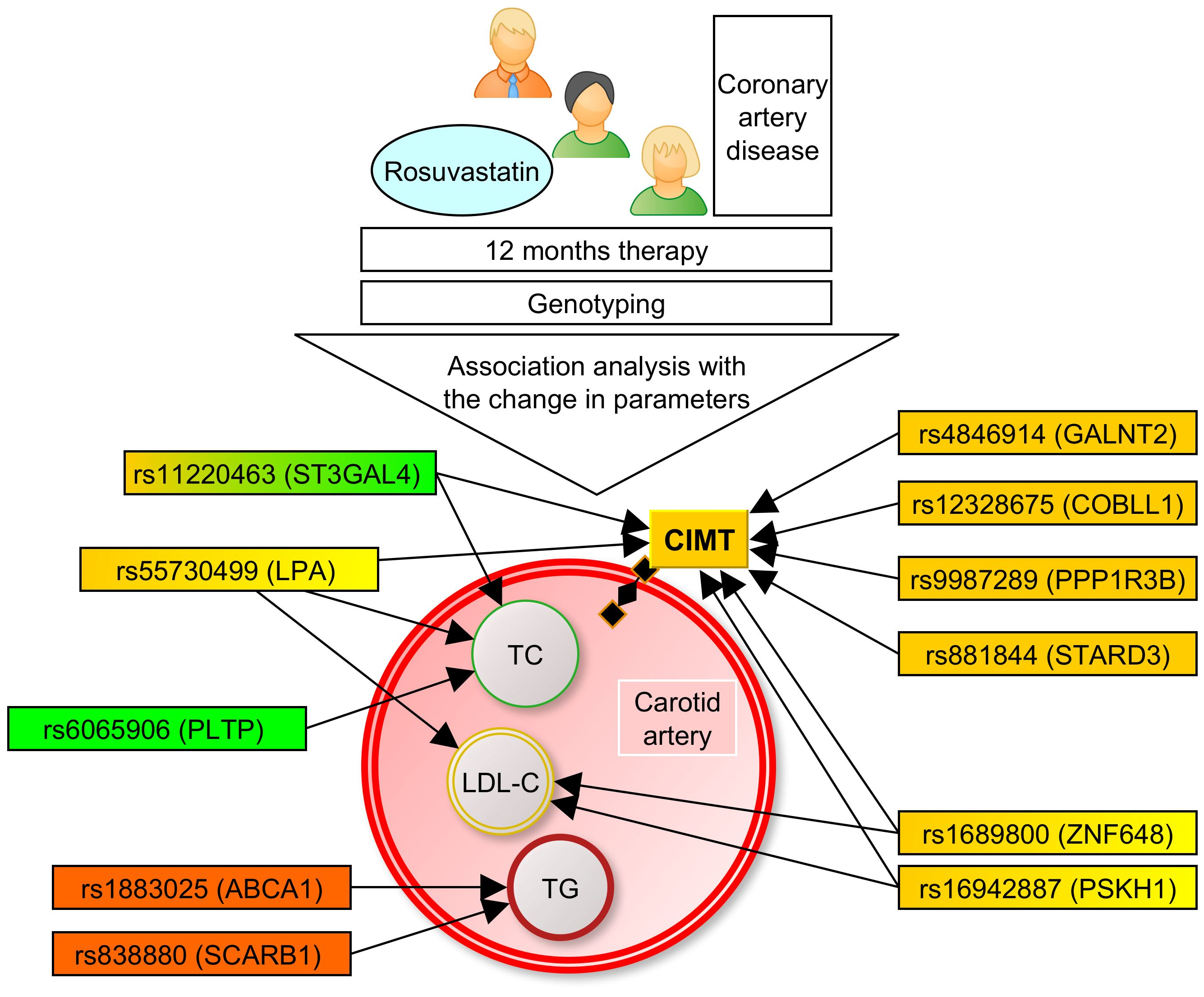
:
1. Introduction
2. Materials and Methods
3. Results
3.1. Associations of the SNPs with Lipid and CIMT Reduction during the 6-Month Therapy by Rosuvastatin
3.2. Associations of SNPs with Lipid and CIMT Reduction during 12-Month Therapy by Rosuvastatin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gupta, R.M.; Schnitzler, G.; Fang, S.; Lee-Kim, V.S.; Barry, A. Multiomic Analysis and CRISPR Perturbation Screens Identify Endothelial Cell Programs and Novel Therapeutic Targets for Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, H. Invited commentary: 30-year perspective on the seven countries study. Am. J. Epidemiol. 2017, 185, 1143–1147. [Google Scholar] [CrossRef]
- McPherson, R.; Tybjaerg-Hansen, A. Genetics of Coronary Artery Disease. Circ. Res. 2016, 118, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Churilin, M.I.; Kononov, S.I.; Mal, G.S.; Polonikov, A.V.; Lazarenko, V.A. Lipid metabolism genes and predisposition to ischemic heart disease. Med. News North Cauc. 2019, 14, 401–407. [Google Scholar] [CrossRef]
- Mutlu, A.S.; Duffy, J.; Wang, M.C. Lipid metabolism and lipid signals in aging and longevity. Dev. Cell. 2021, 56, 1394–1407. [Google Scholar] [CrossRef]
- Walsh, R.; Jurgens, S.J.; Erdmann, J.; Bezzina, C.R. Genome-wide association studies of cardiovascular disease. Physiol. Rev. 2023, 103, 2039–2055. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; Buchkovich, M.L.; Mora, S.; et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 2013, 45, 1274–1283. [Google Scholar]
- Nikpay, M.; Goel, A.; Won, H.H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. A comprehensive 1000 Genomes–based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar] [PubMed] [Green Version]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef] [Green Version]
- Kathiresan, S.; Melander, O.; Guiducci, C.; Surti, A.; Burtt, N.P.; Rieder, M.J.; Cooper, G.M.; Roos, C.; Voight, B.F.; Havulinna, A.S.; et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat. Genet. 2008, 40, 189–197. [Google Scholar] [CrossRef]
- Mack, S.; Coassin, S.; Rueedi, R.; Yousri, N.A.; Seppälä, I.; Gieger, C.; Schönherr, S.; Forer, L.; Erhart, G.; Marques-Vidal, P.; et al. A genome-wide association meta-analysis on lipoprotein (a) concentrations adjusted for apolipoprotein (a) isoforms. J. Lipid Res. 2017, 58, 1834–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gan, W.; Tian, C.; Li, H.; Lin, X.; Chen, Y. Association of PPP1R3B polymorphisms with blood lipid and C-reactive protein levels in a Chinese population. J. Diabetes 2013, 5, 275–281. [Google Scholar] [CrossRef]
