

Comparative Mitochondrial Genomes between the Genera Amiota and Phortica (Diptera: Drosophilidae) with Evolutionary Insights into D-Loop Sequence Variability

Abstract
:1. Introduction
2. Materials and Methods
2.1. Specimen Collection and DNA Extraction
2.2. Library Construction and Sequencing
2.3. Mitogenome Assembly and Annotation
2.4. Mitogenome Analyses
2.5. Phylogenetic Analyses
3. Results and Discussion
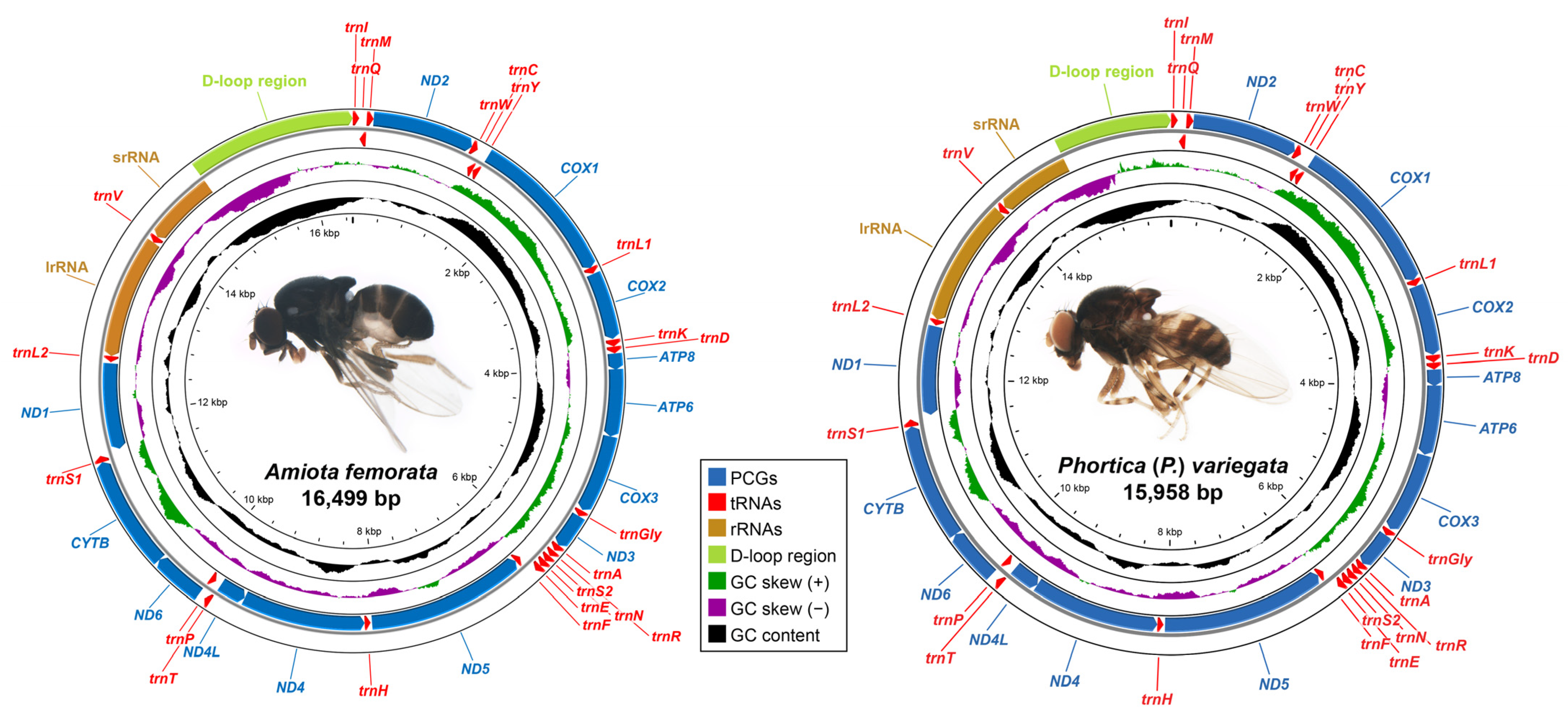
3.1. Mitogenome Organizations and Base Compositions
3.2. Protein-Coding Genes
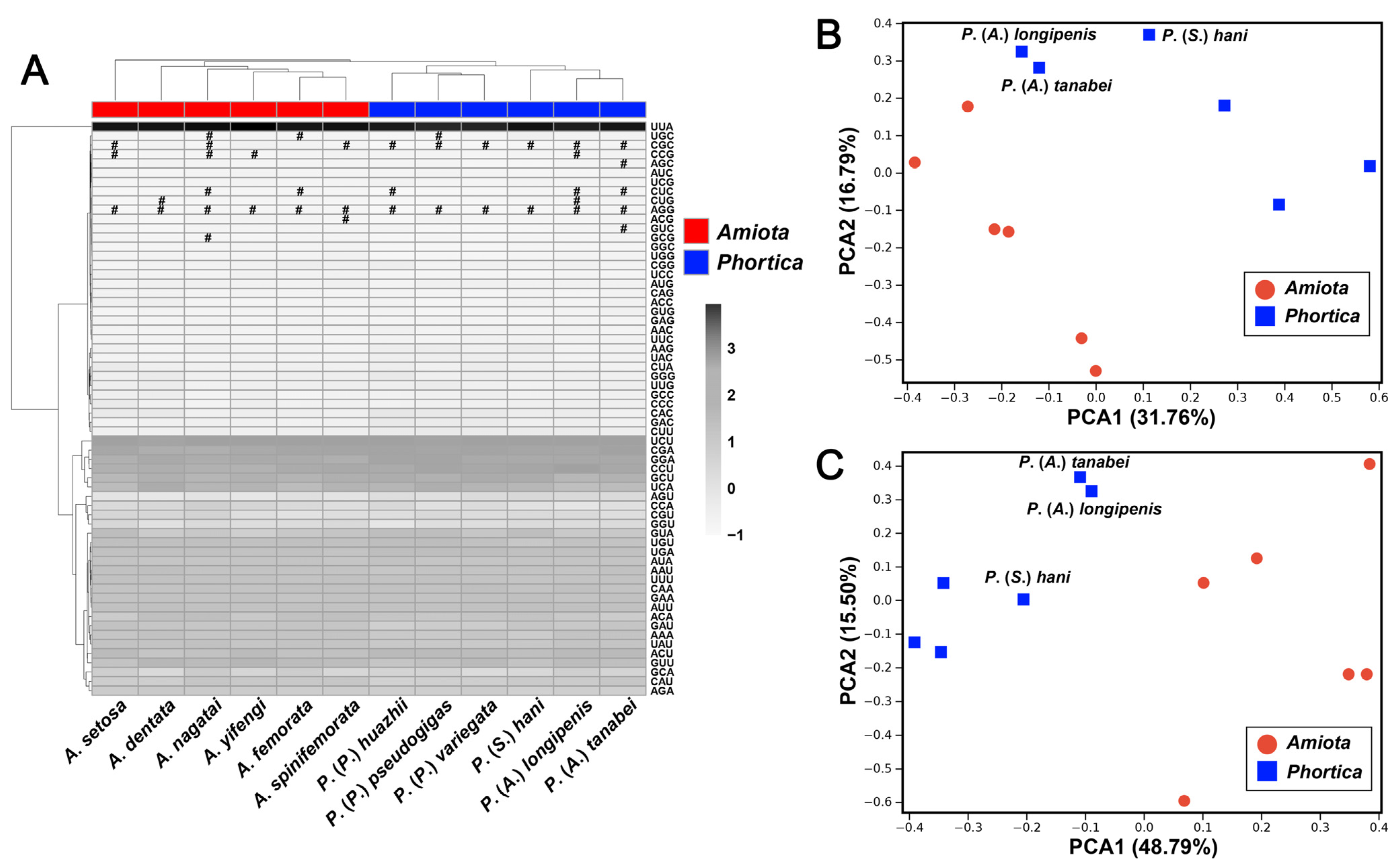
3.3. Transfer RNA and Ribosomal RNA Genes
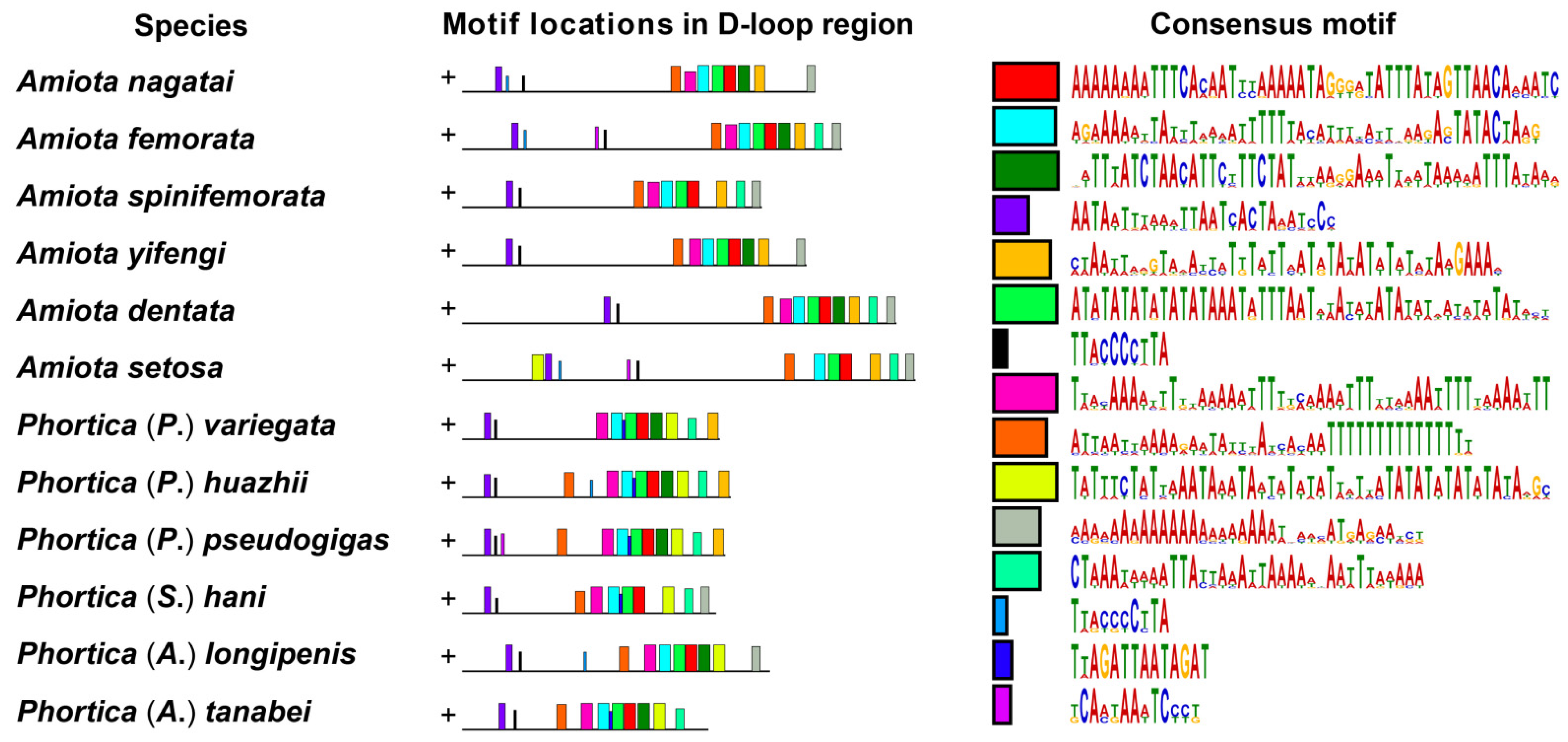
3.4. The D-Loop Regions
3.5. Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kummer, E.; Ban, N. Mechanisms and regulation of protein synthesis in mitochondria. Nat. Rev. Mol. Cell Biol. 2021, 22, 307–325. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Rand, D.M.; Haney, R.A.; Fry, A.J. Cytonuclear coevolution: The genomics of cooperation. Trends Ecol. Evol. 2004, 19, 645–653. [Google Scholar] [CrossRef]
