
Chromosome-Level Genome Assembly and Circadian Gene Repertoire of the Patagonia Blennie Eleginops maclovinus—The Closest Ancestral Proxy of Antarctic Cryonotothenioids
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Specimen Collection and Sample Preservation
2.2. HMW DNA Preparation and CLR Sequencing
2.3. Hi-C Library and Sequencing
2.4. Genome Assembly
2.5. Repeat and Protein-Coding Gene Annotation
2.6. Conserved Synteny Analysis
2.7. Notothenioid Species Phylogeny Reconstruction
2.8. Curation of Predicted Circadian Network Genes from Genome Assemblies
2.9. Phylogenetic Verification of Circadian Gene Orthologs
2.10. Fin RNA Isolation, Iso-Seq Transcriptome Sequencing and Curation of Circadian Network Gene Transcripts
3. Results and Discussion
3.1. Genome Assembly and Annotation
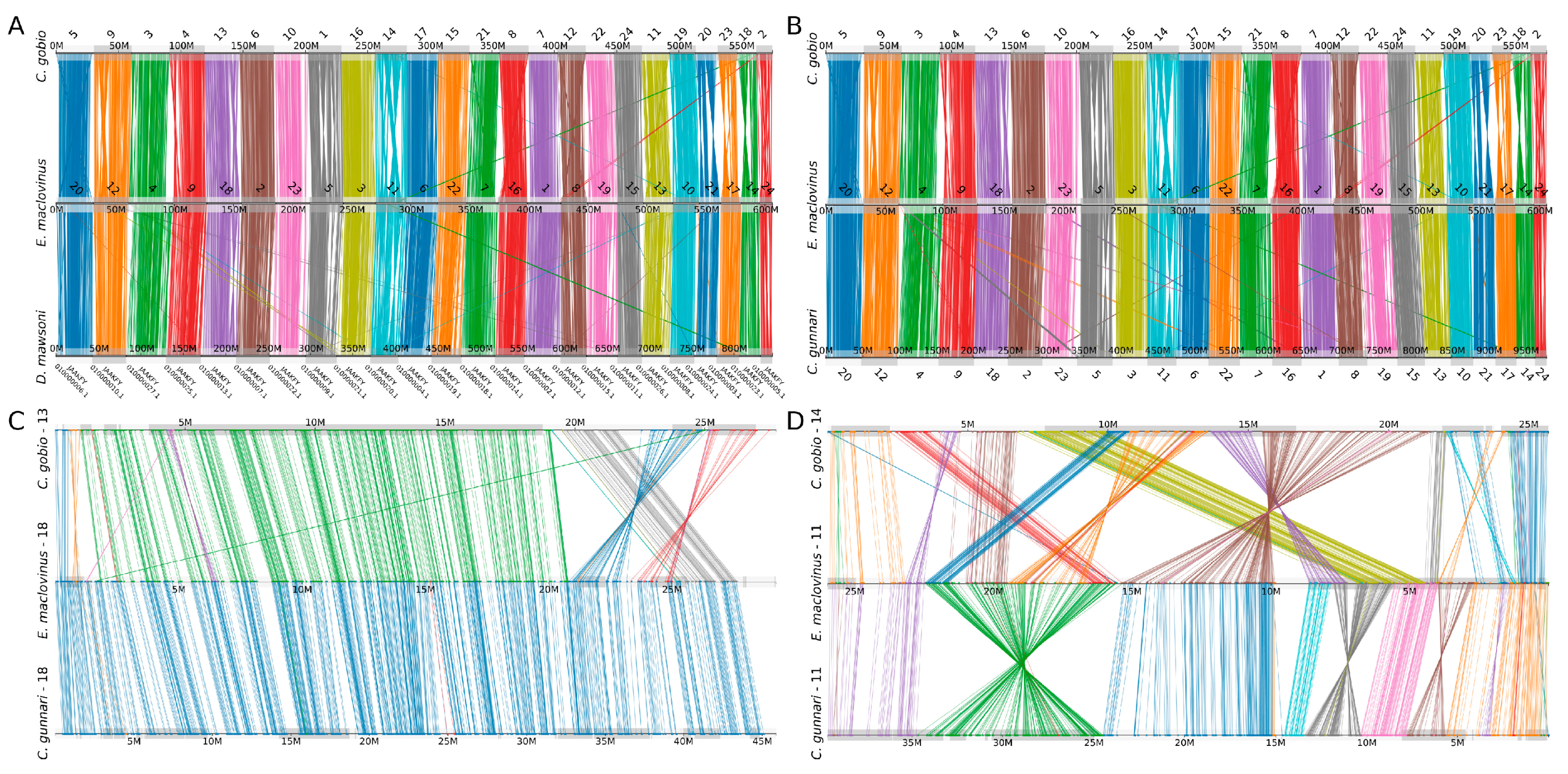
3.2. Conserved Synteny
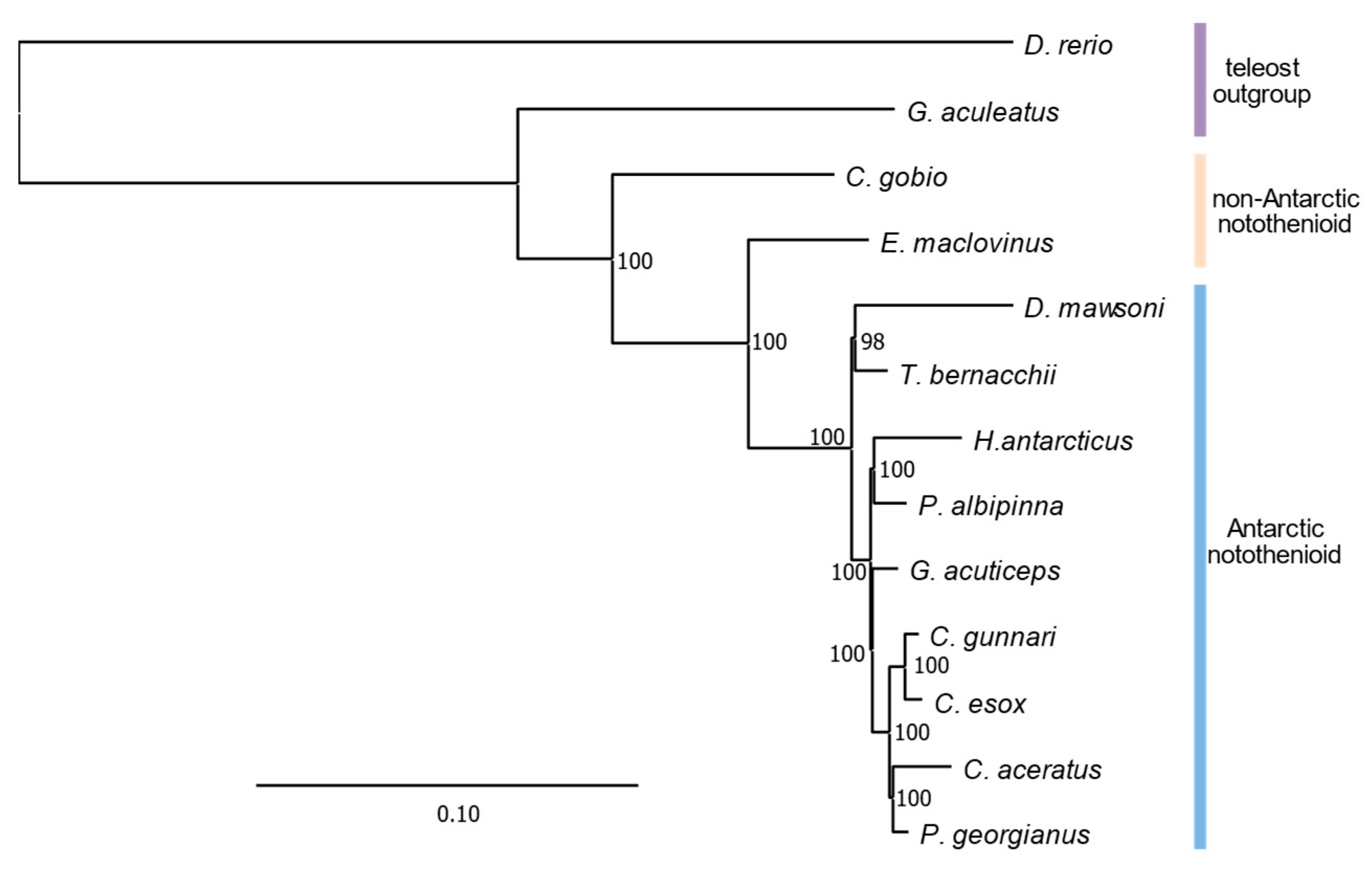
3.3. Phylogenomic Analyeses Affirm E. maclovinus as Immediate Sister to Cryonotothenioids
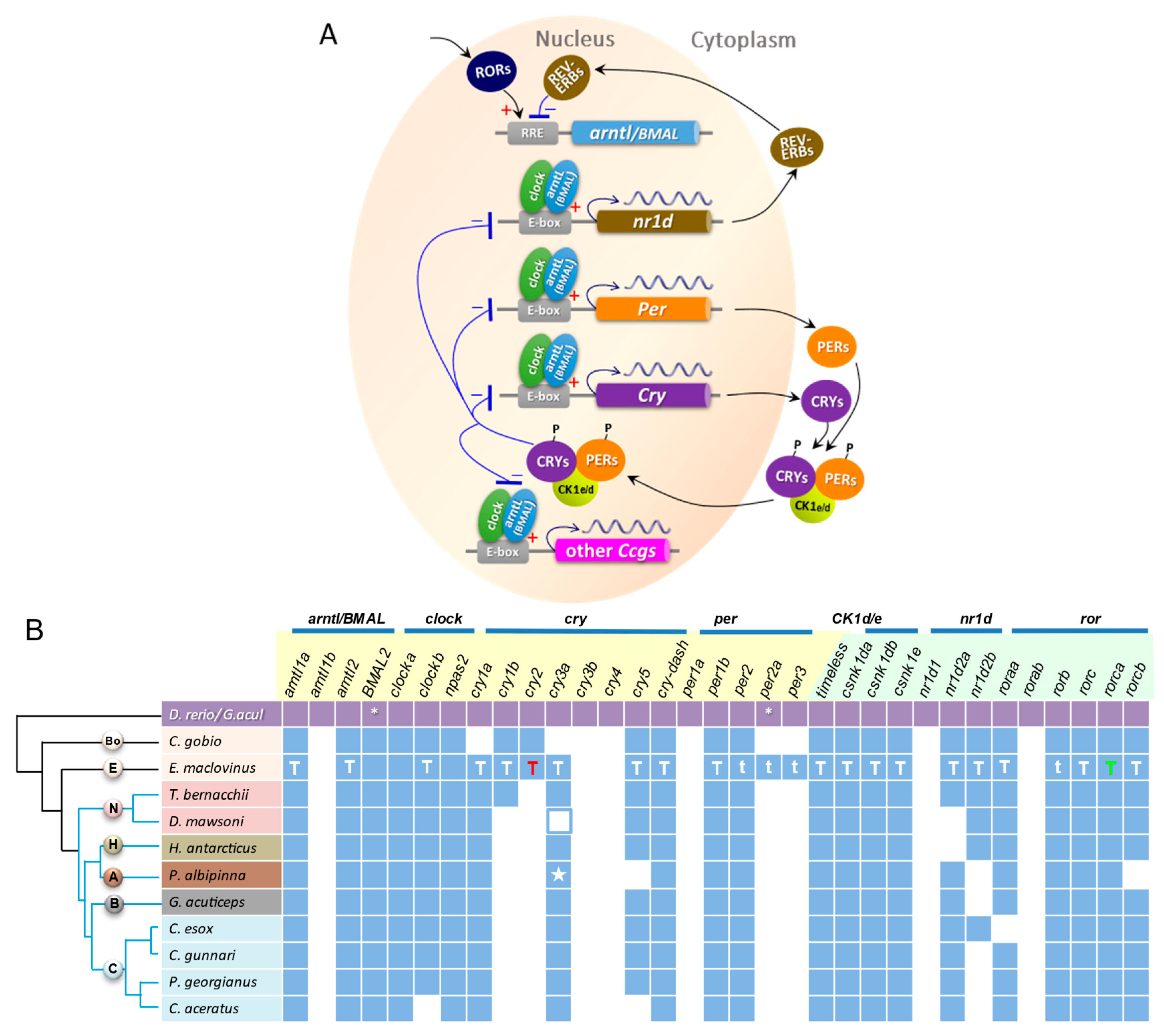
3.4. Circadian Gene Repertoire in E. maclovinus and Ten Notothenioids
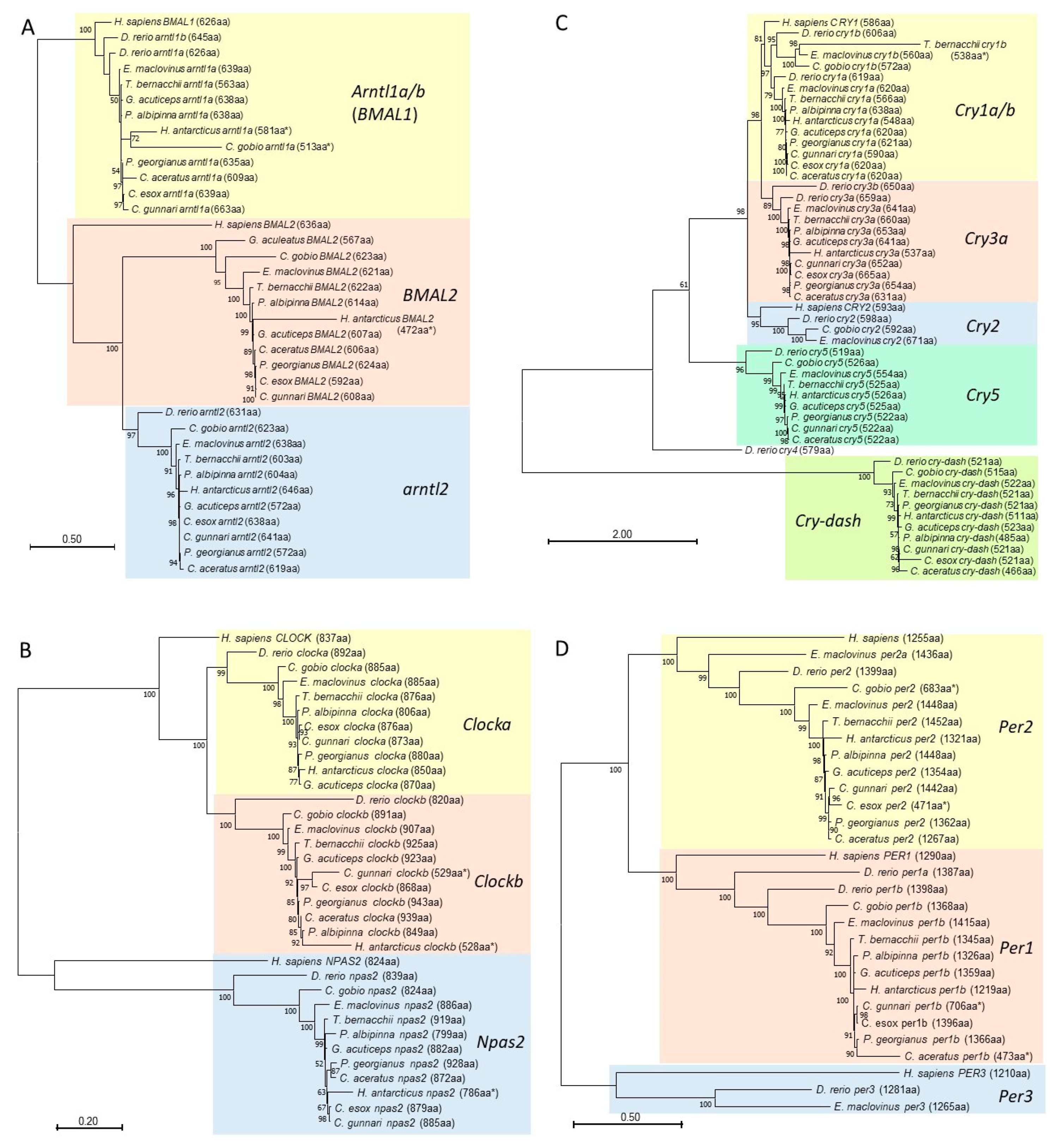
3.5. Phylogenetic Analyses of Core Regulatory Loop Circadian Genes
3.6. Transcriptional Expression of E. maclovinus Circadian Network Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eastman, J.T. The nature of the diversity of Antarctic fishes. Polar Biol. 2005, 28, 94–107. [Google Scholar] [CrossRef]
- Matschiner, M.; Hanel, R.; Salzburger, W. On the Origin and Trigger of the Notothenioid Adaptive Radiation. PLoS ONE 2011, 6, e18911. [Google Scholar] [CrossRef]
- Near, T.J.; Dornburg, A.; Kuhn, K.L.; Eastman, J.T.; Pennington, J.N.; Patarnello, T.; Zane, L.; Fernández, D.A.; Jones, C.D. Ancient climate change, antifreeze, and the evolutionary diversification of Antarctic fishes. Proc. Natl. Acad. Sci. USA 2012, 109, 3434–3439. [Google Scholar] [CrossRef] [Green Version]
- Eastman, J.T.; McCune, A.R. Fishes on the Antarctic continental shelf: Evolution of a marine species flock? J. Fish Biol. 2000, 57 (Suppl. A), 84–102. [Google Scholar] [CrossRef]