- Postmus, I.; Trompet, S.; Deshmukh, H.A.; Arsenault, B.J.; Avery, C.L.; Bis, J.C.; Chasman, D.I.; de Keyser, C.E.; Deshmukh, H.A.; Evans, D.S.; et al. Pharmacogenetic meta-analysis of genome-wide association studies of LDL cholesterol response to statins. Nat. Commun. 2014, 5, 5068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chasman, D.I.; Giulianini, F.; MacFadyen, J.; Barratt, B.J.; Nyberg, F.; Ridker, P.M. Genetic determinants of statin-induced low-density lipoprotein cholesterol reduction: The Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Circ. Cardiovasc. Genet. 2012, 5, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Chasman, D.I.; Giulianini, F.; Demler, O.V.; Udler, M.S. Pleiotropy-Based Decomposition of Genetic Risk Scores: Association and Interaction Analysis for Type 2 Diabetes and CAD. Am. J. Hum. Genet. 2020, 106, 646–658. [Google Scholar] [CrossRef]
- Crouse, J.R., 3rd; Raichlen, J.S.; Riley, W.A.; Evans, G.W.; Palmer, M.K.; O’Leary, D.H.; Grobbee, D.E.; Bots, M.L.; METEOR Study Group. Effect of rosuvastatin on progression of carotid intima-media thickness in low-risk individuals with subclinical atherosclerosis: The METEOR Trial. JAMA 2007, 297, 1344–1353. [Google Scholar] [CrossRef] [Green Version]
- Nezu, T.; Hosomi, N.; Aoki, S.; Matsumoto, M. Carotid Intima-Media Thickness for Atherosclerosis. J. Atheroscler. Thromb. 2016, 23, 18–31. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.; Casas, J.P.; Drenos, F.; Whittaker, J.; Deanfield, J.; Swerdlow, D.I.; Holmes, M.V.; Kivimaki, M.; Langenberg, C.; Wareham, N.; et al. Causal relevance of blood lipid fractions in the development of carotid atherosclerosis: Mendelian randomization analysis. Circ. Cardiovasc. Genet. 2013, 6, 63–72. [Google Scholar] [CrossRef]
- Vargas, J.D.; Manichaikul, A.; Wang, X.Q.; Rich, S.S.; Rotter, J.I.; Post, W.S.; Polak, J.F.; Budoff, M.J.; Bluemke, D.A. Detailed analysis of association between common single nucleotide polymorphisms and subclinical atherosclerosis: The Multi-ethnic Study of Atherosclerosis. Data Brief. 2016, 7, 229–242. [Google Scholar] [CrossRef] [Green Version]
- Guaricci, A.I.; Arcadi, T.; Brunetti, N.D.; Maffei, E.; Montrone, D.; Martini, C.; De Luca, M.; De Rosa, F.; Cocco, D.; Midiri, M.; et al. Carotid intima media thickness and coronary atherosclerosis linkage in symptomatic intermediate risk patients evaluated by coronary computed tomography angiography. Int. J. Cardiol. 2014, 176, 988–993. [Google Scholar] [CrossRef]
- Amato, M.; Montorsi, P.; Ravani, A.; Oldani, E.; Galli, S.; Ravagnani, P.M.; Tremoli, E.; Baldassarre, D. Carotid intima-media thickness by B-mode ultrasound as surrogate of coronary atherosclerosis: Correlation with quantitative coronary angiography and coronary intravascular ultrasound findings. Eur. Heart J. 2007, 28, 2094–2101. [Google Scholar] [CrossRef] [Green Version]