- Basu, U.; Bostwick, A.M.; Das, K.; Dittenhafer-Reed, K.E.; Patel, S.S. Structure, mechanism, and regulation of mitochondrial DNA transcription initiation. J. Biol. Chem. 2020, 295, 18406–18425. [Google Scholar] [CrossRef]
- Stoccoro, A.; Coppedè, F. Mitochondrial DNA methylation and human diseases. Int. J. Mol. Sci. 2021, 22, 4594. [Google Scholar] [CrossRef]
- Latorre-Pellicer, A.; Moreno-Loshuertos, R.; Lechuga-Vieco, A.V.; Sánchez-Cabo, F.; Torroja, C.; Acín-Pérez, R.; Calvo, E.; Aix, E.; González-Guerra, A.; Logan, A.; et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 2016, 535, 561–565. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Gong, L.; Lu, X.; Jiang, L.; Liu, B.; Liu, L.; Lu, Z.; Li, P.; Zhang, X. Gene rearrangements in the mitochondrial genome of Chiromantes eulimene (Brachyura: Sesarmidae) and phylogenetic implications for Brachyura. Int. J. Biol. Macromol. 2020, 162, 704–714. [Google Scholar] [CrossRef]
- Zhang, Z.; Xing, Y.; Cheng, J.; Pan, D.; Lv, L.; Cumberlidge, N.; Sun, H. Phylogenetic implications of mitogenome rearrangements in East Asian potamiscine freshwater crabs (Brachyura: Potamidae). Mol. Phylogenet. Evol. 2020, 143, 106669. [Google Scholar] [CrossRef]