- Lecointre, G.; Améziane, N.; Boisselier, M.-C.; Bonillo, C.; Busson, F.; Causse, R.; Chenuil, A.; Couloux, A.; Coutanceau, J.-P.; Cruaud, C.; et al. Is the species flock concept operational? The Antarctic shelf case. PLoS ONE 2013, 8, e68787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; DeVries, A.L.; Cheng, C.-H.C. Evolution of antifreeze glycoprotein gene from a trypsinogen gene in Antarctic notothenioid fish. Proc. Natl. Acad. Sci. USA 1997, 94, 3811–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.-H.C.; Zhuang, X. Molecular Origins and Mechanisms of Fish Antifreeze Evolution. In Antifreeze Proteins Volume 1: Environment, Systematics and Evolution; Ramløv, H., Friis, D.S., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 275–313. [Google Scholar]
- DeVries, A.L. Glycoproteins as biological antifreeze agents in Antarctic fishes. Science 1971, 172, 1152–1155. [Google Scholar] [CrossRef] [PubMed]
- DeVries, A.L.; Cheng, C.-H.C. Antifreeze proteins and organismal freezing avoidance in polar fishes. In The Physiology of Polar Fishes; Farrell, A.P., Steffensen, J.F., Eds.; Elsevier Academic Press: San Diego, CA, USA, 2005; Volume 22, pp. 155–201. [Google Scholar]
- Todgham, A.E.; Hoaglund, E.A.; Hofmann, G.E. Is cold the new hot? Elevated ubiquitin-conjugated protein levels in tissues of Antarctic fish as evidence for cold-denaturation of proteins in vivo. J. Comp. Physio. B 2007, 177, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Beers, J.M.; Jayasundara, N. Antarctic notothenioid fish: What are the future consequences of ‘losses’ and ‘gains’ acquired during long-term evolution at cold and stable temperatures? J. Exp. Biol. 2015, 218, 1834–1845. [Google Scholar] [CrossRef] [Green Version]
- Daane, J.M.; Detrich, H.W. Adaptations and Diversity of Antarctic Fishes: A Genomic Perspective. Ann. Rev. Anim. Biosci. 2022, 10, 39–62. [Google Scholar] [CrossRef]
- Bilyk, K.T.; DeVries, A.L. Heat tolerance and its plasticity in Antarctic fishes. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2011, 158, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Somero, G.N.; DeVries, A.L. Temperature tolerance of some Antarctic fishes. Science 1967, 156, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Bilyk, K.T.; Vargas-Chacoff, L.; Cheng, C.H.C. Evolution in chronic cold: Varied loss of cellular response to heat in Antarctic notothenioid fish. BMC Evol. Biol. 2018, 18, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, G.E.; Buckley, B.A.; Airaksinen, S.; Keen, J.; Somero, G.N. The Antarctic fish Trematomus bernacchii lacks heat-inducible heat shock protein synthesis. J. Exp. Biol. 2000, 203, 2331–2339. [Google Scholar] [CrossRef]