- Granér, M.; Varpula, M.; Kahri, J.; Salonen, R.M.; Nyyssönen, K.; Nieminen, M.S.; Taskinen, M.R.; Syvänne, M. Association of carotid intima-media thickness with angiographic severity and extent of coronary artery disease. Am. J. Cardiol. 2006, 97, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, Y.; Kawano, H.; Koide, Y.; Baba, T.; Toda, G.; Seto, S.; Yano, K. Relationship of carotid intima-media thickness, pulse wave velocity, and ankle brachial index to the severity of coronary artery atherosclerosis. Clin Cardiol. 2004, 27, 629–634. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Raichlen, J.S.; Nicholls, S.J.; Erbel, R.; Tardif, J.C.; Brener, S.J.; Cain, V.A.; Nissen, S.E.; ASTEROID Investigators. Effect of rosuvastatin therapy on coronary artery stenoses assessed by quantitative coronary angiography: A study to evaluate the effect of rosuvastatin on intravascular ultrasound-derived coronary atheroma burden. Circulation 2008, 117, 2458–2466. [Google Scholar] [CrossRef] [Green Version]
- Puri, R.; Libby, P.; Nissen, S.E.; Wolski, K.; Ballantyne, C.M.; Barter, P.J.; Chapman, M.J.; Erbel, R.; Raichlen, J.S.; Uno, K.; et al. Long-term effects of maximally intensive statin therapy on changes in coronary atheroma composition: Insights from SATURN. Eur. Heart J. Cardiovasc. Imaging 2014, 15, 380–388. [Google Scholar] [CrossRef] [Green Version]
- Rumyantsev, N.A.; Kukes, V.G.; Kazakov, R.E.; Rumyantsev, A.A.; Sychev, D.A. Ispol’zovanie farmakogeneticheskogo testirovaniia dlia predotvrashcheniia nezhelatel’nykh lekarstvennykh reaktsiĭ pri terapii statinami [Use of pharmacogenetic testing to prevent adverse drug reactions during statin therapy]. Ter Arkh. 2017, 89, 82–87. (In Russian) [Google Scholar] [CrossRef]
- Soko, N.D.; Masimirembwa, C.; Dandara, C. Pharmacogenomics of Rosuvastatin: A Glocal (Global + Local) African Perspective and Expert Review on a Statin Drug. OMICS A J. Integr. Biol. 2016, 20, 498–509. [Google Scholar] [CrossRef]
- Alfonsi, J.E.; Hegele, R.A.; Gryn, S.E. Pharmacogenetics of lipid-lowering agents: Precision or indecision medicine? Curr. Atheroscler. Rep. 2016, 18, 24. [Google Scholar] [CrossRef]
- Churilin, M.I.; Kononov, S.I.; Luneva, Y.V.; Kazanov, V.A.; Azarova, I.E.; Klyosova, E.Y.; Bykanova, M.A.; Paschoalini, G.; Kharchenko, A.V.; Zhabin, S.N.; et al. Polymorphisms of Intracellular Cholesterol Transporters Genes: Relationship to Blood Lipid Levels, Carotid Intima-Media Thickness, and the Development of Coronary Heart Disease. Russ. J. Genet. 2020, 56, 234–241. [Google Scholar] [CrossRef]
- Churilin, M.I.; Kononov, S.I.; Luneva, Y.V.; Azarova, Y.E.; Klesova, E.Y.; Kharchenko, A.V.; Zhabin, S.N.; Bushueva, O.Y.; Povetkin, S.V.; Mal, G.S.; et al. Apolipoprotein E gene polymorphisms: A relationship with the risk of coronary artery disease and the effectiveness of lipid-lowering therapy with rosuvastatin. Cardiovasc. Therapy Prev. Russ. Fed. 2020, 19, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Kononov, S.; Mal, G.; Azarova, I.; Klyosova, E.; Bykanova, M.; Churnosov, M.; Polonikov, A. Pharmacogenetic loci for rosuvastatin are associated with intima-media thickness change and coronary artery disease risk. Pharmacogenomics 2022, 23, 15–34. [Google Scholar] [CrossRef]