- Lin, Z.J.; Wang, X.; Wang, J.; Tan, Y.; Tang, X.; Werren, J.H.; Zhang, D.; Wang, X. Comparative analysis reveals the expansion of mitochondrial DNA control region containing unusually high G-C tandem repeat arrays in Nasonia vitripennis. Int. J. Biol. Macromol. 2021, 166, 1246–1257. [Google Scholar] [CrossRef]
- Wang, K.; Li, X.; Ding, S.; Wang, N.; Mao, M.; Wang, M.; Yang, D. The complete mitochondrial genome of the Atylotus miser (Diptera: Tabanomorpha: Tabanidae), with mitochondrial genome phylogeny of lower Brachycera (Orthorrhapha). Gene 2016, 586, 184–196. [Google Scholar] [CrossRef]
- Chen, L.P.; Zheng, F.Y.; Bai, J.; Wang, J.M.; Lv, C.Y.; Li, X.; Zhi, Y.C.; Li, X.J. Comparative analysis of mitogenomes among six species of grasshoppers (Orthoptera: Acridoidea: Catantopidae) and their phylogenetic implications in wing-type evolution. Int. J. Biol. Macromol. 2020, 159, 1062–1072. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar]
- Zhang, N.X.; Yu, G.; Li, T.J.; He, Q.Y.; Zhou, Y.; Si, F.L.; Ren, S.; Chen, B. The complete mitochondrial genome of Delia antiqua and its implications in dipteran phylogenetics. PLoS ONE 2015, 10, e139736. [Google Scholar] [CrossRef]