- Saravia, J.; Paschke, K.; Oyarzún-Salazar, R.; Cheng, C.H.C.; Navarro, J.M.; Vargas-Chacoff, L. Effects of warming rates on physiological and molecular components of response to CTMax heat stress in the Antarctic fish Harpagifer antarcticus. J. Therm. Biol. 2021, 99, 103021. [Google Scholar] [CrossRef]
- di Prisco, G.; Cocca, E.; Parker, S.K.; Detrich, H.W., III. Tracking the Evolutionary Loss of Hemoglobin Expression by the White-blooded Antarctic Icefishes. Gene 2003, 295, 185–191. [Google Scholar] [CrossRef]
- Sidell, B.D.; O’Brien, K.M. When bad things happen to good fish: The loss of hemoglobin and myoglobin expression in Antarctic icefishes. J. Exp. Biol. 2006, 209, 1791–1802. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Lu, Y.; Li, W.; Ren, Y.; Yu, M.; Jiang, S.; Fu, Y.; Wang, J.; Peng, S.; Bilyk, K.T.; et al. The genomic basis for colonizing the freezing Southern Ocean revealed by Antarctic toothfish and Patagonian robalo genomes. GigaScience 2019, 8, giz016. [Google Scholar] [CrossRef] [Green Version]
- Bista, I.; Wood, J.M.D.; Desvignes, T.; McCarthy, S.A.; Matschiner, M.; Ning, Z.; Tracey, A.; Torrance, J.; Sims, Y.; Chow, W.; et al. Genomics of cold adaptations in the Antarctic notothenioid fish radiation. bioRXiv 2022, 494096. [Google Scholar] [CrossRef]
- Rivera-Colón, A.G.; Rayamajhi, N.; Minhas, B.F.; Madrigal, G.; Bilyk, K.T.; Yoon, V.; Hüne, M.; Gregory, S.; Cheng, C.H.C.; Catchen, J.M. Genomics of Secondarily Temperate Adaptation in the Only Non-Antarctic Icefish. Mol. Biol. Evol. 2023, 40, msad029. [Google Scholar] [CrossRef]
- Bilyk, K.T.; Zhuang, X.; Papetti, C. Positive and Relaxed Selective Pressures Have Both Strongly Influenced the Evolution of Cryonotothenioid Fishes during Their Radiation in the Freezing Southern Ocean. Genome Biol. Evol. 2023, 15, evad049. [Google Scholar] [CrossRef]
- Lu, Y.; Li, W.; Li, Y.; Zhai, W.; Zhou, X.; Wu, Z.; Jiang, S.; Liu, T.; Wang, H.; Hu, R.; et al. Population genomics of an icefish reveals mechanisms of glacier-driven adaptive radiation in Antarctic notothenioids. BMC Biol. 2022, 20, 231. [Google Scholar] [CrossRef] [PubMed]