- Lazarenko, V.; Churilin, M.; Azarova, I.; Klyosova, E.; Bykanova, M.; Ob’edkova, N.; Churnosov, M.; Bushueva, O.; Mal, G.; Povetkin, S.; et al. Comprehensive Statistical and Bioinformatics Analysis in the Deciphering of Putative Mechanisms by Which Lipid-Associated GWAS Loci Contribute to Coronary Artery Disease. Biomedicines 2022, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Pignoli, P.; Tremoli, E.; Poli, A.; Oreste, P.; Paoletti, R. Intimal plus medial thickness of the arterial wall: A direct measurement with ultrasound imaging. Circulation 1986, 74, 1399–1406. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudet, P.; Livstone, M.S.; Lewis, S.E.; Thomas, P.D. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief Bioinform. 2011, 12, 449–462. [Google Scholar] [CrossRef] [Green Version]
- Shahid, S.U.; Shabana Humphries, S. The SNP rs10911021 is associated with oxidative stress in coronary heart disease patients from Pakistan. Lipids Health Dis. 2018, 17, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khetarpal, S.A.; Schjoldager, K.T.; Christoffersen, C.; Raghavan, A.; Edmondson, A.C.; Reutter, H.M.; Ahmed, B.; Ouazzani, R.; Peloso, G.M.; Vitali, C.; et al. Loss of Function of GALNT2 Lowers High-Density Lipoproteins in Humans, Nonhuman Primates, and Rodents. Cell Metab. 2016, 24, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Zilmer, M.; Edmondson, A.C.; Khetarpal, S.A.; Alesi, V.; Zaki, M.S.; Rostasy, K.; Madsen, C.G.; Lepri, F.R.; Sinibaldi, L.; Cusmai, R.; et al. Novel congenital disorder of O-linked glycosylation caused by GALNT2 loss of function. Brain 2020, 143, 1114–1126. [Google Scholar] [CrossRef]
- Chen, J.; Guan, L.; Liu, H.; Liu, Q.; Fan, P.; Bai, H. GALNT2 Gene Variant rs4846914 Is Associated with Insulin and Insulin Resistance Depending on BMI in PCOS Patients: A Case-Control Study. Reprod Sci. 2021, 28, 1122–1132. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, H.; Zhang, J.; Han, D.; Zheng, Y.; Guo, X.; He, D.; Guo, J.; Wang, Y. Gene environment interaction of GALNT2 and APOE gene with hypertension in the Chinese Han Population. Biomed. Mater. Eng. 2015, 26 (Suppl. S1), S1977–S2083. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Neilson, L.J.; Zhong, H.; Murray, P.S.; Zanivan, S.; Zaidel-Bar, R. E-cadherin interactome complexity and robustness resolved by quantitative proteomics. Sci. Signal. 2014, 7, rs7. [Google Scholar] [CrossRef] [Green Version]
- Churilin, M.I. Association of RS12328675 COBLL1 polymorphism with coronary heart disease and intermediate phenotypes of atherosclerosis: Validation study in Central Russia. Res. Results Biomed. 2020, 6, 209–218. [Google Scholar] [CrossRef]
- Sakaue, S.; Kanai, M.; Tanigawa, Y.; Karjalainen, J.; Kurki, M.; Koshiba, S.; Narita, A.; Konuma, T.; Yamamoto, K.; Akiyama, M.; et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 2021, 53, 1415–1424. [Google Scholar] [CrossRef]