- Lee, W.; Johnson, J.; Gough, D.J.; Donoghue, J.; Cagnone, G.L.; Vaghjiani, V.; Brown, K.A.; Johns, T.G.; John, J.C.S. Mitochondrial DNA copy number is regulated by DNA methylation and demethylation of POLGA in stem and cancer cells and their differentiated progeny. Cell Death Dis. 2015, 6, e1664. [Google Scholar] [CrossRef] [Green Version]
- Chocron, E.S.; Munkácsy, E.; Pickering, A.M. Cause or casualty: The role of mitochondrial DNA in aging and age-associated disease. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 285–297. [Google Scholar] [CrossRef]
- Ren, J.; Pulakat, L.; Whaley-Connell, A.; Sowers, J.R. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J. Mol. Med. 2010, 88, 993–1001. [Google Scholar] [CrossRef] [Green Version]
- Baccarelli, A.A.; Byun, H.M. Platelet mitochondrial DNA methylation: A potential new marker of cardiovascular disease. Clin. Epigenetics 2015, 7, 44. [Google Scholar] [CrossRef] [Green Version]
- Coppedè, F.; Stoccoro, A. Mitoepigenetics and neurodegenerative diseases. Front. Endocrinol. 2019, 10, 86. [Google Scholar] [CrossRef] [Green Version]
- GBIF. The Global Biodiversity Information Facility. Available online: https://www.gbif.org (accessed on 10 February 2023).