- Bista, I.; McCarthy, S.A.; Wood, J.; Ning, Z.; Detrich Iii, H.W.; Desvignes, T.; Postlethwait, J.; Chow, W.; Howe, K.; Torrance, J.; et al. The genome sequence of the channel bull blenny, Cottoperca gobio (Günther, 1861). Wellcome Open Res. 2020, 5, 148. [Google Scholar] [CrossRef] [PubMed]
- Balushkin, A.V. Classification, phylogenetics, and origins of the families of the suborder Notothenioidei (Perciformes). J. Ichthyol. 1992, 32, 90–110. [Google Scholar]
- Bargelloni, L.; Marcato, S.; Zane, L.; Patarnello, T. Mitochondrial Phylogeny of Notothenioids: A Molecular Approach to Antarctic Fish Evolution and Biogeography. Syst. Biol. 2000, 49, 114–129. [Google Scholar] [CrossRef] [Green Version]
- Lecointre, G.; Bonillo, C.; Ozouf-Costaz, C.; Hureau, J.C. Molecular evidence for the origins of Antarctic fishes: Paraphyly of the Bovichtidae and no indication for the monophyly of the Notothenioidei (Teleostei). Polar Biol. 1997, 18, 193–208. [Google Scholar] [CrossRef]
- Bilyk, K.T.; Zhuang, X.; Vargas-Chacoff, L.; Cheng, C.H.C. Evolution of chaperome gene expression and regulatory elements in the antarctic notothenioid fishes. Heredity 2021, 126, 424–441. [Google Scholar] [CrossRef] [PubMed]
- Rayamajhi, N.; Cheng, C.-H.C.; Catchen, J.M. Evaluating Illumina-, Nanopore-, and PacBio-based genome assembly strategies with the bald notothen, Trematomus borchgrevinki. G3 Genes Genomes Genet. 2022, 12, jkac192. [Google Scholar] [CrossRef]
- Ruan, J.; Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 2020, 17, 155–158. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.P.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Brůna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 2021, 3, lqaa108. [Google Scholar] [CrossRef]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER1: Unsupervised RNA-Seq-Based Genome Annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 2016, 32, 767–769. [Google Scholar] [CrossRef] [Green Version]
- Kriventseva, E.V.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Dias, R.; Simão, F.A.; Zdobnov, E.M. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 2019, 47, D807–D811. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, L.; Hoff, K.J.; Brůna, T.; Borodovsky, M.; Stanke, M. TSEBRA: Transcript selector for BRAKER. BMC Bioinform. 2021, 22, 566. [Google Scholar] [CrossRef]