- Mishra, A.; Malik, R.; Hachiya, T.; Jürgenson, T.; Namba, S.; Posner, D.C.; Kamanu, F.K.; Koido, M.; Le Grand, Q.; Shi, M.; et al. Stroke genetics informs drug discovery and risk prediction across ancestries. Nature 2022, 611, 115–123, Erratum in Nature 2022, 612, E7. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, G.L.; Graff, M.; Nishimura, K.K.; Tao, R.; Haessler, J.; Gignoux, C.R.; Highland, H.M.; Patel, Y.M.; Sorokin, E.P.; Avery, C.L.; et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature 2019, 570, 514–518. [Google Scholar] [CrossRef]
- Sethwala, A.M.; Goh, I.; Amerena, J.V. Combating Inflammation in Cardiovascular Disease. Heart Lung Circ. 2021, 30, 197–206. [Google Scholar] [CrossRef]
- Lind, L. Genome-Wide Association Study of the Metabolic Syndrome in UK Biobank. Metab. Syndr. Relat. Disord. 2019, 17, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Spracklen, C.N.; Marenne, G.; Varshney, A.; Corbin, L.J.; Luan, J.; Willems, S.M.; Wu, Y.; Zhang, X.; Horikoshi, M.; et al. The trans-ancestral genomic architecture of glycemic traits. Nat. Genet. 2021, 53, 840–860. [Google Scholar] [CrossRef] [PubMed]
- Meza, C.A.; La Favor, J.D.; Kim, D.H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. [Google Scholar] [CrossRef] [Green Version]
- Clyne, A.M. Endothelial response to glucose: Dysfunction, metabolism, and transport. Biochem. Soc. Trans. 2021, 49, 313–325. [Google Scholar] [CrossRef]
- Krimbou, L.; Denis, M.; Haidar, B.; Carrier, M.; Marcil, M.; Genest, J., Jr. Molecular interactions between apoE and ABCA1: Impact on apoE lipidation. J. Lipid Res. 2004, 45, 839–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.Q.; Sallah, N.; Dunca, D.; Trivedi, B.; Hunt, K.A.; Hodgson, S.; Lambert, S.A.; Arciero, E.; Wright, J.; Griffiths, C.; et al. Transferability of genetic loci and polygenic scores for cardiometabolic traits in British Pakistani and Bangladeshi individuals. Nat. Commun. 2022, 13, 4664. [Google Scholar] [CrossRef]
- Hoffmann, T.J.; Theusch, E.; Haldar, T.; Ranatunga, D.K.; Jorgenson, E.; Medina, M.W.; Kvale, M.N.; Kwok, P.Y.; Schaefer, C.; Krauss, R.M.; et al. A large electronic-health-record-based genome-wide study of serum lipids. Nat. Genet. 2018, 50, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Byrne, E.M.; Zheng, Z.; Kemper, K.E.; Yengo, L.; Mallett, A.J.; Yang, J.; Visscher, P.M.; Wray, N.R. Genome-wide association study of medication-use and associated disease in the UK Biobank. Nat. Commun. 2019, 10, 1891. [Google Scholar] [CrossRef] [Green Version]
- Mondal, N.; Buffone, A., Jr.; Stolfa, G.; Antonopoulos, A.; Lau, J.T.; Haslam, S.M.; Dell, A.; Neelamegham, S. ST3Gal-4 is the primary sialyltransferase regulating the synthesis of E-, P-, and L-selectin ligands on human myeloid leukocytes. Blood 2015, 125, 687–696. [Google Scholar] [CrossRef] [Green Version]
- Ligthart, S.; Vaez, A.; Hsu, Y.H.; Inflammation Working Group of the CHARGE Consortium; PMI-WG-XCP; LifeLines Cohort Study; Stolk, R.; Uitterlinden, A.G.; Hofman, A.; Alizadeh, B.Z.; et al. Bivariate genome-wide association study identifies novel pleiotropic loci for lipids and inflammation. BMC Genom. 2016, 17, 443. [Google Scholar] [CrossRef] [Green Version]