- Loew, H. Diptera Americae septentrionalis indigena. Descripsit, Centuria secunda. Berl. Entomol. Z. 1862, 6, 185–232. [Google Scholar]
- Schiner, I.R. Vorläufiger Commentar zum dipterologischen Theile der “Fauna Austriaca”. Wien. Ent. Monatschr. 1862, 6, 428–436. [Google Scholar]
- Wheeler, M.R. The Drosophilidae of the Nearctic Region, exclusive of the genus Drosophila. Tex. Univ. Publ. 1952, 5204, 162–218. [Google Scholar]
- Máca, J. Taxonomic notes on the genera previously classified in the genus Amiota Loew (Diptera, Drosophilidae, Steganinae). Acta Univ. Carol. Biol. 2003, 47, 247–274. [Google Scholar]
- Otranto, D.; Stevens, J.R.; Testini, G.; Cantacessi, C.; Máca, J. Molecular characterization and phylogenesis of Steganinae (Diptera, Drosophilidae) inferred by the mitochondrial cytochrome c oxidase subunit 1. Med. Vet. Entomol. 2008, 22, 37–47. [Google Scholar] [CrossRef]
- Cao, H.; Wang, X.; Gao, J.; Prigent, S.R.; Watabe, H.; Zhang, Y.; Chen, H. Phylogeny of the African and Asian Phortica (Drosophilidae) deduced from nuclear and mitochondrial DNA sequences. Mol. Phylogenet. Evol. 2011, 61, 677–685. [Google Scholar] [CrossRef]
- Shao, Z.F.; Li, T.; Jiang, J.J.; Lu, J.M.; Chen, H.W. Molecular phylogenetic analysis of the Amiota taurusata species group within the Chinese species, with descriptions of two new species. J. Insect Sci. 2014, 14, 33. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Gong, L.; Tsaur, S.C.; Zhu, L.; An, K.; Chen, H. Revision of the subgenus Phortica (sensu stricto) (Diptera, Drosophilidae) from East Asia, with assessment of species delimitation using DNA barcodes. Zootaxa 2019, 4678, 1–75. [Google Scholar] [CrossRef]
- Geneious Prime. Version 2020.0.5. Available online: https://www.geneious.com (accessed on 14 January 2020).
- Boratyn, G.M.; Camacho, C.; Cooper, P.S.; Coulouris, G.; Fong, A.; Ma, N.; Madden, T.L.; Matten, W.T.; McGinnis, S.D.; Merezhuk, Y.; et al. BLAST: A more efficient report with usability improvements. Nucleic Acids Res. 2013, 41, W29–W33. [Google Scholar] [CrossRef] [Green Version]
- Donath, A.; Jühling, F.; Al-Arab, M.; Bernhart, S.H.; Reinhardt, F.; Stadler, P.F.; Middendorf, M.; Bernt, M. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 2019, 47, 10543–10552. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Stothard, P.; Grant, J.R.; Domselaar, G.V. Visualizing and comparing circular genomes using the CGView family of tools. Brief. Bioinform. 2019, 20, 1576–1582. [Google Scholar] [CrossRef] [Green Version]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- R Package Pheatmap. Pretty Heatmaps. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 24 July 2022).