- Catchen, J.M.; Conery, J.S.; Postlethwait, J.H. Automated identification of conserved synteny after whole-genome duplication. Genome Res. 2009, 19, 1497–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, C.M.; Bassham, S.; Catchen, J.; Amores, A.; Fuiten, A.M.; Brown, R.S.; Jones, A.G.; Cresko, W.A. The genome of the Gulf pipefish enables understanding of evolutionary innovations. Genome Biol. 2016, 17, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jae Lee, S.; Kim, J.H.; Jo, E.; Choi, E.; Kim, J.; Choi, S.G.; Chung, S.; Kim, H.W.; Park, H. Chromosomal assembly of the Antarctic toothfish ( Dissostichus mawsoni) genome using third-generation DNA sequencing and Hi-C technology. Zool. Res. 2021, 42, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amores, A.; Catchen, J.; Nanda, I.; Warren, W.; Walter, R.; Schartl, M.; Postlethwait, J.H. A RAD-Tag Genetic Map for the Platyfish (Xiphophorus maculatus) Reveals Mechanisms of Karyotype Evolution Among Teleost Fish. Genetics 2014, 197, 625–641. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. STAG: Species Tree Inference from All Genes. bioRXiv 2018, 267914. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. STRIDE: Species Tree Root Inference from Gene Duplication Events. Mol. Biol. Evol. 2017, 34, 3267–3278. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Beale, A.; Guibal, C.; Tamai, T.K.; Klotz, L.; Cowen, S.; Peyric, E.; Reynoso, V.H.; Yamamoto, Y.; Whitmore, D. Circadian rhythms in Mexican blind cavefish Astyanax mexicanus in the lab and in the field. Nat. Commun. 2013, 4, 2769. [Google Scholar] [CrossRef] [Green Version]
- Foulkes, N.S.; Whitmore, D.; Vallone, D.; Bertolucci, C. Chapter One—Studying the Evolution of the Vertebrate Circadian Clock: The Power of Fish as Comparative Models. In Advances in Genetics; Foulkes, N.S., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 95, pp. 1–30. [Google Scholar]
- Mazzei, F.; Ghigliotti, L.; Coutanceau, J.-P.; Detrich, H.W.; Prirodina, V.; Ozouf-Costaz, C.; Pisano, E. Chromosomal characteristics of the temperate notothenioid fish Eleginops maclovinus (Cuvier). Polar Biol. 2008, 31, 629. [Google Scholar] [CrossRef]