- Ozbeyaz, N.B.; Gokalp, G.; Algul, E.; Kilic, P.; Saricam, O.; Aydinyilmaz, F.; Guliyev, I. Could Systemic Inflammation in Healthy Individuals with Obesity Indicate Subclinical Atherosclerosis? Angiology 2023, 74, 62–69. [Google Scholar] [CrossRef]
- Kivimäki, M.; Lawlor, D.A.; Smith, G.D.; Kumari, M.; Donald, A.; Britton, A.; Casas, J.P.; Shah, T.; Brunner, E.; Timpson, N.J.; et al. Does high C-reactive protein concentration increase atherosclerosis? The Whitehall II Study. PLoS ONE 2008, 3, e3013. [Google Scholar] [CrossRef] [Green Version]
- Moran, C.A.; Sheth, A.N.; Mehta, C.C.; Hanna, D.B.; Gustafson, D.R.; Plankey, M.W.; Mack, W.J.; Tien, P.C.; French, A.L.; Golub, E.T.; et al. The association of C-reactive protein with subclinical cardiovascular disease in HIV-infected and HIV-uninfected women. AIDS 2018, 32, 999–1006. [Google Scholar] [CrossRef]
- Brede, G.; Solheim, J.; Prydz, H. PSKH1, a novel splice factor compartment-associated serine kinase. Nucleic Acids Res. 2002, 30, 5301–5309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, L.P.; Wendling, C.; Védie, B.; Kobayashi, T.; Chenard, M.P.; Tomasetto, C.; Drin, G.; Alpy, F. STARD3 mediates endoplasmic reticulum-to-endosome cholesterol transport at membrane contact sites. EMBO J. 2017, 36, 1412–1433. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, Y.; Liu, X.; Zheng, N.; Gu, Y.; Song, Y.; Wang, X. Regulatory roles of external cholesterol in human airway epithelial mitochondrial function through STARD3 signalling. Clin. Transl. Med. 2022, 12, e902. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhai, X.; Li, J.; Albers, J.J.; Vuletic, S.; Ren, G. Structural basis of the lipid transfer mechanism of phospholipid transfer protein (PLTP). Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1082–1094. [Google Scholar] [CrossRef]
- Albers, J.J.; Vuletic, S.; Cheung, M.C. Role of plasma phospholipid transfer protein in lipid and lipoprotein metabolism. Biochim. Biophys. Acta 2012, 1821, 345–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Baseline Parameter | Mean ± Standard Deviation/ Median (Q1; Q3) |
|---|---|
| Age (years) | 61.0 ± 7.25 |
| Body mass index (kg/m2) | 28.77 ± 4.18 |
| Hypertension (%) | 97.5 |
| Past myocardial infarction (%) | 57.6 |
| Systolic blood pressure (mmHg) | 131.1 ± 8.1 |
| Diastolic blood pressure (mmHg) | 74.9 ± 4.4 |
| Total cholesterol (mmol/L) | 5.28 (4.60; 6.06) |
| LDL-C (mmol/L) | 3.27 (2.70; 4.08) |
| HDL-C (mmol/L) | 1.06 (0.97; 1.29) |
| TG (mmol/L) | 1.71 (1.22; 2.37) |
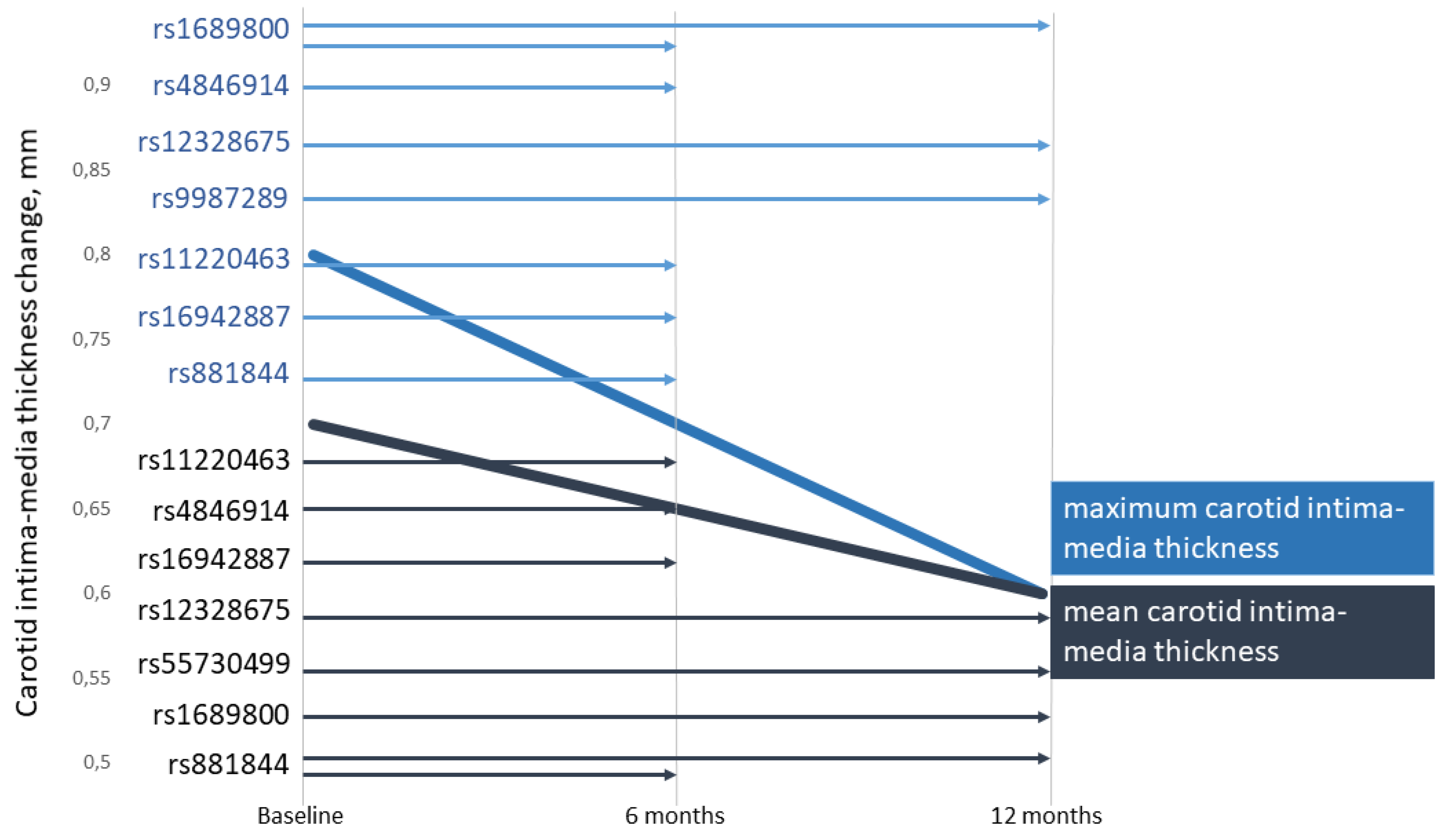
| CIMT, maximum (mm) | 0.80 (0.60; 1.00) |
| CIMT, mean (mm) | 0.70 (0.55; 0.85) |
| Chr | Gene (SNP ID) | Effect Allele | EAF | N | Total Cholesterol | LDL-C | CIMT, Maximum | CIMT, Mean | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Beta * | Pperm # | Beta * | Pperm # | Beta * | Pperm # | Beta * | Pperm # | |||||
| 1 | ZNF648 (rs1689800) | G | 0.392 | 115 | 0.020 | 0.1786 | 0.046 | 0.0493 A | −0.084 | 0.0234 R | −0.021 | 0.2308 |