- R Package Ggplot2. Create Elegant Data Visualisations Using the Grammar of Graphics. Available online: https://ggplot2.tidyverse.org (accessed on 24 July 2022).
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- FigTree. A Graphical Viewer of Phylogenetic Trees and for Producing Publication-Ready Figures. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 30 August 2022).
- Chen, Z.T.; Du, Y.Z. The first two mitochondrial genomes from Taeniopterygidae (Insecta: Plecoptera): Structural features and phylogenetic implications. Int. J. Biol. Macromol. 2018, 111, 70–76. [Google Scholar] [CrossRef]
- Zhang, D.; Yan, L.; Zhang, M.; Chu, H.; Cao, J.; Li, K.; Hu, D.; Pape, T. Phylogenetic inference of calyptrates, with the first mitogenomes for Gasterophilinae (Diptera: Oestridae) and Paramacronychiinae (Diptera: Sarcophagidae). Int. J. Biol. Sci. 2016, 12, 489–504. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Du, Z.; Luo, X.; Zhang, F.; Zhang, J.; Li, H. The conserved mitochondrial genomes of Drosophila mercatorum (Diptera: Drosophilidae) with different reproductive modes and phylogenetic implications. Int. J. Biol. Macromol. 2019, 138, 912–918. [Google Scholar] [CrossRef]
- Ren, L.; Zhang, X.; Li, Y.; Shang, Y.; Chen, S.; Wang, S.; Qu, Y.; Cai, J.; Guo, Y. Comparative analysis of mitochondrial genomes among the subfamily Sarcophaginae (Diptera: Sarcophagidae) and phylogenetic implications. Int. J. Biol. Macromol. 2020, 161, 214–222. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Hu, K.; Zhao, Y.; Lin, R.; Li, Y.; Huang, Z.; Zhang, X.; Geng, X.; Ding, J. Mitochondrial genome characteristics of two Sphingidae insects (Psilogramma increta and Macroglossum stellatarum) and implications for their phylogeny. Int. J. Biol. Macromol. 2018, 113, 592–600. [Google Scholar] [CrossRef]
- Lu, H.F.; Su, T.J.; Luo, A.R.; Zhu, C.D.; Wu, C.S. Characterization of the complete mitochondrion genome of diurnal moth Amata emma (Butler) (Lepidoptera: Erebidae) and its phylogenetic implications. PLoS ONE 2013, 8, e72410. [Google Scholar] [CrossRef]
- Yuan, M.L.; Zhang, Q.L.; Guo, Z.L.; Wang, J.; Shen, Y.Y. Comparative mitogenomic analysis of the superfamily Pentatomoidea (Insecta: Hemiptera: Heteroptera) and phylogenetic implications. BMC Genom. 2015, 16, 460. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Huang, M. Characterization of the complete mitochondrial genome of Simulium (Byssodon) maculatum (Diptera: Simuliidae) and its phylogenetic implications. Int. J. Biol. Macromol. 2019, 121, 152–160. [Google Scholar] [CrossRef]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.R.A.; Yalvac, M.E.; Khoo, B.; Eckardt, S.; Mclaughlin, K.J. Adapting CRISPR/Cas9 system for targeting mitochondrial genome. Front. Genet. 2021, 12, 627050. [Google Scholar] [CrossRef]