- Kim, B.-M.; Amores, A.; Kang, S.; Ahn, D.-H.; Kim, J.-H.; Kim, I.-C.; Lee, J.H.; Lee, S.G.; Lee, H.; Lee, J.; et al. Antarctic blackfin icefish genome reveals adaptations to extreme environments. Nat. Ecol. Evol. 2019, 3, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Auvinet, J.; Graça, P.; Belkadi, L.; Petit, L.; Bonnivard, E.; Dettaï, A.; Detrich, W.H.; Ozouf-Costaz, C.; Higuet, D. Mobilization of retrotransposons as a cause of chromosomal diversification and rapid speciation: The case for the Antarctic teleost genus Trematomus. BMC Genom. 2018, 19, 339. [Google Scholar] [CrossRef] [Green Version]
- Ghigliotti, L.; Cheng, C.-H.C.; Pisano, E. Sex determination in Antarctic notothenioid fish: Chromosomal clues and evolutionary hypotheses. Polar Biol. 2016, 39, 11–22. [Google Scholar] [CrossRef]
- Morescalchi, A.; Hureau, J.C.; Olmo, E.; Ozouf-Costaz, C.; Pisano, E.; Stanyon, R. A multiple sex-chromosome system in Antarctic ice-fishes. Polar Biol. 1992, 11, 655–661. [Google Scholar] [CrossRef]
- Morescalchi, A.; Pisano, E.; Stanyon, R.; Morescalchi, M.A. Cytotaxonomy of antarctic teleosts of the Pagothenia/Trematomus complex (Nototheniidae, Perciformes). Polar Biol. 1992, 12, 553–558. [Google Scholar] [CrossRef]
- Amores, A.; Wilson, C.A.; Allard, C.A.H.; Detrich, H.W.; Postlethwait, J.H. Cold Fusion: Massive Karyotype Evolution in the Antarctic Bullhead Notothen Notothenia coriiceps. G3 Genes Genomes Genet. 2017, 7, 2195–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccolin, F.; Pitzschler, L.; Biscontin, A.; Kawaguchi, S.; Meyer, B. Circadian regulation of diel vertical migration (DVM) and metabolism in Antarctic krill Euphausia superba. Sci. Rep. 2020, 10, 16796. [Google Scholar] [CrossRef]
- Teschke, M.; Wendt, S.; Kawaguchi, S.; Kramer, A.; Meyer, B. A circadian clock in Antarctic krill: An endogenous timing system governs metabolic output rhythms in the euphausid species Euphausia superba. PLoS ONE 2011, 6, e26090. [Google Scholar] [CrossRef] [Green Version]
- Höring, F.; Biscontin, A.; Harms, L.; Sales, G.; Reiss, C.S.; De Pittà, C.; Meyer, B. Seasonal gene expression profiling of Antarctic krill in three different latitudinal regions. Mar. Genom. 2021, 56, 100806. [Google Scholar] [CrossRef]