| 1 | GALNT2 (rs4846914) | G | 0.388 | 115 | 0.004 | 0.7778 | −0.005 | 0.8571 | −0.045 | 0.0133 A | −0.038 | 0.0344 A |
| 2 | COBLL1 (rs12328675) | C | 0.170 | 111 | −0.003 | 0.8571 | −0.003 | 1.0000 | 0.035 | 0.2647 | 0.041 | 0.1000 |
| 6 | LPA (rs55730499) | T | 0.056 | 115 | 0.323 | 0.0022 R | 0.504 | 0.0224 R | −0.010 | 0.8571 | 0.028 | 0.8571 |
| 7 | NPC1L1 (rs217406) | G | 0.203 | 115 | 0.017 | 0.2308 | 0.033 | 0.3556 | −0.001 | 1.0000 | −0.003 | 1.0000 |
| 8 | PPP1R3B (rs9987289) | A | 0.086 | 115 | 0.019 | 0.3404 | −0.013 | 0.8571 | 0.041 | 0.3404 | 0.051 | 0.1550 |
| 9 | ABCA1 (rs1883025) | T | 0.263 | 115 | 0.001 | 1.0000 | −0.004 | 0.8571 | −0.024 | 0.2982 | −0.025 | 0.2535 |
| 11 | F2 (rs3136441) | C | 0.180 | 102 | 0.011 | 0.6429 | 0.025 | 0.7778 | 0.021 | 0.6923 | 0.023 | 0.2982 |
| 11 | ST3GAL4 (rs11220463) | T | 0.190 | 115 | −0.031 | 0.1280 | −0.068 | 0.0803 | 0.066 | 0.0159 D | 0.061 | 0.0243 A |
| 12 | SCARB1 (rs838880) | C | 0.336 | 115 | 0.002 | 1.0000 | 0.005 | 1.0000 | −0.014 | 0.7273 | −0.015 | 0.6923 |
| 16 | CETP (rs3764261) | A | 0.147 | 115 | 0.007 | 0.6250 | 0.027 | 0.8571 | −0.045 | 0.2466 | −0.042 | 0.1900 |
| 16 | PSKH1 (rs16942887) | A | 0.116 | 115 | 0.009 | 0.6429 | 0.025 | 0.4643 | 0.066 | 0.0421 D | 0.068 | 0.0483 D |
| 17 | STARD3 (rs881844) | C | 0.310 | 115 | 0.003 | 0.8571 | −0.038 | 0.1667 | 0.048 | 0.0086 A | 0.057 | 0.0033 A |
| 19 | LILRA3 (rs386000) | C | 0.203 | 115 | 0.006 | 0.5455 | −0.001 | 1.0000 | 0.015 | 0.7778 | 0.003 | 0.8571 |
| 20 | PLTP (rs6065906) | C | 0.160 | 115 | 0.140 | 0.0135 R | −0.008 | 1.0000 | −0.022 | 0.6923 | −0.022 | 0.5455 |
| Chr | Gene (SNP ID) | Effect Allele | EAF | N | 6-Month Period | 12-Month Period | ||
|---|---|---|---|---|---|---|---|---|
| Beta * | Pperm # | Beta * | Pperm # | |||||
| 1 | ZNF648 (rs1689800) | G | 0.392 | 114 | −0.6495 | 0.1148 | −0.1946 | 0.4643 |
| 1 | GALNT2 (rs4846914) | G | 0.388 | 114 | 0.4816 | 0.1919 | 0.05046 | 0.7778 |
| 2 | COBLL1 (rs12328675) | C | 0.170 | 110 | −0.2948 | 0.6250 | −0.1431 | 0.7778 |
| 6 | LPA (rs55730499) | T | 0.056 | 114 | −0.5923 | 0.4242 | 0.1825 | 0.6923 |
| 7 | NPC1L1 (rs217406) | G | 0.203 | 114 | 0.4106 | 0.4118 | −0.1208 | 0.6429 |
| 8 | PPP1R3B (rs9987289) | A | 0.086 | 114 | −0.9197 | 0.1887 | −0.7977 | 0.0756 |
| 9 | ABCA1 (rs1883025) | T | 0.263 | 114 | −0.177 | 0.5789 | −0.7246 | 0.0160 D |
| 11 | F2 (rs3136441) | C | 0.180 | 101 | −0.185 | 0.5789 | 0.02512 | 1.0000 |
| 11 | ST3GAL4 (rs11220463) | T | 0.190 | 114 | −0.774 | 0.1587 | 3.624 | 0.0503 R |
| 12 | SCARB1 (rs838880) | C | 0.336 | 114 | 1.736 | 0.0478 R | 0.1063 | 0.6429 |
| 16 | CETP (rs3764261) | A | 0.147 | 114 | 0.695 | 0.2931 | 0.02338 | 1.0000 |
| 16 | PSKH1 (rs16942887) | A | 0.116 | 114 | −0.6306 | 0.1439 | −0.2626 | 0.3947 |
| 17 | STARD3 (rs881844) | C | 0.310 | 114 | 0.4075 | 0.4815 | −0.2337 | 0.2647 |
| 19 | LILRA3 (rs386000) | C | 0.203 | 114 | −0.1847 | 0.8571 | −0.1448 | 0.6923 |