- Yacoub, H.A.; Fathi, M.M. Phylogenetic analysis using d-loop marker of mtDNA of Saudi native chicken strains. Mitochondrial DNA 2013, 24, 538–551. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, Y.; Yoshikawa, Y.; Takano, T.; Mori, M.; Hondo, R.; Mochizuki, M.; Ueda, F. Genetic variation of mitochondrial DNA in Phalacrocorax carbo in Japan. J. Vet. Med. Sci. 2020, 82, 735–739. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Subgenus/Species Group | Species | Collection Site | Mitogenome Accession Number | SRA Accession Number |
|---|---|---|---|---|---|
| Amiota | / | dentata Okada | Fengtongzhai, Baoxing, Sichuan, China | OP381033 | SRR21438455 |
| Amiota | taurusata | femorata Chen & Takamori | Dafengding, Mabian, Sichuan, China | OP381034 | SRR21438454 |
| Amiota | nagatai | nagatai Okada | Conghua, Guangzhou, Guangdong, China | OP381035 | SRR21438452 |
| Amiota | alboguttata | setosa Zhang & Chen | Dafengding, Mabian, Sichuan, China | OP381036 | SRR21438451 |
| Amiota | taurusata | spinifemorata Li & Chen | Gexigou, Yajiang, Sichuan, China | OP381037 | SRR21438450 |
| Amiota | basdeni | yifengi Zhang & Chen | Gexigou, Yajiang, Sichuan, China | OP381038 | SRR21438449 |
| Phortica | Ashima | longipenis Chen & Gao | Hesong, Menghai, Yunnan, China | OP381039 | SRR21438448 |
| Phortica | Ashima | tanabei Chen & Toda | Muyiji Park, Ximeng, Yunnan, China | OP381040 | SRR21438447 |
| Phortica | Phortica | huazhii Cheng & Chen | Ruili Park, Yunnan, China | OP381041 | SRR21438446 |
| Phortica | Phortica | pseudogigas Zhang & Gan | Mangshi, Yunnan, China | OP381042 | SRR21438445 |
| Phortica | Phortica | variegata (Fallén) | / | OP381043 | a SRR826812 |
| Phortica | Shangrila | hani Zhang & Shi | Gexigou, Yajiang, Sichuan, China | OP381044 | SRR21438453 |
| Gene | Strand | A. femorata | P. (P.) variegata | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Position (bp) | Size (bp) | Anti- or Start/Stop Codons | IGN(s) (bp) | Position (bp) | Size (bp) | Anti- or Start/Stop Codons | IGN(s) (bp) | ||
| trnI (Ile) | + | 1–65 | 65 | GAU | 7 | 1–65 | 65 | GAU | 15 |
| trnQ (Gln) | − | 73–141 | 69 | UUG | −1 | 81–149 | 69 | UUG | −1 |
| trnM (Met) | + | 141–209 | 69 | CAU | 0 | 149–217 | 69 | CAU | 0 |
| ND2 | + | 210–1232 | 1023 | ATT/TAA | 0 | 218–1243 | 1026 | ATT/TAA | 0 |
| trnW (Trp) | + | 1231–1298 | 68 | UCA | 6 | 1242–1309 | 68 | UCA | 6 |
| trnC (Cys) | − | 1291–1353 | 63 | GCA | 2 | 1302–1364 | 63 | GCA | 2 |
| trnY (Tyr) | − | 1356–1421 | 66 | GUA | 0 | 1367–1432 | 66 | GUA | 0 |
| COX1 | + | 1420–2958 | 1539 | TCG/TAA | 3 | 1431–2969 | 1539 | TCG/TAA | 3 |
| trnL1 (Leu) (UUR) | + | 2954–3019 | 66 | UAA | 3 | 2965–3030 | 66 | UAA | 2 |
| COX2 | + | 3023–3703 | 681 | ATG/TAA | 7 | 3033–3717 | 685 | ATG/T- | 3 |
| trnK (Lys) | + | 3711–3781 | 71 | CUU | −1 | 3721–3791 | 71 | CUU | 0 |
| trnD (Asp) | + | 3781–3850 | 70 | GUC | 0 | 3792–3858 | 67 | GUC | 0 |