- Häfker, N.S.; Andreatta, G.; Manzotti, A.; Falciatore, A.; Raible, F.; Tessmar-Raible, K. Rhythms and Clocks in Marine Organisms. Annu. Rev. Mar. Sci. 2023, 15, 509–538. [Google Scholar] [CrossRef]
- Cziko, P.A.; DeVries, A.L.; Evans, C.W.; Cheng, C.-H.C. Antifreeze protein-induced superheating of ice inside Antarctic notothenioid fishes inhibits melting during summer warming. Proc. Natl. Acad. Sci. USA 2014, 111, 14583–14588. [Google Scholar] [CrossRef] [Green Version]
- Hunt, B.M.; Hoefling, K.; Cheng, C.-H.C. Annual warming episodes in seawater temperatures in McMurdo Sound in relationship to endogenous ice in notothenioid fish. Antarct. Sci. 2003, 15, 333–338. [Google Scholar] [CrossRef] [Green Version]
- Dalrymple, R.W.; Padman, L. Are Tides Controlled by Latitude? In Latitudinal Controls on Stratigraphic Models and Sedimentary Concepts; Fraticelli, C.M., Markwick, P.J., Martinius, A.W., Suter, J.R., Eds.; Society for Sedimentary Geology: Tulsa, OK, USA, 2019; Volume 108. [Google Scholar] [CrossRef]
- Scully, A.L.; Kay, S.A. Time Flies for Drosophila. Cell 2000, 100, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Minami, Y.; Ode, K.L.; Ueda, H.R. Mammalian Circadian Clock: The Roles of Transcriptional Repression and Delay. In Circadian Clocks; Kramer, A., Merrow, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 359–377. [Google Scholar] [CrossRef]
- Cox, K.H.; Takahashi, J.S. Circadian clock genes and the transcriptional architecture of the clock mechanism. J. Mol. Endocrinol. 2019, 63, R93–R102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buhr, E.D.; Takahashi, J.S. Molecular Components of the Mammalian Circadian Clock. In Circadian Clocks; Kramer, A., Merrow, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 3–27. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2017, 18, 164–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toloza-Villalobos, J.; Arroyo, J.I.; Opazo, J.C. The Circadian Clock of Teleost Fish: A Comparative Analysis Reveals Distinct Fates for Duplicated Genes. J. Mol. Evol. 2015, 80, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Bolton, C.M.; Bekaert, M.; Eilertsen, M.; Helvik, J.V.; Migaud, H. Rhythmic Clock Gene Expression in Atlantic Salmon Parr Brain. Front. Physiol. 2021, 12, 761109. [Google Scholar] [CrossRef]