| 20 | PLTP (rs6065906) | C | 0.160 | 114 | −0.4654 | 0.5789 | −0.0718 | 0.8571 |
| Chr | Gene (SNP ID) | Effect Allele | EAF | N | Total Cholesterol | LDL-C | CIMT, Maximum | CIMT, Mean | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Beta * | Pperm # | Beta * | Pperm # | Beta * | Pperm # | Beta * | Pperm # | |||||
| 1 | ZNF648 (rs1689800) | G | 0.392 | 113 | 0.010 | 0.3091 | 0.024 | 0.2043 | −0.093 | 0.0105 R | −0.036 | 0.0282 A |
| 1 | GALNT2 (rs4846914) | G | 0.388 | 113 | −0.004 | 0.7273 | −0.016 | 0.4118 | 0.002 | 0.8571 | −0.002 | 0.8571 |
| 2 | COBLL1 (rs12328675) | C | 0.170 | 109 | −0.011 | 0.4643 | −0.018 | 0.4643 | 0.059 | 0.0213 A | 0.075 | 0.0056 D |
| 6 | LPA (rs55730499) | T | 0.056 | 113 | 0.364 | 0.0001 R | 0.367 | 0.0415 R | 0.018 | 0.8571 | 0.414 | 0.0146 R |
| 7 | NPC1L1 (rs217406) | G | 0.203 | 113 | −0.002 | 1.0000 | 0.003 | 0.2043 | 0.019 | 0.3636 | 0.018 | 0.4516 |
| 8 | PPP1R3B (rs9987289) | A | 0.086 | 113 | 0.004 | 0.8571 | −0.068 | 0.4118 | 0.072 | 0.0359 D | 0.079 | 0.0109 D |
| 9 | ABCA1 (rs1883025) | T | 0.263 | 113 | 0.010 | 0.3148 | 0.001 | 0.4643 | 0.011 | 0.8571 | 0.019 | 0.2687 |
| 11 | F2 (rs3136441) | C | 0.180 | 100 | 0.013 | 0.3478 | 0.018 | 0.2043 | −0.011 | 0.6429 | −0.005 | 1.0000 |
| 11 | ST3GAL4 (rs11220463) | T | 0.190 | 113 | −0.181 | 0.0273 R | −0.027 | 0.4118 | 0.001 | 1.0000 | 0.032 | 0.1709 |
| 12 | SCARB1 (rs838880) | C | 0.336 | 113 | −0.001 | 1.0000 | −0.005 | 0.4643 | −0.003 | 1.0000 | −0.004 | 1.0000 |
| 16 | CETP (rs3764261) | A | 0.147 | 113 | 0.017 | 0.3478 | 0.021 | 0.2043 | −0.053 | 0.0833 | −0.043 | 0.0880 |
| 16 | PSKH1 (rs16942887) | A | 0.116 | 113 | 0.006 | 0.5789 | 0.086 | 0.0175 D | −0.011 | 0.5217 | −0.004 | 0.7778 |
| 17 | STARD3 (rs881844) | C | 0.310 | 113 | 0.009 | 0.4375 | 0.010 | 0.8571 | 0.028 | 0.1852 | 0.040 | 0.0223 A |
| 19 | LILRA3 (rs386000) | C | 0.203 | 113 | 0.019 | 0.2400 | 0.015 | 0.5200 | 0.026 | 0.2982 | 0.010 | 0.7778 |
| 20 | PLTP (rs6065906) | C | 0.160 | 113 | 0.013 | 0.4815 | 0.031 | 0.2571 | −0.029 | 0.3478 | −0.006 | 1.0000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kononov, S.; Azarova, I.; Klyosova, E.; Bykanova, M.; Churnosov, M.; Solodilova, M.; Polonikov, A. Lipid-Associated GWAS Loci Predict Antiatherogenic Effects of Rosuvastatin in Patients with Coronary Artery Disease. Genes 2023, 14, 1259. https://doi.org/10.3390/genes14061259
Kononov S, Azarova I, Klyosova E, Bykanova M, Churnosov M, Solodilova M, Polonikov A. Lipid-Associated GWAS Loci Predict Antiatherogenic Effects of Rosuvastatin in Patients with Coronary Artery Disease. Genes. 2023; 14(6):1259. https://doi.org/10.3390/genes14061259
Chicago/Turabian StyleKononov, Stanislav, Iuliia Azarova, Elena Klyosova, Marina Bykanova, Mikhail Churnosov, Maria Solodilova, and Alexey Polonikov. 2023. "Lipid-Associated GWAS Loci Predict Antiatherogenic Effects of Rosuvastatin in Patients with Coronary Artery Disease" Genes 14, no. 6: 1259. https://doi.org/10.3390/genes14061259