| ATP8 | + | 3851–4012 | 162 | ATT/TAA | 5 | 3859–4020 | 162 | ATC/TAA | 5 |
| ATP6 | + | 4006–4683 | 678 | ATG/TAA | −1 | 4014–4691 | 678 | ATG/TAA | 6 |
| COX3 | + | 4683–5471 | 789 | ATG/TAA | 8 | 4698–5486 | 789 | ATG/TAA | 9 |
| trnG (Gly) | + | 5480–5543 | 64 | UCC | 0 | 5496–5560 | 65 | UCC | 0 |
| ND3 | + | 5544–5897 | 354 | ATT/TAG | 0 | 5561–5914 | 354 | ATT/TAG | 0 |
| trnA (Ala) | + | 5896–5962 | 67 | UGC | 8 | 5913–5977 | 65 | UGC | −1 |
| trnR (Arg) | + | 5971–6034 | 64 | UCG | 3 | 5977–6039 | 63 | UCG | 2 |
| trnN (Asn) | + | 6038–6102 | 65 | GUU | 0 | 6042–6106 | 65 | GUU | 0 |
| trnS2 (Ser) (AGN) | + | 6103–6170 | 68 | GCU | 0 | 6107–6173 | 67 | GCU | 0 |
| trnE (Glu) | + | 6171–6238 | 68 | UUC | 18 | 6174–6239 | 66 | UUC | 18 |
| trnF (Phe) | − | 6257–6322 | 66 | GAA | 0 | 6258–6324 | 67 | GAA | 0 |
| ND5 | − | 6323–8042 | 1720 | ATT/T- | 15 | 6325–8044 | 1720 | ATT/T- | 15 |
| trnH (His) | − | 8058–8122 | 65 | GUG | 0 | 8060–8124 | 65 | GUG | 0 |
| ND4 | − | 8123–9461 | 1339 | ATG/T- | 2 | 8125–9463 | 1339 | ATG/T- | 0 |
| ND4L | − | 9464–9754 | 291 | ATG/TAA | 2 | 9464–9754 | 291 | ATG/TAA | 2 |
| trnT (Thr) | + | 9757–9821 | 65 | UGU | 0 | 9757–9821 | 65 | UGU | 0 |
| trnP (Pro) | − | 9822–9887 | 66 | UGG | 3 | 9822–9887 | 66 | UGG | 2 |
| ND6 | + | 9891–10,415 | 525 | ATT/TAA | −1 | 9890–10,414 | 525 | ATT/TAA | −1 |
| CYTB | + | 10,415–11,551 | 1137 | ATG/TAG | 0 | 10,414–11,550 | 1137 | ATG/TAG | 0 |
| trnS1 (Ser) (UCN) | + | 11,550–11,616 | 67 | UGA | 15 | 11,549–11,615 | 67 | UGA | 18 |
| ND1 | − | 11,632–12,568 | 937 | ATA/T- | 10 | 11,634–12,570 | 937 | ATT/T- | 10 |
| trnL2 (Leu) (CUN) | − | 12,579–12,644 | 66 | UAG | 0 | 12,581–12,644 | 64 | UAG | 0 |
| lrRNA | − | 12,645–13,971 | 1327 | / | 0 | 12,645–13,964 | 1320 | / | 0 |
| trnV (Val) | − | 13,972–14,043 | 72 | UAC | 0 | 13,965–14,036 | 72 | UAC | 0 |
| srRNA | − | 14,044–14,827 | 784 | / | 0 | 14,037–14,824 | 788 | / | 0 |
| D-loop region | + | 14,828–16,499 | 1672 | / | 0 | 14,825–15,958 | 1134 | / | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Wang, Y.; Chen, H.; Huang, J. Comparative Mitochondrial Genomes between the Genera Amiota and Phortica (Diptera: Drosophilidae) with Evolutionary Insights into D-Loop Sequence Variability. Genes 2023, 14, 1240. https://doi.org/10.3390/genes14061240
Zhang C, Wang Y, Chen H, Huang J. Comparative Mitochondrial Genomes between the Genera Amiota and Phortica (Diptera: Drosophilidae) with Evolutionary Insights into D-Loop Sequence Variability. Genes. 2023; 14(6):1240. https://doi.org/10.3390/genes14061240
Chicago/Turabian StyleZhang, Caihong, Yalian Wang, Hongwei Chen, and Jia Huang. 2023. "Comparative Mitochondrial Genomes between the Genera Amiota and Phortica (Diptera: Drosophilidae) with Evolutionary Insights into D-Loop Sequence Variability" Genes 14, no. 6: 1240. https://doi.org/10.3390/genes14061240