- Martins, R.S.T.; Gomez, A.; Zanury, S.; Carrillo, M.; Canario, A.V.M. Photoperiodic Modulation of Circadian Clock and Reproductive Axis Gene Expression in the Pre-Pubertal European Sea Bass Brain. PLoS ONE 2015, 10, e0144158. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly Contiguity | ||
|---|---|---|
| Scale | Contig | Scaffold |
| Total Size (bp) | 606,099,673 | 606,289,673 |
| Number of Fragments | 406 | 26 |
| Largest Fragment (bp) | 19,993,184 | 37,057,500 |
| N50 (bp) | 7,582,354 | 26,674,500 |
| L50 | 27 | 11 |
| Number of Chromosome-Scale Scaffolds | -- | 24 |
| Total Bases in Chromosomes (bp) | -- | 606,192,173 |
| Percent of Assembly in Chromosomes | -- | 99.98% |
| BUSCO v5.3.1 Gene Completeness | ||
| Complete | 3513 (96.5%) | |
| Complete and Single Copy | 3476 (95.5%) | |
| Complete and Duplicated | 37 (1.0%) | |
| Fragmented | 12 (0.3%) | |
| Missing | 115 (3.2%) | |
| Total | 3640 | |
| Reference Gene Set | actinopterygii_odb10 | |
| Gene Annotation | ||
| Number of Protein Coding Genes | 25,081 | |
| Repeat Annotation | ||
| Total Repeats | 33.18% | |
| LTR | 2.92% | |
| SINE | 0.64% | |
| LINE | 5.06% | |
| DNA | 15.84 | |
| Unclassified | 5.72% | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, C.-H.C.; Rivera-Colón, A.G.; Minhas, B.F.; Wilson, L.; Rayamajhi, N.; Vargas-Chacoff, L.; Catchen, J.M. Chromosome-Level Genome Assembly and Circadian Gene Repertoire of the Patagonia Blennie Eleginops maclovinus—The Closest Ancestral Proxy of Antarctic Cryonotothenioids. Genes 2023, 14, 1196. https://doi.org/10.3390/genes14061196
Cheng C-HC, Rivera-Colón AG, Minhas BF, Wilson L, Rayamajhi N, Vargas-Chacoff L, Catchen JM. Chromosome-Level Genome Assembly and Circadian Gene Repertoire of the Patagonia Blennie Eleginops maclovinus—The Closest Ancestral Proxy of Antarctic Cryonotothenioids. Genes. 2023; 14(6):1196. https://doi.org/10.3390/genes14061196
Chicago/Turabian StyleCheng, Chi-Hing Christina, Angel G. Rivera-Colón, Bushra Fazal Minhas, Loralee Wilson, Niraj Rayamajhi, Luis Vargas-Chacoff, and Julian M. Catchen. 2023. "Chromosome-Level Genome Assembly and Circadian Gene Repertoire of the Patagonia Blennie Eleginops maclovinus—The Closest Ancestral Proxy of Antarctic Cryonotothenioids" Genes 14, no. 6: 1196. https://doi.org/10.3390/genes14061196